

Abstract
Enterotoxigenic Bacteroides fragilis is the most frequent disease-causing anaerobe in the intestinal tract of humans and livestock and its specific virulence factor is fragilysin, also known as B. fragilis toxin. This is a 21-kDa zinc-dependent metallopeptidase existing in three closely related isoforms that hydrolyze E-cadherin and contribute to secretory diarrhea, and possibly to inflammatory bowel disease and colorectal cancer. Here we studied the function and zymogenic structure of fragilysin-3 and found that its activity is repressed by a ∼170-residue prodomain, which is the largest hitherto structurally characterized for a metallopeptidase. This prodomain plays a role in both the latency and folding stability of the catalytic domain and it has no significant sequence similarity to any known protein. The prodomain adopts a novel fold and inhibits the protease domain via an aspartate-switch mechanism. The catalytic fragilysin-3 moiety is active against several protein substrates and its structure reveals a new family prototype within the metzincin clan of metallopeptidases. It shows high structural similarity despite negligible sequence identity to adamalysins/ADAMs, which have only been described in eukaryotes. Because no similar protein has been found outside enterotoxigenic B. fragilis, our findings support that fragilysins derived from a mammalian adamalysin/ADAM xenolog that was co-opted by B. fragilis through a rare case of horizontal gene transfer from a eukaryotic cell to a bacterial cell. Subsequently, this co-opted peptidase was provided with a unique chaperone and latency maintainer in the time course of evolution to render a robust and dedicated toxin to compromise the intestinal epithelium of mammalian hosts.
Keywords: bacterial endotoxin, human pathogen, zymogen activation
The gastrointestinal tract is that part of the interface between the organism and its external environment where food digestion and nutrient uptake occur. The tract hosts bacteria that are beneficial for the host by controlling invasion and proliferation of pathogens, enhancing the immune system, processing indigestible food and providing essential nutrients. The most populated section of the tract is the large intestine, which is anaerobic and contains ten times more bacterial cells than the number of human cells in the entire body (1). However, in certain circumstances, this beneficial relationship can be disrupted and pathogenic bacteria can invade and proliferate, causing a number of disturbances. Members of the genus Bacteroides comprise the majority of intestinal obligate anaerobes, of which Bacteroides fragilis is most frequently associated with disease. Enterotoxigenic B. fragilis (ETBF) strains colonize and affect humans and livestock, and they have been linked to intraabdominal abscesses, diarrhea, inflammatory bowel disease, anaerobic bacteremia, and colon cancer (1–3). In addition to the bacterial capsule, which induces abscess formation, the only identified virulence factor for ETBF is a 21-kDa zinc-dependent metallopeptidase (MP), termed fragilysin alias B. fragilis toxin (BFT) (4–6). It is synthesized as a preproprotein of 397 residues, with an 18-residue signal peptide for secretion, a ∼170-residue prodomain (PD) flanked by flexible segments, and a ∼190-residue catalytic domain (CD). The latter encompasses two sequence elements that ascribe it to the metzincin clan of MPs: (i) an extended zinc-binding consensus sequence (ZBCS), HEXXHXXG/NXXH/D, which comprises three histidines that bind the catalytic zinc ion plus the general base/acid glutamate involved in catalysis; and (ii) a conserved methionine within a tight 1,4-β-turn, the Met-turn (5–8). However, upstream of the ZBCS there is no significant sequence similarity to any other metzincin, which suggests that fragilysin is a unique metzincin prototype (7).
The enzyme exists in three closely related isoforms of identical length: fragilysin-1, -2, and -3 alias BFT-1, -2, and -3, which display pairwise sequence identities of 93–96% (9). Analysis of clinical isolates reveals that the three isoforms are generally present simultaneously (2) and that fragilysin-1 is the most abundant (see table 9 in ref. 2). The three fragilysin isoforms are encoded by a chromosomal pathogenicity islet that is absent in nonenterotoxigenic strains. In addition to fragilysin, this island contains a second gene, mpII, which is countertranscribed and encodes a potential MP of similar size and moderate sequence identity (28–30%) to the three fragilysin isoforms. However, its potential role in ETBF pathogenesis remains to be established (2).
The only proven substrate for fragilysin-1 in vivo is E-cadherin, an intercellular adhesion molecule. Shedding of E-cadherin by fragilysin-1 led to increased permeability of the epithelium and, ultimately, cell proliferation, which supported a role for this MP in colorectal carcinoma (10). In vitro, fragilysin-1 was shown to cleave type-IV collagen, gelatin, actin, fibrinogen, myosin, tropomyosin, human complement C3, and α1-proteinase inhibitor (5, 6). The protein is stable at room temperature and below, but it undergoes rapid autodigestion above 37 °C. Fragilysin-3, alias BFT-3 and BFT-Korea, was shown to cleave E-cadherin in HT29/C1 cells similarly to isoforms 1 and 2 (9), but no further biochemical studies have been reported.
Orally administered broad-spectrum antibiotics may remove enteropathogens from the gastrointestinal tract but they also affect the beneficial and commensal flora. In the absence of this flora, opportunistic microorganisms may colonize the intestine and lead to severe digestion alterations and gastrointestinal diseases. In addition, ETBF can be resistant to antibiotics such as penicillin, ampicillin, clindamycin, tetracycline, and metronidazole (2). Accordingly, there is a substantiated need for better understanding ETBF targets such as fragilysin and for the design of highly specific antimicrobials to tackle them, and detailed structural information greatly contributes to these aims. To explore the mechanisms of fragilysin activation and activity, we examined the proteolytic capacity of fragilysin-3 in vitro and the X-ray crystal structure of its zymogen, profragilysin-3, which provided a high-resolution scaffold for the design of specific inhibitors. Taken together with the results of phylogenetic studies, these data enabled us to propose a mechanism for latency maintenance and activation of this enterotoxigenic MP, as well as a plausible hypothesis for its evolutionary origin based on xenology (from  , Greek for “stranger” or “alien”), which is gene homology that is the result of horizontal gene transfer (HGT) between species and not of Darwinian evolution.
, Greek for “stranger” or “alien”), which is gene homology that is the result of horizontal gene transfer (HGT) between species and not of Darwinian evolution.
Results and Discussion
Autolytic vs. Heterolytic Activation.
Wild-type profragilysin-3 was recombinantly overexpressed in Escherichia coli and purified to homogeneity (Fig. S1A). Stepwise autolytic processing of profragilysin-3 via various intermediates was observed, which resulted in an apparently stable 21-kDa form (Fig. S1B). This protein started at the same residue, Ala212, as authentic fragilysin-1 purified from natural sources. In contrast, the Glu349Ala-mutant of profragilysin-3, which ablated the general base/acid required for catalysis, was more stable over time and only slight autolysis was observed (Fig. S1C). As to the possible implications of such autolysis for in vivo activation, previous studies on profragilysin-1 had shown that mutation of essential residues for structure and function eliminated proteolytic activity of the toxin but not PD processing; i.e., the latter process was exerted by another protease (11). Therefore, autolysis may occur in the purified recombinant proenzyme in vitro only, and a heterolytic activation mechanism would be responsible for activation in vivo. The most likely candidate is trypsin, which is widely expressed in stomach and small intestine, and also in stomach- and colon-cancer cell lines (12, 13). In addition, the final activation cleavage site, Arg211-Ala212, matches trypsin’s substrate specificity and, in our hands, limited proteolysis generated mature wild-type fragilysin-3—indistinguishable from the autolytically activated form—and the mature Glu349Ala-mutant, both starting at Ala212. This provided a means of reproducibly obtaining a homogeneous sample for subsequent studies (Fig. S1A). To assess the importance of the N-terminus of the mature peptidase, limited proteolysis experiments were further carried out on the zymogen with three serine proteinases of distinct specificity, which all rendered forms with comparable activity to that of the autolytically processed or trypsin-activated enzyme.
Role of the Prodomain.
The fragilysin-3 CD alone was difficult to express in E. coli even though a number of fusion constructs were assayed. It could be obtained in soluble form with an N-terminal His6-Z-tag. However, after fusion protein removal, the CD was observed to aggregate in size-exclusion chromatography (Fig. S2A). In addition, the protein did not show activity and, contrary to the mature form obtained by tryptic processing, it was completely digested by trypsin (Fig. S2B). Moreover, a comparative thermal shift assay was performed to measure the temperature of midtransition (Tm) of the two CD forms and the proenzyme. The Tm for trypsin-activated fragilysin-3 was ∼5 °C lower than that of profragilysin-3 (Tm values of 51.4 ± 0.9 °C and 56.1 ± 0.1 °C, respectively), revealing the significantly higher stability of the zymogen due to the presence of the PD. In contrast, it was not possible to determine Tm for the directly expressed variant due to the anomalous behavior of the curve (Fig. S2C), which we attribute to a labile conformational state and aggregation. This hypothesis was further confirmed by circular dichroism: the spectrum of directly expressed fragilysin-3 was very similar to that of the trypsin-activated variant at 50 °C; i.e., under conditions of thermal denaturation of the latter (Fig. S2D). Taken together, all these results indicate that directly expressed fragilysin-3 is unstable and strongly point to a role for the PD, in addition to latency maintenance, as a chaperone that assists in the folding and stabilization of CD, as previously reported for other MP zymogens (14, 15). In the absence of this chaperone, fragilysin-3 CD is not able to fold correctly in the environment provided by the bacterial expression host.
Proteolytic Activity and its Inhibition.
Mature wild-type fragilysin-3 cleaved casein, fibrinogen, actin, and fibronectin (Fig. S1 D–G), as well as a casein fluorescein-conjugate, azocollagen, and azocasein, but not azoalbumin in the conditions assayed. In addition, eight standard fluorogenic peptides were analyzed for cleavage, two of which were efficiently cleaved, including a matrix metalloproteinase (MMP) probe, NFF3 (16). The rest were cleaved moderately or not at all (Fig. S1I). Analysis of protein and peptide cleavage fragments revealed broad substrate specificity for fragilysin-3 (see Table S1), and maximal activity was recorded at pH5.5 (Fig. S1H). As histidine side chains normally tend to be in a double protonated state at pH < ∼ 6 and this species is incompatible with cation binding, this maximum of activity entails that the catalytic zinc ion must be in an overall hydrophobic environment and very firmly bound to the protein. This is in agreement with binding constants exceeding 1012 M-1 reported for extracellular zinc enzymes (17). Activity was completely abolished by standard zinc-chelating agents and significantly impaired by excess zinc and the broad-spectrum small-molecule MMP inhibitor, CT1746 (18). No inhibition was exerted by inhibitors of serine, cysteine, or aspartic proteases, or by inhibitors of other MPs (Table S2). In contrast, only residual activity was found for the trypsin-processed Glu349Ala-mutant against fibrinogen and fluorophore-labeled casein. Together, these results indicate that fragilysin-3 is an active, broad-spectrum MP in vitro.
Overall Structure of Profragilysin-3.
The structure of profragilysin-3 was solved using data to 1.8 Å resolution (see Table S3). The zymogen is a bilobal molecule and shows the PD and the CD as two delimitated globular moieties (Fig. 1A). The PD is constituted by a large twisted antiparallel β-sheet (strands β4-β8, β10, and β9), which vertically traverses the whole domain (obliquely back-to-front in Fig. 1 A and B) and has a concave and a convex side. Preceding strand β4, an N-terminal ∼40-residue segment nestles at the convex side of the β-sheet and folds into a small three-stranded β-sheet of mixed parallel (β1 and β2) and antiparallel (β3) connectivity plus an adjacent antiparallel helix, α1. The latter is inserted roughly coplanar to the sheet between strands β1 and β2. This sheet is orthogonal to the large β-sheet and both give rise to a β-sandwich structure (Fig. 1 A and B). The strands of the large β-sheet are linked by simple vertical connectivity from β4 to β8. Thereafter, the chain enters helix α2, which runs along the front surface, roughly top-to-bottom (Fig. 1A), and flows into a short 310-helix, η1, before entering the last strand of the sheet, β9, at the front of the molecule. After this strand, the polypeptide folds back and enters the penultimate strand of the sheet, β10, which leads to helix α3, at the domain interface. After this helix, the chain adopts an extended conformation that includes strand β11 and runs across the front surface of the CD (see below). The last residue defined by electron density of PD varies from Pro199 to Pro202 in the four independent molecules found in the crystal asymmetric unit, on the outermost left surface of the catalytic moiety (Fig. 1A).
Fig. 1.

Overall structure of profragilysin-3. (A) Richardson-type plot of profragilysin-3 in stereo with helices (labeled α1–α7 and η1) as ribbons and β-strands (β1–β15) as arrows. The polypeptide chain is flexible and thus interrupted between the end of the PD (depicted in magenta) and the beginning of the CD (cyan). The latter is displayed approximately in the standard orientation characteristic for MPs (i.e., with the view into the active-site cleft) that runs from left (nonprimed side) to right (primed side) according to ref. 46. (B) Topology of profragilysin-3 PD displaying the regular secondary structure elements, which comprise strands β1–β11 (arrows) and helices α1–α3 plus η1 (rods). Strands β4 and β11 establish parallel β-sheet interactions with CD strands β13 and β15 (in cyan), respectively. (D) Zinc-binding site of profragilysin-3 with the metal ion in magenta and the coordinating residues from the PD (Asp349) and the CD (His348, His352, and His358). The distance ranges for each bond found in the four molecules of the asymmetric unit of the wild-type structure are depicted.
The polypeptide chain is again defined by electron density from Thr210 or Arg211, which features the beginning of the roughly spherical CD. This domain is divided by the active-site cleft into a large upper subdomain (Thr210/Arg211-Gly355) and a small lower subdomain (Ala356-Asp397). The former consists of a twisted five-stranded β-sheet, whose first four strands from back to front in Fig. 1A (β13, β12, β14, and β16) run parallel and from left to right, whereas the outermost front strand, β15, runs antiparallel to the previous strands and creates the upper-rim of the active-site cleft. Loop Lβ14β15 protrudes from the molecular surface and delimitates the active-site cleft on its primed side. Two helices, the “backing helix” α4 and helix α5 are found on the concave and convex sides of the sheet, respectively. The upper subdomain ends after the “active-site helix” α6, which comprises the first part of the ZBCS and thus the first two zinc-binding histidines, His348 and His352, and the catalytic general base/acid, Glu349. At Gly355, still within the ZBCS, the polypeptide chain undergoes a sharp turn downward and enters the lower subdomain, which only spans 42 residues and mainly comprises the third zinc-binding histidine, His358; the methionine containing Met-turn (Asp364-Leu365-Met366-Tyr367); and the “C-terminal helix” α7, which forms part of the CD moiety (Fig. 1A). In this way, our structure confirms earlier hypotheses based on homology modeling of fragilysin-1 (19), which suggested that this helix formed part of the domain rather than protruding from it to interact with the eukaryotic membrane, as had been proposed by other authors (20). Interestingly, the polypeptide chain does not finish after helix α7 but adopts a loop structure that lies below the N-terminal segment of upper subdomain. The chain C-terminus at Asp397 is anchored to the domain moiety through interactions with the side chains of His252 and Arg256 from Lα4β13. It is conceivable that C-terminal deletion mutants disrupt this interaction network and thus impair overall stability and activity of the CD. This is in accordance with previous biochemical studies showing that ablation of the two C-terminal residues of fragilysin-1 greatly reduced activity and eight missing residues even abolished it, giving rise to an unstable molecule (21).
Molecular Base for Zymogenicity.
PDs prevent access of substrates to active-site clefts in zymogens. In profragilysin-3, the PD interaction with the CD exhibits good shape complementarity and covers an area of 1,996 Å2 at the protein interface. The interaction involves 63 residues and comprises 98 close contacts (see Table S4). In contrast to metalloprocarboxypeptidases of the funnelin tribe of MPs (22) and to pro-MMPs (23), the PD does not cap the CD but is attached to its right lateral surface (Fig. 1A). It prevents access to the active-site cleft through the C-terminal segment, which runs in extended conformation across the entire CD front in the opposite orientation to a substrate. The most important segment for latency is that encompassing α3, Lα3β11, and β11, which traverses the front of the CD from right to left. It establishes a parallel β-sheet interaction on the nonprimed side of the cleft through strand β11 with “upper-rim strand” β15 of the CD (Fig. 2 A and B and Table S4). It also approaches the beginning of helix α5 above the cleft, as well as Lβ14β15, Lβ15β16, Lβ16α6, active-site helix α6, and the segment connecting the Met-turn with C-terminal helix α7. Additional segments involved in PD/CD interaction include the solvent-accessible part of the backing helix α4 of the CD and the concave side of the large PD β-sheet, which contributes with residues from β5-β8 and β10; a parallel β-sheet consisting of strands β13 (CD) and β4 (PD); and segments α3 and Lα3β11 (PD) and helix α5 and loops Lβ15β16, Lα6α7, and Lβ16α4 (CD). A prominent bulge preceding β11 gives rise to a tight 1,4-turn spanning Tyr191-Asp194 (Fig. 2 A and B). This bulge causes the side chains of Tyr191 and Asp194 to penetrate the catalytic moiety whereas the residues embraced, Ile192 and Asn193, point to the bulk solvent. The aromatic residue occupies the S1’ site of the cleft, which is framed by atoms provided by Ile313, Leu314, Gly344, Val345, His352, His358, Leu365, and Tyr370, as well as by backbone atoms of segment Leu365-Leu374 (Fig. 2B). This is a large pocket (360 Å3) and Tyr191 is a long way from filling it. Although most of the framing side chains are hydrophobic, the pocket could also accommodate short side chains and large hydrophilic residues, which could be bound by the main-chain carbonyl groups of Leu365, Asp364, Tyr367, or Tyr373, or by Thr371 Oγ1. Accordingly, this pocket would be compatible with a broad specificity at this site. Asp194, in turn, coordinates the catalytic zinc ion in substitution of the solvent molecule usually found in mature CDs primed for catalysis (24). The binding is bidentate and exerted from the top by the two terminal carboxylate oxygen atoms, which together with the Nϵ2 atoms of His348, His352, and His358 give rise to a tetrahedral + 1 coordination sphere of the catalytic zinc (Fig. 1C). In addition, Asp194 Oδ2 points to the Oϵ1 atom of the general base/acid, Glu349, at 2.69–3.04 Å in the four monomers in the asymmetric unit (Fig. 2B). This implies that one of the two oxygens must be protonated. Downstream of Asp194, Tyr195 most likely occupies cleft subsite S2, framed by His352, Leu319, and His358; and Ile196 is probably in S3, shaped by Trp318, Leu280, As277, and Ile316 (Fig. 2 A and B). As observed for 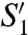 , these sites could harbor several types of residue, thus supporting the broad substrate specificity of fragilysin-3 observed in vitro. This inhibitory mechanism follows an “aspartate switch,” in analogy to MMPs and ADAMs/adamalysins, for which the term “cysteine switch” was coined. The name was based on a cysteine Sγ atom replacing the catalytic solvent molecule in the zymogen (25, 26). Such an aspartate-switch mechanism has been described for proastacin (27). In this zymogen, however, the PD is much shorter (34 residues), the zinc-binding aspartate is provided by a wide loop immediately downstream of a prodomain helix that occupies the primed side of the cleft, and no interactions are observed between the PD and the upper-rim strand.
, these sites could harbor several types of residue, thus supporting the broad substrate specificity of fragilysin-3 observed in vitro. This inhibitory mechanism follows an “aspartate switch,” in analogy to MMPs and ADAMs/adamalysins, for which the term “cysteine switch” was coined. The name was based on a cysteine Sγ atom replacing the catalytic solvent molecule in the zymogen (25, 26). Such an aspartate-switch mechanism has been described for proastacin (27). In this zymogen, however, the PD is much shorter (34 residues), the zinc-binding aspartate is provided by a wide loop immediately downstream of a prodomain helix that occupies the primed side of the cleft, and no interactions are observed between the PD and the upper-rim strand.
Fig. 2.

Active-site cleft of profragilysin-3. (A) Detail of the active-site cleft of fragilysin-3 superimposed with its Connolly surface colored according to electrostatic potential. PD segment Cys189-Thr198 is shown as a stick model. (B) Close-up view of Fig. 1A in stereo showing the active-site environment, the specificity pocket, and the residues involved. All residues except those already tagged in Fig. 1C are labeled.
Structural Similarities.
We found no overall similarity between the PD and any other structure deposited with the Protein Data Bank (PDB). Only the functionally unrelated bacteriochlorophyll A binding protein from the green sulfur bacterium, Prosthecochloris aestuarii 2K, showed similarity with the central part of the large β-sheet and helix α2 (PDB 3EOJ; (28); Z-score 4.3, rmsd 5.3 Å, 106 common residues; 6% sequence identity) (Fig. S3A). However, in the Prosthecochloris protein, the β-strands are much longer and form part of an overall open-barrel structure with no resemblance to profragilysin-3. In addition, topological relatedness includes only about 50% of PD residues: the small orthogonal β-sheet and helix α1, on the convex side of the large sheet, as well as helices η1 and α3 of PD, do not have structural equivalents in the Prosthecochloris protein. Accordingly, we conclude that profragilysin-3 PD conforms to a new fold, understood as a domain with a structure formed by regular secondary structure elements in an orientation and connectivity not found in previously reported molecules.
In contrast, searches with the CD unambiguously identified members of the adamalysin/ADAMs family as close structural relatives of fragilysin-3, with Z-scores of 13–17, rmsd values of 2.5–2.9 Å, and common sequence stretches of 160–166 residues (out of 188 total residues in fragilysin-3 and 200–260 in adamalysin/ADAM catalytic moieties) but only ∼15% sequence identity. Next in similarity was ulilysin, the structural prototype of the pappalysin family, followed by MMPs, serralysins, and astacins, which are all members of the metzincin clan of MPs (7, 8). Fig. S3B depicts the superposition of fragilysin-3 with adamalysin II [PDB 1IAG (29, 30)], which reveals that almost all their regular secondary structure elements colocalize. Both CDs display a large upper subdomain of 3/4 and a small lower subdomain of 1/4 of the total size. With helix α5, fragilysin-3 has one of the most characteristic elements for adamalysins/ADAMs not present in other metzincin prototypes, the large “adamalysin helix” preceding the central strand of the upper subdomain β-sheet (7). Furthermore, the upper-rim strand and the preceding bulge on top of the cleft on its primed side are very similar in both structures, both in length and conformation.
The adamalysin/ADAM family is named after the first family member to be structurally characterized, adamalysin II from Crotalus adamanteus snake venom, and mammalian reproductive-tract proteins (31, 32). The family has 40 members in humans (http://degradome.uniovi.es/met.html#M10) and has only been found in metazoans and some fungi, which include two opportunistic human pathogens, Pneumocystis carinii and Aspergillus fumigatus; and fission yeast, Schizosaccharomyces pombe (7, 33–35). Sequence similarity searches within bacteria and archaea revealed potential adamalysin-like sequences only in Marinobacter aquaeolei and Marinobacter algicola, which, however, have yet to be characterized. Because fragilysins showed only 15% sequence identity with adamalysins/ADAMs, we performed a phylogenetic analysis including four human, two ophidian, and two fungal adamalysins/ADAMs, in addition to the putative Marinobacter relatives and fragilysin-3 (Fig. S3C). This study revealed that the metazoan forms cluster together. Next in divergence would be the two Marinobacter relatives, followed by the fungal forms. Fragilysin-3 would be clearly farthest away in evolution, indicating that fragilysins are not true adamalysins/ADAMs anymore despite the close overall structure due to the action of evolution in a particular environment. Consistent with this separation, there are several subtle differences between fragilysin-3 and adamalysin II (Fig. S3B).
Implications for Fragilysin Isoforms.
The sequence identity values indicate that the three profragilysin isoforms have identical structure. They all span 397 residues, and differences in sequence of profragilysin-3 (UniProt O86049) with profragilysin-1 (Q9S5W0) and -2 (O05091) are found at just five positions in PD and at 26 positions in CD (Table S5). Interestingly, almost half of the mutations (12/26) cluster at the lower subdomain of the CD. All changes are compatible with the current structure as they mostly affect surface-exposed segments. Only three mutants, Asp277Lys, Asn312Lys, and Lys331Glu, only present in fragilysin-1, could affect substrate binding and alter distant subsites at both ends of the cleft; i.e., beyond S5 and 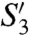 . In any case, no mutation affects the residues shaping pockets S1 and
. In any case, no mutation affects the residues shaping pockets S1 and  , which were reported to preferentially accommodate hydrophobic residues in the case of fragilysin-1 (5, 6). As fragilysin-3 shows broad specificity (Table S1), we do not have a structural explanation for these differences. Overall, the conclusions of this structural analysis can be extrapolated to all three isoforms, which is consistent with a common function in vivo.
, which were reported to preferentially accommodate hydrophobic residues in the case of fragilysin-1 (5, 6). As fragilysin-3 shows broad specificity (Table S1), we do not have a structural explanation for these differences. Overall, the conclusions of this structural analysis can be extrapolated to all three isoforms, which is consistent with a common function in vivo.
Conclusions.
Until the discovery of fragilysins, the main molecular determinants of ETBF virulence, no MP had been reported to act as a potent enterotoxin (19). We show here that fragilysin-3 is a functional broad-spectrum MP in vitro with a large 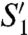 pocket that could be targeted by specific inhibitors to treat ETBF infection and associated diseases. In vivo, this MP is inhibited by a unique PD via an aspartate-switch mechanism until it is secreted to the gut lumen. Fragilysin-3 is one of three isoforms present in ETBF and no further related proteins have been described. They are encoded by a pathogenicity islet, which is absent in nonvirulent B. fragilis strains, that must have been acquired by HGT from an exogenous source (36). HGT may occur through nucleic-acid transduction, conjugation, or transformation, and it contributes to evolution of life complementing the Darwinistic tree-based mechanism. It results in the acquisition of xenologs (i.e., homologs that do not result from common ancestor inheritance) and confers adaptive advantages that can change the relationship of bacterial species with the environment and their pathogenic character (37). Such gene shuffling is common within and across bacteria and archaea and examples include the transfer of resistance plasmids (38). Contrary, documented transfer from bacteria to eukaryotic cells is restricted to few examples, among them the interaction between Agrobacterium tumefaciens and plant cells, which leads to Crown–Gall disease (39, 40). Lastly, gene transfer from eukaryotes to bacteria is extremely uncommon and only detectable through phylogenetic and comparative genome analyses (41, 42). It is of great potential relevance for bacterial pathogenicity (37, 41). Direct evidence for such HGT does not exist as there is no footprint of such evolutionary processes and, therefore, sequence-independent structural similarity provides a complementary tool to unveil potential cases of xenolog exaptation. In this sense, the CD structure, but not the sequence, of fragilysin-3 strongly resembles adamalysins/ADAMs only and it is conceivable that fragilysins derived from a xenolog of adamalysins/ADAMs co-opted long ago during the intimate coexistence between B. fragilis and mammalian intestinal tracts. Fragilysins would subsequently have evolved in a bacterial environment, thus giving rise to small structural changes and a different protein sequence expressed as three isoforms, and, putatively, to the gene product of mpII. In this context, it has been shown that human colon cell lines express mRNA of ADAM-10, -12, and -15 (43), which could potentially be incorporated by competent intestinal bacteria by transformation and subsequent action of DNA polymerase I, which exerts RNA-dependent DNA polymerase activity in several bacteria (44). In addition, mammalian adamalysin/ADAM CDs are difficult to produce as recombinant proteins and they require PDs that work as intramolecular chaperones (45). The same is the case for fragilysin-3, which could only be produced in a functional form if fused to its unique and tailor-made PD. All these lines of evidence support a relation between the aforementioned mammalian MPs and fragilysins and, thus, the development of a functional protein that eventually became toxic for the intestinal wall: putatively the origin of its own ancestor.
pocket that could be targeted by specific inhibitors to treat ETBF infection and associated diseases. In vivo, this MP is inhibited by a unique PD via an aspartate-switch mechanism until it is secreted to the gut lumen. Fragilysin-3 is one of three isoforms present in ETBF and no further related proteins have been described. They are encoded by a pathogenicity islet, which is absent in nonvirulent B. fragilis strains, that must have been acquired by HGT from an exogenous source (36). HGT may occur through nucleic-acid transduction, conjugation, or transformation, and it contributes to evolution of life complementing the Darwinistic tree-based mechanism. It results in the acquisition of xenologs (i.e., homologs that do not result from common ancestor inheritance) and confers adaptive advantages that can change the relationship of bacterial species with the environment and their pathogenic character (37). Such gene shuffling is common within and across bacteria and archaea and examples include the transfer of resistance plasmids (38). Contrary, documented transfer from bacteria to eukaryotic cells is restricted to few examples, among them the interaction between Agrobacterium tumefaciens and plant cells, which leads to Crown–Gall disease (39, 40). Lastly, gene transfer from eukaryotes to bacteria is extremely uncommon and only detectable through phylogenetic and comparative genome analyses (41, 42). It is of great potential relevance for bacterial pathogenicity (37, 41). Direct evidence for such HGT does not exist as there is no footprint of such evolutionary processes and, therefore, sequence-independent structural similarity provides a complementary tool to unveil potential cases of xenolog exaptation. In this sense, the CD structure, but not the sequence, of fragilysin-3 strongly resembles adamalysins/ADAMs only and it is conceivable that fragilysins derived from a xenolog of adamalysins/ADAMs co-opted long ago during the intimate coexistence between B. fragilis and mammalian intestinal tracts. Fragilysins would subsequently have evolved in a bacterial environment, thus giving rise to small structural changes and a different protein sequence expressed as three isoforms, and, putatively, to the gene product of mpII. In this context, it has been shown that human colon cell lines express mRNA of ADAM-10, -12, and -15 (43), which could potentially be incorporated by competent intestinal bacteria by transformation and subsequent action of DNA polymerase I, which exerts RNA-dependent DNA polymerase activity in several bacteria (44). In addition, mammalian adamalysin/ADAM CDs are difficult to produce as recombinant proteins and they require PDs that work as intramolecular chaperones (45). The same is the case for fragilysin-3, which could only be produced in a functional form if fused to its unique and tailor-made PD. All these lines of evidence support a relation between the aforementioned mammalian MPs and fragilysins and, thus, the development of a functional protein that eventually became toxic for the intestinal wall: putatively the origin of its own ancestor.
Materials and Methods
A detailed description of procedures is provided in SI Materials and Methods. Briefly, profragilysin-3 was amplified from genomic B. fragilis DNA and cloned into a modified pET-28a vector for overexpression in E. coli Origami-2 (DE3) cells. The protein was purified by nickel-affinity chromatography, digested with tobacco-etch virus protease to remove the N-terminal hexahistidine tag, and polished by gel filtration. The selenomethionine variant was obtained in the same way, except that cells were grown in minimal medium with selenomethionine replacing methionine. The Glu349Ala mutant was obtained by site-directed mutagenesis and produced as aforementioned. The active wild-type enzyme was obtained by time-dependent autolysis or tryptic limited proteolysis of the zymogen and subsequent gel-filtration purification. Thermal shift and circular dichroism assays, as well as proteolytic activity assays against protein and peptide substrates and inhibitory assays, were performed according to standard protocols. The 1.8 Å-crystal structure of profragilysin-3 was solved by single-wavelength anomalous diffraction by using orthorhombic selenomethionine-derivatized crystals obtained by sitting-drop vapor diffusion. Program SHELXD was used to identify all 20 selenium sites of the dimer present in the asymmetric unit and the noncrystallographic symmetry operator could be derived. Subsequent phasing with SHELXE and density modification with DM under 2-fold averaging rendered an electron density map that enabled manual tracing of roughly 3/4 of each protomer. These coordinates were refined with REFMAC5 and used to solve the structure of monoclinic native crystals, which contained a tetramer per asymmetric unit, by Patterson search with PHASER. A subsequent run with ARP/wARP rendered an excellent electron density map. Thereafter, manual model building alternated with crystallographic refinement until the model was complete. Phylogenetic analyses were performed with MULTALIN and PHYLIP after optimal superposition of the available crystal structures to derive a sequence alignment.
Supplementary Material
Acknowledgments.
We thank M. J. Avila-Campos (Brazil) for providing B. fragilis genomic DNA, J. L. Reymond (Switzerland) for providing fluorogenic peptides, T. Guevara for excellent laboratory assistance, Robin Rycroft for helpful suggestions to the manuscript, and to the Crystallography Platform (PAC) at Barcelona Science Park (PCB) for assistance during crystallization experiments. This study was supported in part by grants from European, Spanish, and Catalan agencies (FP7-HEALTH-F3-2009-223101 “AntiPathoGN,” FP7-HEALTH-2010-261460 “Gums&Joints,” BIO2008-04080-E, BIO2009-10334, CSD2006-00015, PSE-010000-2009-8, and 2009SGR1036). We acknowledge the help provided by European Molecular Biology Laboratory (EMBL) at Grenoble, France and European Synchrotron Radition Facility (ESRF) synchrotron local contacts. Funding for data collection was provided in part by ESRF.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 3P24).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1012173108/-/DCSupplemental.
References
- 1.Rabizadeh S, Sears C. New horizons for the infectious diseases specialist: How gut microflora promote health and disease. Curr Infect Dis Rep. 2008;10:92–98. doi: 10.1007/s11908-008-0017-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sears CL. Enterotoxigenic Bacteroides fragilis: A rogue among symbiotes. Clin Microbiol Rev. 2009;22:349–369. doi: 10.1128/CMR.00053-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holton J. Enterotoxigenic Bacteroides fragilis. Curr Infect Dis Rep. 2008;10:99–104. doi: 10.1007/s11908-008-0018-7. [DOI] [PubMed] [Google Scholar]
- 4.Myers LL, Firehammer BD, Shoop DS, Border MM. Bacteroides fragilis: A possible cause of acute diarrheal disease in newborn lambs. Infect Immun. 1984;44:241–244. doi: 10.1128/iai.44.2.241-244.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moncrief JS, et al. The enterotoxin of Bacteroides fragilis is a metalloprotease. Infect Immun. 1995;63:175–181. doi: 10.1128/iai.63.1.175-181.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vines RR, Wilkins TD. In: Handbook of Proteolytic Enzymes. Barrett AJ, Rawlings ND, Woessner JF Jr, editors. London: Elsevier; 2004. pp. 588–591. [Google Scholar]
- 7.Gomis-Rüth FX. Structural aspects of the metzincin clan of metalloendopeptidases. Mol Biotechnol. 2003;24:157–202. doi: 10.1385/MB:24:2:157. [DOI] [PubMed] [Google Scholar]
- 8.Gomis-Rüth FX. Catalytic domain architecture of metzincin metalloproteases. J Biol Chem. 2009;284:15353–15357. doi: 10.1074/jbc.R800069200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chung GT, et al. Identification of a third metalloprotease toxin gene in extraintestinal isolates of Bacteroides fragilis. Infect Immun. 1999;67:4945–4949. doi: 10.1128/iai.67.9.4945-4949.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu S, Rhee KJ, Zhang M, Franco A, Sears CL. Bacteroides fragilis toxin stimulates intestinal epithelial cell shedding and γ-secretase-dependent E-cadherin cleavage. J Cell Sci. 2007;120:1944–1952. doi: 10.1242/jcs.03455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Franco AA, Buckwold SL, Shin JW, Ascon M, Sears CL. Mutation of the zinc-binding metalloprotease motif affects Bacteroides fragilis toxin activity but does not affect propeptide processing. Infect Immun. 2005;73:5273–5277. doi: 10.1128/IAI.73.8.5273-5277.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koshikawa N, et al. Expression of trypsin by epithelial cells of various tissues, leukocytes, and neurons in human and mouse. Am J Pathol. 1998;153:937–944. doi: 10.1016/S0002-9440(10)65635-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miyata S, et al. Expression of trypsin in human cancer cell lines and cancer tissues and its tight binding to soluble form of Alzheimer amyloid precursor protein in culture. J Biochem. 1999;125:1067–1076. doi: 10.1093/oxfordjournals.jbchem.a022388. [DOI] [PubMed] [Google Scholar]
- 14.Khan AR, James MN. Molecular mechanisms for the conversion of zymogens to active proteolytic enzymes. Protein Sci. 1998;7:815–836. doi: 10.1002/pro.5560070401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marie-Claire C, Roques BP, Beaumont A. Intramolecular processing of prothermolysin. J Biol Chem. 1998;273:5697–5701. doi: 10.1074/jbc.273.10.5697. [DOI] [PubMed] [Google Scholar]
- 16.Nagase H, Fields CG, Fields GB. Design and characterization of a fluorogenic substrate selectively hydrolyzed by stromelysin 1 (matrix metalloproteinase-3) J Biol Chem. 1994;269:20952–20957. [PubMed] [Google Scholar]
- 17.Fraústoda Silva JJR, Williams RJP. The Biological Chemistry of the Elements: The Inorganic Chemistry of Life. New York: Oxford University Press; 2001. [Google Scholar]
- 18.Chander SK, et al. An in vivo model for screening peptidomimetic inhibitors of gelatinase A. J Pharm Sci. 1995;84:404–409. doi: 10.1002/jps.2600840405. [DOI] [PubMed] [Google Scholar]
- 19.Obiso RJJ, Bevan DR, Wilkins TD. Molecular modeling and analysis of fragilysin, the Bacteroides fragilis toxin. Clin Infect Dis. 1997;25(Suppl. 2):S153–S155. doi: 10.1086/516240. [DOI] [PubMed] [Google Scholar]
- 20.Saidi RF, Sears CL. Bacteroides fragilis toxin rapidly intoxicates human intestinal epithelial cells (HT29/C1) in vitro. Infect Immun. 1996;64:5029–5034. doi: 10.1128/iai.64.12.5029-5034.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sears CL, Buckwold SL, Shin JW, Franco AA. The C-terminal region of Bacteroides fragilis toxin is essential to its biological activity. Infect Immun. 2006;74:5595–5601. doi: 10.1128/IAI.00135-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gomis-Rüth FX. Structure and mechanism of metallocarboxypeptidases. Crit Rev Biochem Mol Biol. 2008;43:319–345. doi: 10.1080/10409230802376375. [DOI] [PubMed] [Google Scholar]
- 23.Tallant C, Marrero A, Gomis-Rüth FX. Matrix metalloproteinases: Fold and function of their catalytic domains. BBA-Mol Cell Res. 2010;1803:20–28. doi: 10.1016/j.bbamcr.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 24.Auld DS. In: Handbook of Proteolytic Enzymes. Barrett AJ, Rawlings ND, Woessner JF Jr, editors. London: Elsevier Academic Press; 2004. pp. 268–289. [Google Scholar]
- 25.Springman EB, Angleton EL, Birkedal-Hansen H, Van Wart HE. Multiple modes of activation of latent human fibroblast collagenase: Evidence for the role of a Cys73 active-site zinc complex in latency and a “cysteine switch” mechanism for activation. Proc Natl Acad Sci USA. 1990;87:364–368. doi: 10.1073/pnas.87.1.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rosenblum G, et al. Molecular structures and dynamics of the stepwise activation mechanism of a matrix metalloproteinase zymogen: Challenging the cysteine switch dogma. J Am Chem Soc. 2007;129:13566–13574. doi: 10.1021/ja073941l. [DOI] [PubMed] [Google Scholar]
- 27.Guevara T, et al. Proenzyme structure and activation of astacin metallopeptidase. J Biol Chem. 2010;285:13958–13965. doi: 10.1074/jbc.M109.097436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tronrud DE, Wen J, Gay L, Blankenship RE. The structural basis for the difference in absorbance spectra for the FMO antenna protein from various green sulfur bacteria. Photosynth Res. 2009;100:79–87. doi: 10.1007/s11120-009-9430-6. [DOI] [PubMed] [Google Scholar]
- 29.Gomis-Rüth FX, et al. Refined 2.0 Å X-ray crystal structure of the zinc-endopeptidase adamalysin II. Primary and tertiary structure determination, refinement, molecular structure and comparison with astacin, collagenase and thermolysin. J Mol Biol. 1994;239:513–544. doi: 10.1006/jmbi.1994.1392. [DOI] [PubMed] [Google Scholar]
- 30.Gomis-Rüth FX, Meyer EF, Kress LF, Politi V. Structures of adamalysin II with peptidic inhibitors. Implications for the design of tumor necrosis factor α convertase inhibitors. Protein Sci. 1998;7:283–292. doi: 10.1002/pro.5560070207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gomis-Rüth FX, Kress LF, Bode W. First structure of a snake venom metalloproteinase: A prototype for matrix metalloproteinases/collagenases. EMBO J. 1993;12:4151–4157. doi: 10.1002/j.1460-2075.1993.tb06099.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schlondorff J, Blobel CP. Metalloprotease-disintegrins: Modular proteins capable of promoting cell-cell interactions and triggering signals by protein-ectodomain shedding. J Cell Sci. 1999;112:3603–3617. doi: 10.1242/jcs.112.21.3603. [DOI] [PubMed] [Google Scholar]
- 33.Kennedy CC, Kottom TJ, Limper AH. Characterization of a novel ADAM protease expressed by Pneumocystis carinii. Infect Immun. 2009;77:3328–3336. doi: 10.1128/IAI.01383-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakamura T, Abe H, Hirata A, Shimoda C. ADAM family protein Mde10 is essential for development of spore envelopes in the fission yeast Schizosaccharomyces pombe. Eukaryot Cell. 2004;3:27–39. doi: 10.1128/EC.3.1.27-39.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lavens SE, Rovira-Graells N, Birch M, Tuckwell D. ADAMs are present in fungi: Identification of two novel ADAM genes in Aspergillus fumigatus. FEMS Microbiol Lett. 2005;248:23–30. doi: 10.1016/j.femsle.2005.05.017. [DOI] [PubMed] [Google Scholar]
- 36.Franco AA, et al. Molecular evolution of the pathogenicity island of enterotoxigenic Bacteroides fragilis strains. J Bacteriol. 1999;181:6623–6633. doi: 10.1128/jb.181.21.6623-6633.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ochman H, Lawrence JG, Groisman EA. Lateral gene transfer and the nature of bacterial innovation. Nature. 2000;405:299–304. doi: 10.1038/35012500. [DOI] [PubMed] [Google Scholar]
- 38.Macrina FL, Archer GL. In: Bacterial Conjugation. Clewell DB, editor. New York: Plenum; 1993. pp. 313–329. [Google Scholar]
- 39.Sprague GF., Jr Genetic exchange between kingdoms. Curr Opin Genet Dev. 1991;1:530–533. doi: 10.1016/s0959-437x(05)80203-5. [DOI] [PubMed] [Google Scholar]
- 40.Gomis-Rüth FX, Solà M, de la Cruz F, Coll M. Coupling factors in macromolecular type-IV secretion machineries. Curr Pharm Design. 2004;10:1551–1565. doi: 10.2174/1381612043384817. [DOI] [PubMed] [Google Scholar]
- 41.Koonin EV, Makarova KS, Aravind L. Horizontal gene transfer in prokaryotes: Quantification and classification. Annu Rev Microbiol. 2001;55:709–742. doi: 10.1146/annurev.micro.55.1.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Doolittle WF. Lateral genomics. Trends Cell Biol. 1999;9:M5–M8. [PubMed] [Google Scholar]
- 43.Charrier L, et al. ADAM-15 inhibits wound healing in human intestinal epithelial cell monolayers. Am J Physiol-Gastr L. 2005;288:G346–G353. doi: 10.1152/ajpgi.00262.2004. [DOI] [PubMed] [Google Scholar]
- 44.Harada F, et al. RNA-dependent DNA polymerase (RT) activity of bacterial DNA polymerases. Bull Osaka Med Coll. 2005;51:35–41. [Google Scholar]
- 45.Gonzales PE, Galli JD, Milla ME. Identification of key sequence determinants for the inhibitory function of the prodomain of TACE. Biochemistry. 2008;47:9911–9919. doi: 10.1021/bi801049v. [DOI] [PubMed] [Google Scholar]
- 46.Abramowitz N, Schechter I, Berger A. On the size of the active site in proteases. II. Carboxypeptidase-A. Biochem Bioph Res Co. 1967;29:862–867. doi: 10.1016/0006-291x(67)90299-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.