

Abstract
The amide III region of the peptide infrared and Raman spectra has been used to determine the relative populations of the three major backbone conformations (PII, β, and αR) in 19 amino acid dipeptides. The results provide a benchmark for force field or other methods of predicting backbone conformations in flexible peptides. There are three resolvable backbone bands in the amide III region. The major population is either PII or β for all dipeptides except Gly, whereas the αR population is measurable but always minor (≤ 10%) for 18 dipeptides. (The Gly φ,ψ map is complex and so is the interpretation of the amide III bands of Gly.) There are substantial differences in the relative β and PII populations among the 19 dipeptides. The band frequencies have been assigned as PII, 1,317–1,306 cm-1; αR, 1,304–1,294 cm-1; and β, 1,294–1,270 cm-1. The three bands were measured by both attenuated total reflection spectroscopy and by Raman spectroscopy. Consistent results, both for band frequency and relative population, were obtained by both spectroscopic methods. The β and PII bands were assigned from the dependence of the 3J(HN,Hα) coupling constant (known for all 19 dipeptides) on the relative β population. The PII band assignment agrees with one made earlier from Raman optical activity data. The temperature dependences of the relative β and PII populations fit the standard model with Boltzmann-weighted energies for alanine and leucine between 30 and 60 °C.
Keywords: vibrational spectroscopy, backbone conformations, spectral populations, aqueous solution
A basic unsolved problem in protein folding is making accurate calculations of the folding energetics of flexible peptides. Accurate experimental results for the major backbone conformations are needed to test prediction methods. Even the simple problem of calculating the φ,ψ map of the alanine dipeptide is beyond the reach of standard force fields used in molecular dynamics simulations (1). There are two probable reasons: one is that the energy differences between the major backbone conformations are small, and the second is that standard force fields contain so many parameters that errors cannot be found readily by comparing simulations with experimental results. The ability to calculate accurately the relative energies of the various backbone conformations is needed for simulating early stages of the protein folding process, which is an important current problem in molecular biophysics.
When simulated by standard force fields, the φ,ψ map of the alanine dipeptide has three major conformational basins, PII, β, and αR. Different force fields agree on this point but give widely varying results for the populations in the three basins (1). The φ,ψ maps of the 19 amino acid residues (Pro excluded) from the protein structure database show the same three basins and are similar in outline for the various residues if data for Gly and Pre-Pro are excluded in addition to Pro (2). The three basins are centered approximately at PII (-75°, 145°), β (-120°, 120°), and αR (-60°, -40°). However, quantitative differences in relative populations among the amino acid residues are evident. The “Coil Library” of residue conformations outside helices and sheets (3–5) also contains regions outside the three major basins and specific structures, minor in amounts, have been dissected (6).
Our strategy for determining experimentally the populations of the three major basins in short flexible peptides is based on the following two premises. First, the single-residue dipeptides (N-acetyl-X-N′-methylamide) provide the optimal starting point because they represent the simplest system. The 3J(HN,Hα) coupling constants of 19 dipeptides (Pro excluded) have been measured (7) and the dipeptide coupling constants are the same as those of the central residue in GGXGG peptides when the latter values are measured in 6 M guanidinium chloride (GdmCl) (8) and when results are excluded for four polar residues able to make side-chain–main-chain H bonds. Quantitative differences are expected between dipeptide coupling constants and those of longer peptides because there is a substantial neighboring residue effect on backbone conformation in flexible peptides (9–11). The second premise of our study is that conformer populations must be measured by a fast spectroscopic method so that conformational averaging over different conformers does not occur. The infrared and Raman methods used here have a timescale < 0.1 ps (12), whereas backbone conformer lifetimes lie in the range 10–200 ps (12).
A preliminary study (13) showed that the amide III region of the peptide group, observed by vibrational spectroscopy, is a promising choice for determining backbone conformers. The amide III region contains three resolvable bands that arise from different backbone conformers, as shown earlier (13) and confirmed here. Preliminary values of the conformer populations were obtained earlier by attenuated total reflection (ATR) spectroscopy for Ala and Val in a study of 13 dipeptides (13). The weak absorption in the amide III region is compensated by using ATR spectroscopy to increase sensitivity (13). To obtain the absorbance spectrum by ATR spectroscopy, it is necessary to eliminate the reflectance contribution to the ATR spectrum; the procedure was described earlier (13). After this correction is made, the resulting spectrum is termed the ATR-absorbance spectrum. Absorption in the amide III region results chiefly from N-H in-plane bending but also mixing with C-C and C-N stretching, C═O in-plane bending and Cα-H bending (13).
Our approach is to resolve and assign the three bands (A, B, C) within the amide III region that meet criteria described below for being equivalent in both ATR and Raman spectra; these three bands can be assigned to three backbone conformers, PII, β, and αR. Accurate fitting is required to resolve the A, B, C bands and more than one fitting is usually possible because side chains contribute additional, overlapping bands. The relative intensities of the side-chain and backbone bands are different in the ATR-absorbance and Raman spectra, and this property is used to subtract the side-chain bands. The band shapes of the backbone conformers are different in the ATR-absorbance and Raman spectra because different selection rules apply in infrared and Raman spectra. The shape and intensity of a band reflect the broken selection rules and lower symmetry found in solution compared with the gas phase.
Our fitting procedure requires that each of the A, B, C bands has closely the same frequency in the fitted ATR-absorbance and Raman spectra. This requirement allows choosing a unique pair of ATR and Raman fittings for all but a few dipeptides. Chiefly because of low solubility, ATR results are missing for Trp and Phe, whereas Raman results are missing for Glu-, Gln, Asp0, and Asp- (Table 1). For the other dipeptides, the match between the populations of the A, B, C conformers obtained from the ATR and Raman spectra is used as a test of goodness of fit. The 3J(HN,Hα) coupling constants of the 19 dipeptides have been measured (7) and their values are determined chiefly by the relative populations of PII and β, which are the two major conformers. This property allows assignment of the main bands A and C to PII and β, based on comparison between the dipeptide coupling constants and the relative populations in bands A and C.
Table 1.
Fractional populations of the three backbone conformations obtained by fitting Raman and ATR-absorbance spectra
| Infrared | Raman | |||||
| f(PII) | f(αR) | f(β) | f(PII) | f(αR) | f(β) | |
| Gly | 0.22 | 0.66 | 0.12 | 0.22 | 0.63 | 0.15 |
| Ala | 0.60 | 0.11 | 0.29 | 0.62 | 0.09 | 0.29 |
| Lys | 0.55 | 0.04 | 0.41 | 0.54 | 0.05 | 0.41 |
| Arg | 0.54 | 0.07 | 0.39 | 0.53 | 0.06 | 0.41 |
| Leu | 0.55 | 0.10 | 0.35 | 0.56 | 0.10 | 0.34 |
| Trp | — | — | — | 0.54 | 0.02 | 0.44 |
| Met | 0.50 | 0.03 | 0.47 | 0.53 | 0.04 | 0.43 |
| Ser | 0.49 | 0.04 | 0.47 | 0.50 | 0.04 | 0.46 |
| Glu0 | 0.47 | 0.05 | 0.48 | 0.46 | 0.05 | 0.49 |
| Glu- | 0.59 | 0.05 | 0.36 | — | — | — |
| Gln | 0.44 | 0.08 | 0.48 | — | — | — |
| Tyr | 0.46 | 0.07 | 0.47 | 0.46 | 0.07 | 0.47 |
| Phe | — | — | — | 0.45 | 0.06 | 0.49 |
| Val | 0.47 | 0.02 | 0.51 | 0.47 | 0.02 | 0.51 |
| Cys | 0.43 | 0.03 | 0.54 | 0.41 | 0.04 | 0.55 |
| Ile | 0.46 | 0.02 | 0.52 | 0.46 | 0.02 | 0.52 |
| Thr | 0.39 | 0.03 | 0.58 | 0.39 | 0.05 | 0.56 |
| Asn | 0.40 | 0.02 | 0.58 | 0.38 | 0.03 | 0.59 |
| Asp0 | 0.43 | 0.02 | 0.55 | — | — | — |
| Asp- | 0.49 | 0.05 | 0.46 | — | — | — |
| His | 0.38 | 0.04 | 0.58 | 0.39 | 0.04 | 0.57 |
Glu- and Asp- are measured at pH 4.9, and Glu0 and Asp0 are measured at pH 2.9.
Results and Discussion
Comparison Between ATR-Absorbance and Raman Results for the Band Frequencies and Relative Populations of the A, B, C Bands.
Fig. 1 compares, for four dipeptides, the fitted A, B, C bands found by ATR and Raman spectroscopy. The corresponding fittings for the other 15 dipeptides are given in Fig. S1. The proline dipeptide was not studied because its φ backbone angle is fixed by the chemical linkage of the proline ring. Results for Glu and Asp depend on the extent of ionization and results for both pH 2.9 (E0, D0) and pH 4.9 (E-, D-) are given. The relative populations obtained from the ATR-absorbance and Raman fittings are given in Table 1 and the band frequencies are given in Table S1.
Fig. 1.

Fitting the ATR-absorbance (Upper) and Raman (Lower) spectra (amide III region) for four dipeptides: A, glycine; B, arginine; C, methionine; and D, isoleucine. The black line is the experimental spectrum, the red line is the fitted curve, and the blue line is the difference between the experimental and fitted curves. The three colored components are gray (band A) PII; blue (band B) αR; red (band C) β. The uncolored components represent side-chain vibrational bands and main-chain vibrational bands that are independent on backbone conformation.
Bands A, B, and C are found in characteristic segments of the amide III region (Fig. 2): Band A occurs within 1,317–1,306 cm-1, band B is within 1,304–1,294 cm-1, and band C is within 1,294–1,270 cm-1. For band A, the band frequencies differ by only ≤ 1 cm-1 between the ATR-absorbance and Raman fittings whereas, for bands B and C, slightly larger differences (≤ 3 cm-1) between the ATR-absorbance and Raman fittings are found. (However, in one case—the Ala dipeptide—the ATR and Raman fittings differ by 7 cm-1.) Bands A and C have large populations whereas band B has a minor population, typically ≤ 7% but larger for Ala and Leu, 10%. In protein structures, Gly has a different φ,ψ map from the other amino acid residues (2) and for the Gly dipeptide, band B is the major band; we do not attempt to assign it to conformers. The relative population of each conformer agrees between the ATR-absorbance and Raman fittings within 2% for both bands A and B and within 4% for band C (Table 1). This good overall agreement between ATR-absorbance and Raman results indicates that the fittings are reliable: See discussion of errors below.
Fig. 2.

Diagram of the amide III regions in which the three backbone conformer bands [PII (band A), αR (band B), and β (band C)] are found for the 19 dipeptides.
Use of NMR Coupling Constants to Assign Bands A and C.
The 3J(HN,Hα) coupling constant is determined by the backbone angle φ; the measured value for a dipeptide is an average over all conformers present. The value of 3J(HN,Hα) varies widely among the 19 dipeptides (7) because the (PII/β) ratio varies widely. As a result, the known values of 3J(HN,Hα) for the 19 dipeptides are used here to assign the two major bands (A and C) to PII and β. Values of 3J(HN,Hα) for the PII (φ ∼ -75°) and β conformers (φ ∼ -120°) differ almost by a factor of 2, whereas the minor αR (φ ∼ -60°) conformer has a value of 3J similar to PII. Consequently, the plot of 〈3J〉 values for individual dipeptides versus the fraction of β conformer, f(β), is expected to be approximately linear:
| [1] |
The difference between values of 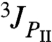 and
and  is neglected because the difference between them is small; moreover, the αR conformer has only a minor relative population.
is neglected because the difference between them is small; moreover, the αR conformer has only a minor relative population.
This coupling constant test (Fig. 3) shows that band C contains the β conformer. A good straight line (R = 0.942) for Eq. 1 is found when values of f(β) are taken from band C and 〈3J〉 values are taken from ref. 7. The intercept values of 〈3J〉 at f(β) = 1.00 and 0 agree with expected values whereas linearity of the plot tests the basic assumption that each major band (A, C) arises from either the PII or β backbone conformer. The value of 〈3J〉 at f(β) = 1.00 (9.3 Hz) is within the range 8.5–9.6 Hz prescribed for the β conformer whereas the value of 〈3J〉 at f(β) = 0 (5.1 Hz) is close to the range 5.2–5.8 Hz for the PII conformer described below. The average measured value of 3J(HN,Hα) for β conformers in BioMagResBank data is 8.5 Hz, whereas the average value found by the Karplus relation from φ-values of residues in a Coil Library is 9.6 Hz (7). The average value of 3J(HN,Hα) for PII is 5.2 Hz in BioMagResBank data, whereas 5.8 Hz is the average value found by using the Karplus relation with data from a Coil Library. The band frequency values for a given backbone conformer vary among the 19 dipeptides (Table S1) within a narrow range (Fig. 2).
Fig. 3.

Plot of the measured 〈3J(Hα,HN)〉 coupling constants of the 19 dipeptides, from ref. 7, versus the relative population of the β-conformer, taken from the average of the ATR-absorbance and Raman values.
The PII band frequency in the amide III region has been assigned earlier by others. Because the PII conformation is helical, it gives a strong Raman optical activity (ROA) signal. Thus, ROA has been used to assign PII in the amide III region to 1,319 cm-1 for disordered poly-l-glutamate (14), to 1,310 cm-1 for cationic (Ala)4, and to 1,312 cm-1 for cationic (Ala)5 (14). These values agree satisfactorily with our assignment of PII in dipeptides to the segment 1,317–1,306 cm-1. The band frequencies for PII, αR, and β shown in Fig. 2 and Table S1 do not support the proposal (15) that amide III frequencies of backbone conformers depend chiefly on their ψ-values and only slightly on their φ-values: The band frequencies of PII and β, which have similar ψ-values but different φ-values, are the farthest apart, whereas the band frequency of αR, which has a different ψ-value, is in between PII and β.
Effect of Temperature.
ATR-absorbance spectra of the Ala and Leu dipeptides were recorded as a function of temperature between 30 and 80 °C and the relative populations of the three conformers are shown in Table 2. For both Ala and Leu, the population of PII (the major conformer) decreases at the high temperature. The results can be compared with the Boltzmann-weighted model in which the fraction of conformer i, fi, is given by its energy Ei relative to the most stable conformer
| [2] |
T is temperature (kelvin), k is Boltzmann’s constant, and the sum is over the three backbone conformers. If the values of Ei are independent of temperature, then the expected temperature dependences for the three conformers can be calculated from the observed populations at 30 °C.
Table 2.
Temperature dependence of the fractional populations of PII, αR, and β conformations compared with predicted values
| Temp, °C | Alanine | Leucine | ||||
| f(PII) | f(αR) | f(β) | f(PII) | f(αR) | f(β) | |
| 30 | 0.60 (0.60) | 0.11 (0.11) | 0.29 (0.29) | 0.55 (0.55) | 0.10 (0.10) | 0.35 (0.35) |
| 40 | 0.60 (0.59) | 0.10 (0.12) | 0.30 (0.29) | 0.55 (0.54) | 0.09 (0.10) | 0.36 (0.35) |
| 50 | 0.58 (0.59) | 0.10 (0.12) | 0.32 (0.30) | 0.53 (0.54) | 0.10 (0.11) | 0.37 (0.35) |
| 60 | 0.58 (0.58) | 0.09 (0.12) | 0.33 (0.30) | 0.52 (0.53) | 0.11 (0.11) | 0.37 (0.35) |
| 70 | 0.55 (0.57) | 0.11 (0.13) | 0.34 (0.30) | 0.49 (0.53) | 0.12 (0.12) | 0.39 (0.35) |
| 80 | 0.54 (0.57) | 0.12 (0.13) | 0.34 (0.30) | 0.50 (0.52) | 0.11 (0.12) | 0.39 (0.36) |
Measured values are taken from the ATR-absorbance spectra. Predicted values (values in brackets) are found from the Boltzmann-weighted model (Eq. 2) using the data at 30 °C to find the relative Ei values.
This model (Table 2) gives predicted values that fit the data for f(PII) and f(β) within the estimated experimental error (see below) for the Ala and Leu dipeptides between 30 and 60 °C but the predicted values fall below the experimental values of f(PII) at 70 and 80 °C (Table 2). Further work is needed to determine if the discrepancy between the model and experiment at 70 and 80 °C is real or arises from an experimental difficulty at high temperatures. Thus, the Boltzmann-weighted model with constant Ei values (Eq. 2) gives a quantitatively correct explanation between 30 and 60 °C for why f(PII) decreases and f(β) increases with temperature (note that PII is the major species). A decrease in f(PII) and an increase in f(β) with increasing temperature was deduced earlier for short Ala peptides (2–7 Ala residues) from measurements of the 3J(HN,Hα) coupling constant versus temperature (16).
Effects of Solvent and Ionization State.
Earlier studies found large changes in the values of 3J(HN,Hα) for dipeptides when the solvent was changed from water to a nonaqueous solvent (7, 17, 18). Consequently, we determined the values of f(β) and f(PII) from the ATR-absorbance spectrum of the Ala dipeptide in DMSO at 30 °C (Fig. S2). A major decrease in f(PII) from 0.60 in water to 0.31 in DMSO was found, with an accompanying increase in f(β) from 0.29 in water to 0.58 in DMSO. The value of f(αR) remained the same (0.11) in DMSO as in water. The band frequencies of the three conformers in DMSO (1,313 cm-1 for PII, 1,277 cm-1 for β, and 1,296 cm-1 for αR) fell within the same ranges as in water (see above). The value of the 〈3J(HN,Hα)〉 coupling constant of the Ala dipeptide in DMSO (7.6 Hz, room temperature) (17) was substantially larger than the value in water (6.06 Hz, 30 °C) (7) and the increase was attributed to a major increase in f(β) (17). Similar large increases in the value of 3J(HN,Hα) in DMSO were found also for other dipeptides (18).
Likewise, large changes in the values of 3J(HN,Hα) for Asp and Glu were found when the pH was changed from 2.9 (∼nonionized) to 4.9 (∼ionized) (7). Consequently the values of f(β) and f(PII) were measured for Asp and Glu at pH 2.9 and at pH 4.9 by both ATR-absorbance and Raman spectroscopy (Table 1). When plotted as f(β) versus 3J(HN,Hα) (Fig. 4), the results show the correlation expected from the known strong dependence of 3J(HN,Hα) on the relative population of the β backbone conformer. The success of this correlation provides a check on the correctness of the analysis of relative populations.
Fig. 4.

Correlation between 〈3J(Hα,HN)〉 and f(β) for Glu and Asp at two pH values (2.9 and 4.9) at which the degree of ionization changes and causes a large change in the value of each variable. The line is taken from Fig. 3.
Molar Absorption Coefficients.
Measurement of molar absorption coefficients is rarely attempted in peptide vibrational spectroscopy and the relative intensities of the various conformer bands are usually regarded as a direct measure of the relative conformer populations. We measured the molar absorption coefficients to learn if there are measurable differences among the various dipeptides and, if so, why. An obvious possible cause is that PII and β might have different molar absorption coefficients. The PII/β population ratio varies from 60/29 (ATR-absorbance values) for Ala to 38/58 for His at 30 °C. Because the band shapes differ among the A, B, C conformers and among the 19 dipeptides, we determined the integrated molar absorption coefficient (IMAC) given by the area under the curve of absorbance versus frequency. The results show no significant variation in IMAC values among the 19 dipeptides (Table S1); the SD is 6.2%.
Estimation of Errors and Comparison with Literature Results.
In both the ATR-absorbance spectroscopy and Raman spectroscopy methods used here, the major source of error is in choosing the correct fitting of each spectrum. In making this choice, the main problem is to remove the side-chain vibrational bands. For the ATR-absorbance method, it is also necessary to remove the reflectance contribution from the measured ATR spectrum. For the Raman spectroscopy method, it is also necessary to take account of any fluorescence background in choosing the correct baseline. The side-chain vibrational bands have different intensities in the ATR-absorbance and Raman spectra and they can be identified by comparing the two spectra. The band frequencies of the three backbone conformers are required to be closely the same in the Raman and ATR-absorbance fittings in order to decide between different possible fittings of the Raman and ATR-absorbance spectra. (The Raman and ATR-absorbance band frequencies are compared in Table S1.) Then the errors associated with the values for the relative populations are estimated by comparing the values given by the ATR-absorbance and Raman methods. The two fitting methods are independent of each other because the relative intensity contributions from the side-chain vibrational bands are different in the ATR-absorbance and Raman spectra. Also, the band shapes are different in the ATR-absorbance and Raman spectra because the selection rules are different.
When these two relationships between fitting the ATR-absorbance and Raman spectra are put in place (constraining the band frequencies to be the same and identifying the side-chain bands by their different relative intensities), then the relative populations of the three conformers found by the ATR-absorbance and Raman methods are remarkably consistent (Table 1). There are 14 cases in Table 1 where the relative populations can be compared between the Raman and ATR-absorbance results. (The results for Gly are not considered here because its φ,ψ map cannot be divided into the same three conformer basins as those of the other amino acid residues.) The two values of f(β) agree within 0.00 in six cases, 0.01 in seven cases, and 0.02 in one case; the two values of f(PII) agree within 0.00 in five cases, within 0.01 in seven cases, and within 0.02 or 0.03 in one case each; and the two values of f(αR) agree within 0.00 in seven cases and within 0.01 in seven cases. Thus, the relative population of each conformer is determined consistently within about 0.01, or 2% for f(β) and 2% for f(PII), but 10–20% for f(αR).
The only other results, as far as we know, for resolved vibrational bands of backbone conformers have been reported for Raman skeletal vibrations in the region 810–950 cm-1 (13, 19). Results for the Ala dipeptide from two laboratories are in good agreement with each other, both in regard to band frequencies and relative populations. The minor αR band is well resolved from the major PII band, but the minor β-band overlaps the PII band and is resolved only by curve fitting. The relative populations are αR, 0.18 (13), 0.17 (19); PII, 0.76 (13), 0.79 (19); β, 0.06 (13), 0.06 (19); the values from ref. 19 were obtained from their published spectra. A minor band (0.08) attributed to C7eq was also reported (19). However, the relative populations found here (and previously; ref. 13) by ATR-absorbance (αR 0.11, PII 0.60, β 0.29) differ significantly, as reported earlier (13). The reason remains to be found; a possible explanation is that the Raman scattering coefficients have different values for the various conformers. This argument is not likely to apply, however, to the Raman scattering results in the amide III region reported here, because the same relative populations were obtained by Raman scattering and ATR-absorbance, and no significant differences among the absorptivity coefficients of the 19 dipeptides (IMAC values, Table S1) were found.
Most earlier studies of the relative populations of backbone conformers in short peptides have relied on measured properties that are averages over all conformers present and on interpreting these average values with the aid of additional spectroscopic data. Thus, 3J(HN,Hα) coupling constants were used to show that short alanine peptides have predominantly the PII conformer at 0 °C but that partial conversion to the β-conformer occurs between 0 and 60 °C, and far-UV CD spectroscopy was used to confirm both points (16). No direct test for the presence of a minor αR conformer could be made using these techniques. Different kinds of vibrational spectroscopy have been used in combination to analyze the mixed spectra that result when bands from individual backbone conformers are not resolved but instead are superimposed (20, 21). Hopefully this type of analysis can be tested in future work by comparison with the dipeptide results reported here. There are large differences between the relative populations reported here and results (chiefly for the Ala and Gly dipeptides) reported from various molecular dynamics simulations, but the underlying problem remains to be addressed that the energy differences between conformers are probably no larger than the errors of standard force fields (1).
Related work was reviewed briefly in 2008 (13). New papers have come out in 2009 (20) and also 2010 (21) that assess the conformational preferences of the various amino acid residues in short peptides by combining information either from different types of vibrational spectroscopy or from including also NMR coupling constants, and these papers give references to other recent work.
Materials and Methods
General properties of the ATR-absorbance and Raman spectroscopy methods used here have been described previously (13) and additional information is given in SI Materials and Methods.
Supplementary Material
Acknowledgments.
This work was supported by a grant from the Slovenian Research Agency. We thank Neville Kallenbach and Robert Woody for discussion.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1017317108/-/DCSupplemental.
References
- 1.Hu H, Elstner M, Hermans J. Comparison of a QM/MM force field and molecular mechanics force fields in simulations of alanine and glycine dipeptides (Ace-Ala-Nme and Ace-Gly-Nme) in water in relation to the problem of modeling the unfolded peptide backbone in solution. Proteins: Struct, Funct, Genet. 2003;50:451–463. doi: 10.1002/prot.10279. [DOI] [PubMed] [Google Scholar]
- 2.Lovell SC, et al. Structure validation by Cα geometry: φ, ψ and Cβ deviation. Proteins: Struct, Funct, Genet. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 3.Avbelj FA, Baldwin RL. Role of backbone solvation and electrostatics in generating preferred peptide backbone conformations: Distributions of phi. Proc Natl Acad Sci USA. 2003;100:5742–5747. doi: 10.1073/pnas.1031522100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jha AK, et al. Helix, sheet and polyproline II frequencies and strong nearest-neighbor effects in a restricted coil library. Biochemistry. 2005;44:9691–9702. doi: 10.1021/bi0474822. [DOI] [PubMed] [Google Scholar]
- 5.Fitzkee NC, Fleming PJ, Rose GD. The Protein Coil Library: A structural database of nonhelix, nonstrand fragments derived from the PDB. Proteins: Struct, Funct, Bioinf. 2005;58:852–854. doi: 10.1002/prot.20394. [DOI] [PubMed] [Google Scholar]
- 6.Perskie LL, Street TO, Rose GD. Structures, basins and energies: A deconstruction of the Protein Coil Library. Protein Sci. 2008;17:1151–1161. doi: 10.1110/ps.035055.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Avbelj F, Golic Grdadolnik S, Grdadolnik J, Baldwin RL. Intrinsic backbone preferences are fully present in blocked amino acids. Proc Natl Acad Sci USA. 2006;103:1272–1277. doi: 10.1073/pnas.0510420103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Plaxco KW, et al. The effects of guanidine hydrochloride on the “random coil” conformations and NMR chemical shifts of the peptide series GGXGG. J Biomol NMR. 1997;10:221–230. doi: 10.1023/A:1018340217891. [DOI] [PubMed] [Google Scholar]
- 9.Penkett CJ, et al. NMR analysis of main-chain conformational preferences in an unfolded fibronectin-binding protein. J Mol Biol. 1997;274:152–159. doi: 10.1006/jmbi.1997.1369. [DOI] [PubMed] [Google Scholar]
- 10.Avbelj F, Baldwin RL. Origin of the neighboring residue effect on peptide backbone conformation. Proc Natl Acad Sci USA. 2004;101:10967–10972. doi: 10.1073/pnas.0404050101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen K, Liu Z, Zhou C, Shi Z, Kallenbach NR. Neighbor effect on PPII conformation in alanine peptides. J Am Chem Soc. 2005;127:10146–10147. doi: 10.1021/ja052094o. [DOI] [PubMed] [Google Scholar]
- 12.Mu Y, Kosov DS, Stock G. Conformational dynamics of trialanine in water. 2. Comparison of AMBER, CHARMM, GROMOS and OPLS force fields to NMR and infrared experiments. J Phys Chem B. 2003;107:5064–5073. [Google Scholar]
- 13.Grdadolnik J, Golic Grdadolnik S, Avbelj F. Determination of conformational preferences of dipeptides using vibrational spectroscopy. J Phys Chem B. 2008;112:2712–2718. doi: 10.1021/jp7096313. [DOI] [PubMed] [Google Scholar]
- 14.McColl IH, et al. Vibrational Raman optical activity characterization of poly (L-proline) II helix. J Am Chem Soc. 2004;126:5076–5077. doi: 10.1021/ja049271q. [DOI] [PubMed] [Google Scholar]
- 15.Mikhonin AV, Bykov SV, Myshkina NS, Asher SA. Peptide secondary structure folding reaction coordinate: Correlation between UV Raman amide III frequency, ψ Ramachandran angle, and hydrogen bonding. J Phys Chem B. 2006;110:1928–1943. doi: 10.1021/jp054593h. [DOI] [PubMed] [Google Scholar]
- 16.Shi Z, Olson CA, Rose GD, Baldwin RL, Kallenbach NR. Polyproline II structure in a sequence of seven alanine residues. Proc Natl Acad Sci USA. 2002;99:9190–9195. doi: 10.1073/pnas.112193999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Madison V, Kopple KD. Solvent-dependent conformational distributions of some dipeptides. J Am Chem Soc. 1980;102:4855–4863. [Google Scholar]
- 18.Fermandjian S, et al. Local interactions in peptides: H-H and 13C-H coupling constants for the conformational analysis of N-acetyl-N′-methylamides of aliphatic amino acids. Int J Pept Protein Res. 1990;35:473–480. [PubMed] [Google Scholar]
- 19.Takekiyo T, Imai T, Kato M, Taniguchi Y. Temperature and pressure effects on conformational equilibria of alanine dipeptide in aqueous solution. Biopolymers. 2004;73:283–290. doi: 10.1002/bip.10548. [DOI] [PubMed] [Google Scholar]
- 20.Schweitzer-Stenner R. Distribution of conformations sampled by the central amino acid residue in tripeptides inferred from amide I band profiles and NMR scalar coupling constants. J Phys Chem B. 2009;113:2922–2932. doi: 10.1021/jp8087644. [DOI] [PubMed] [Google Scholar]
- 21.Hagarman A, Measy TJ, Mathieu D, Schwalbe H, Schweitzer-Stenner R. Intrinsic propensities of amino acid residues in GxG peptides inferred from amide I′ band profiles and NMR scalar coupling constants. J Am Chem Soc. 2010;132:540–551. doi: 10.1021/ja9058052. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.