

Abstract
The protective antigen (PA) moiety of anthrax toxin forms oligomeric pores that translocate the enzymatic moieties of the toxin—lethal factor (LF) and edema factor (EF)—across the endosomal membrane of mammalian cells. Here we describe site-directed spin-labeling studies that identify interactions of LF with the prepore and pore conformations of PA. Our results reveal a direct interaction between the extreme N terminus of LF (residues 2–5) and the Φ-clamp, a structure within the lumen of the pore that catalyzes translocation. Also, consistent with a recent crystallographic model, we find that, upon binding of the translocation substrate to PA, LF helix α1 separates from helices α2 and α3 and binds in the α-clamp of PA. These interactions, together with the binding of the globular part of the N-terminal domain of LF to domain 1′ of PA, indicate that LF interacts with the PA pore at three distinct sites. Our findings elucidate the state from which translocation of LF and EF proceeds through the PA pore.
Anthrax toxin, in addition to its importance in regard to the pathogenesis of Bacillus anthracis, is one of the simplest and most tractable systems for studying protein translocation across membranes. The toxin is composed of two intracellularly acting enzymes—edema factor (EF) (1) and lethal factor (LF) (2)—and a receptor-binding (3, 4), pore-forming (5) protein, termed protective antigen (PA). These three proteins are secreted from the Bacillus anthracis as monomeric units. Monomeric PA83 is activated by furin-family proteases (6) on the host cell surface and spontaneously assembles into homoheptamers, [PA63]7 (7), and homooctamers, [PA63]8 (8) (collectively termed prepores). [PA63]7 binds up to three, and [PA63]8 binds up to four, molecules of LF and/or EF (8, 9), forming noncovalent receptor-bound complexes at the cell surface. These complexes are taken into the cell through endocytosis (10) and trafficked to the endosome, where, upon exposure to low pH, the prepores insert into the membrane (5, 11, 12), forming pores through which LF and/or EF translocate to the cytosol (13).
LF and EF bind to the surface of PA (on domain 1′) (14) through their homologous, ∼250-residue N-terminal domains (LFN and EFN, Fig. 1E) (15). Each has a positively charged, unstructured N terminus (16, 17) (∼20–30 residues), which is positioned at or near the entry of the lumen when the proteins bind to the prepore (18). PA pores have a negatively charged lumen (7) and form cation-selective channels in planar lipid bilayers (19). LF or LFN blocks current flow through these channels (20), whereas N-terminally truncated LFN mutants do not (21). These observations have led to the hypothesis that upon binding of LFN to domain 1′ of a pore, the N terminus is drawn into the lumen by electrostatic forces, initiating N- to C-terminal translocation. However, data have been lacking to indicate precisely how the unstructured LF or EF N terminus interacts with the lumen of the pore.
Fig. 1.

Site-directed spin labeling reveals three LFN-PA interactions. (A) Attachment of the MTSL spin label to cysteine forms the R1 side chain. (B) Crystal structure of LFN (16) with spin-labeled residues marked in red. The N terminus (residues 1–26) is sketched in black. Helix α1 is shown in magenta, helix α2 in orange, and helix α3 in yellow. (C) PA prepore crystal structure (37) with three subunits removed, showing the His-tagged, spin-labeled subunit in purple and the R178A/K214E subunits in green. Spin-labeled residues are shown in red. (D) Schematic showing PA heteroheptamer with a single LFN binding site and a single spin label. Aerial view of the seven domains 1′ of PA prepore; in green are subunits incapable of binding LFN due to their R178A/K214E mutations (shown as black “X’s”). In purple is the spin-labeled (red star) subunit capable of binding LFN. An LFN molecule is shown docked on the surface with “thumb” representing the N terminus (38). (E) An EM reconstruction of the PA pore (25) with a cartoon representation of LFN (in blue) bound. The globular domain of LFN is represented as a circle, helix α1 is represented as a rectangle, and the unstructured N terminus is shown in yellow. The PA domain 1′, α-clamp, and Φ-clamp binding sites are shown in red.
The Phe427 residues of the PA prepore, located in the lumen and near the base of the structure, converge upon formation of the pore, generating a structure termed the Φ-clamp. The Φ-clamp is necessary for efficient translocation of LF and EF (22, 23). Mutation of Phe427 to all but the most conservative substitutions abolishes translocation and, in some cases, severely inhibits current block by LF/LFN in planar lipid bilayers (22, 23). These data suggest that the putative interaction between the LFN N terminus and the lumen of the PA pore may occur at the Φ-clamp; however, no direct evidence for such an interaction has been reported. Furthermore, which residues within the LFN N terminus would contribute to such an interaction is unknown. Recently, a crystal structure of PA prepore bound to LFN was solved (24), which confirmed binding of LFN to PA domain 1′ and revealed a previously uncharacterized interaction between LFN helix α1 (residues 31–42) and a newly identified entity of PA, termed the α-clamp, located at the “mouth” of the PA lumen. However, the first 28 N-terminal residues of LFN were disordered in this crystal structure, as they are in the crystal structure of LF alone (16), providing no additional evidence as to where, if at all, these residues bind in the lumen of the PA pore.
Characterizing interactions between membrane-inserted PA pore and LF has been challenging. No crystal structure of the pore exists, and the resolution of electron micrograph images remains low (25, 26). Site-directed spin labeling and EPR (SDSL-EPR) represents an attractive approach to studying PA pore-LF interactions, as it allows molecular-level resolution of interactions and can be performed with detergent-bound (27), liposome-inserted (27), or aggregated forms (22) of PA pore. Here we employ SDSL-EPR to examine PA-LFN binding interactions. We constructed a PA heteroheptamer containing a single LFN binding site and used this mutant to identify and characterize three sites of PA-LFN interaction. We report a stable, direct interaction between the Φ-clamp in the PA pore and the extreme N terminus of LFN (residues 2–5). Our results also confirm that the interaction between the PA α-clamp and LFN helix α1 observed recently in the octameric PA prepore-LFN crystal structure (24) occurs in solution with a predominantly heptameric form of PA. Further, we show that the previously characterized interaction between the LFN globular domain and PA domain 1′ (14) exists in both the prepore and pore state of PA and is independent of a functional Φ-clamp. Collectively, our results show that the PA-LFN binding interaction is more complex than originally described and consists of three sites within PA: the Φ-clamp, the α-clamp, and domain 1′, which interact with the extreme N terminus, helix α1, and the globular domain of LFN, respectively (Fig. 1E). The 3-site model has implications for understanding the processes of pore-mediated unfolding and translocation of the enzymatic cargo proteins of anthrax toxin.
Results and Discussion
LFN Residues 2, 5, and 10 Interact with PA Pore.
To investigate binding of residues within the unstructured N-terminal segment of LFN to the lumen of the PA prepore and pore, we introduced an individual cysteine into LFN at residue 2, 5, 10, 20, or 30 (Fig. 1B) and attached a nitroxide spin label (R1, Fig. 1A) (28, 29). We recorded the spectrum of each spin-labeled LFN variant alone and in the presence of PA prepore (pH 8.5; Fig. 2, Left) or pore (pH 5.5; Fig. 2, Right). Studies with both the prepore and the pore were performed in solution in the absence of lipid. Earlier studies indicate that the structure of the PA pore is essentially the same whether aggregated in solution or inserted into membranes. Thus, EM images of the pore collected in the absence of lipid (25) are not significantly different from images of the pore inserted into nanodiscs (26); in particular, the cap region, the region of focus in this study, is nearly identical in the two sets of images, with each showing a constriction point at the putative Φ-clamp site (25, 26). Moreover, EPR studies reported here and previously (27) indicate that the PA Φ-clamp constricts and the β-barrel forms in pores either aggregated in solution (Fig. S1) or inserted into liposomes (ref. 27 and Fig. S1).
Fig. 2.

LFN residues 2, 5, and 10 interact with PA pore. Shown are EPR spectra of spin-labeled LFN variants with and without WT PA. Black line—LFN alone. Red line—with WT PA (1∶1 molar ratio 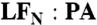 heptamer). (Left) Spectra collected at pH 8.5 (when PA is in the prepore conformation). (Right) Spectra collected at pH 5.5 (when PA is in the pore conformation). Spectra were collected at room temperature and normalized to the intensity of the central peak.
heptamer). (Left) Spectra collected at pH 8.5 (when PA is in the prepore conformation). (Right) Spectra collected at pH 5.5 (when PA is in the pore conformation). Spectra were collected at room temperature and normalized to the intensity of the central peak.
Binding of LFN spin-labeled variants G2R1, G5R1, H10R1, E20R1, and K30R1 to PA prepore induced only negligible spectral broadening (Fig. 2, Left), consistent with the crystal structure of the PA prepore-LFN complex, in which these residues lie in a disordered region (24). Similarly, binding to the pore caused no significant change in the spectra of LFN E20R1 and K30R1. With LFN variants G2R1, G5R1, and H10R1, however, a new and relatively immobilized state appeared in the spectra, suggesting direct interaction between these residues and the PA pore (Fig. 2, Right). The small population of a highly mobile state (sharp lines) could be due to unbound R1 or unbound LFN.
Interaction Between PA Pore and LFN Residues 2, 5, and 10 Is Dependent upon a Functional Φ-Clamp.
We hypothesized that the interactions observed between LFN residues 2, 5, and 10 and the pore (Fig. 2, Right) occurred in the lumen of PA, specifically at or near the Φ-clamp. To test this hypothesis, we introduced mutations (F427H or F427S) into the Φ-clamp that are known to allow for formation of ion-conductive pores but inhibit protein translocation (23). Pore-induced spectral broadening of the LFN G2R1, G5R1, and H10R1 spectra was completely abolished with PA F427H or F427S mutant pores (Fig. 3 and data not shown). In contrast, the conservative, translocation-competent mutant, PA F427W (22, 23), moderately “rescued” the spectral broadening of LFN G2R1, G5R1, and H10R1 (Fig. 3 and data not shown), further supporting the idea that broadening is caused by Φ-clamp interactions within the pore.
Fig. 3.

Interaction between PA pore and LFN residues 2 and 5 is dependent upon functional Φ-clamp. Shown are EPR spectra of LFN G2R1 or G5R1 with and without PA F427H, F427S, or F427W collected at pH 5.5 (when PA is in the pore conformation). Black line—LFN alone. Red line—with PA (1∶1 molar ratio 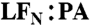 heptamer). Spectra were collected at room temperature and normalized to the intensity of the central peak.
heptamer). Spectra were collected at room temperature and normalized to the intensity of the central peak.
Construction of a PA Heteroheptamer to Assess PA-LFN Spin–Spin Interactions.
After identifying LFN residues involved in PA pore binding (Fig. 2), we sought to identify PA residues involved in these interactions by assessing LFN-PA spin–spin interactions. In order to assess a single LFN-PA spin–spin interaction, we needed to minimize LFN-LFN and PA-PA spin–spin interactions; this was achieved by constructing a PA heteroheptamer containing a single spin-labeled residue and a single LFN binding site (30). Briefly, we mixed His-tagged, spin-labeled PA83 monomer containing an unaltered domain 1′ LF binding site in a 1∶20 ratio with PA83 R178A/K214E, a domain 1′ mutant shown to be deficient in binding LF (refs. 14 and 18 and Fig. S2). We then trypsinized the mixture to allow heptamerization and isolated heteroheptamers containing a His-tagged, spin-labeled, LF-binding-competent subunit using a Ni column (Fig. S3). Assuming stochastic association of monomeric units, the average number of spin-labeled, LF-binding-competent subunits per heteroheptamer would equal 1.15. The average number of spin labels per heteroheptamer was measured as 1.2 ± 0.2, consistent with stochastic mixing and indicating that the heteroheptamers contained, on average, one functional binding site. The LFN binding ability of the heteroheptamers was tested in planar lipid bilayers. Whereas PA [R178A/K214E]7 showed drastically reduced LFN binding relative to WT PA, the PA  mutant showed LFN binding similar to WT PA (Table S1). Together, these data indicate that the PA heteroheptamers we constructed contained a single spin-labeled subunit and a single LFN binding site.
mutant showed LFN binding similar to WT PA (Table S1). Together, these data indicate that the PA heteroheptamers we constructed contained a single spin-labeled subunit and a single LFN binding site.
LFN Residues 2 and 5 Interact Directly with the Φ-Clamp in the PA Pore.
Upon finding that the interaction between the PA pore and LFN residues 2 and 5 is dependent upon a functional Φ-clamp (Fig. 3), we asked whether these LFN residues bind directly to the Φ-clamp in the pore. We were not able to introduce a spin label directly into the Φ-clamp, as mutation of even a single F427 residue causes severe defects in LFN binding and translocation (30, 31). However, it was shown in earlier work that residue S429, just 2 residues C-terminal to the Φ-clamp, can be mutated to Cys without significant effect on the function of PA (32). We therefore chose to construct the PA [S429R1]1[R178A/K214E]6 heteroheptamer (termed PA S429R1 hereafter; Fig. 1 C and D) and probed interaction of this heteroheptamer with LFN G2R1 and LFN G5R1 by measuring spin–spin interaction. In agreement with our mobility shift studies (Fig. 2) and the PA prepore-LFN crystal structure (24), LFN residues G2R1 and G5R1 did not interact with PA residue S429R1 in the prepore conformation (Fig. 4). However, both LFN residues G2R1 and G5R1 interacted with PA residue S429R1 in the pore, with a spin–spin distance estimated to be 12–13 Å (Fig. 4 and Table S2). LFN H10R1 showed very weak spin–spin interaction with PA S429R1 pore, whereas no spin–spin interaction was detected between LFN E20R1 and PA S429R1 pore.
Fig. 4.

LFN residues 2 and 5 directly interact with Φ-clamp in PA pore. Black—additive EPR spectra of spin-labeled LFN + spin-labeled PA. Red—EPR spectra of 1∶1 molar ratio spin-labeled LFN:spin-labeled PA complex. Spectra were collected at 233 K at either pH 8.5 (when PA is in the prepore state) or pH 5.5 (when PA is in the pore state) and normalized to the same number of spins.
To ensure that the LFN G2R1-PA S429R1 pore and LFN G5R1-PA S429R1 pore spin–spin interactions were also present in membrane-inserted pore, we tested LFN G2R1 and G5R1 spin–spin interaction with PA S429R1 pore inserted into liposomes. The spectra looked nearly identical to those collected in the absence of lipid, with the spectra collected in liposomes showing spin–spin interaction distances of 12–13 Å for both LFN G2R1-PA S429R1 pore and LFN G5R1-PA S429R1 pore (Fig. S4 and Table S2). To test whether this interaction was dependent upon a functional Φ-clamp, we constructed PA S429R1 heteroheptamers in which the Φ-clamp was homogeneously mutated to either F427H or F427S. For both Φ-clamp mutants, the LFN G2R1 and G5R1 spin–spin interactions with PA S429R1 pore were completely abolished (Fig. 4). These results, along with our mobility shift data (Figs. 2 and 3), indicate that LFN residues 2–5 interact with the Φ-clamp in the pore state of PA.
LFN Residue 40 Interacts with PA Prepore and Pore Independently of a Functional Φ-Clamp.
The recently solved PA prepore-LFN crystal structure (24) revealed a unique interaction between LFN helix α1 and the PA α-clamp. To test for such an interaction in solution, we constructed the LFN M40R1 variant (Fig. 1B) and examined the mobility shift of the R1 spin label upon binding PA prepore or pore. Binding to either form of heptameric PA induced broadening of the LFN M40R1 spectrum (Fig. 5A, Left), although the broadening was much more significant upon prepore binding. Mutation of PA F427 to H, S, or W (Fig. 5A, Right, and data not shown) did not affect broadening of the spectra with either prepore or pore. These results indicate that LFN residue 40 interacts with both the prepore and the pore in a Φ-clamp-independent manner. The greater spectral broadening observed with the prepore is consistent with the finding that LFN adopts a molten globular conformation under the acidic conditions that were employed in the measurements with the pore (33).
Fig. 5.

LFN residue 40 interacts with PA α-clamp. (A) EPR spectra of LFN M40R1 with and without WT PA or PA F427H. Black line—LFN M40R1 alone. Red line—with WT PA or PA F427H (1∶1 molar ratio LFN∶PA heptamer). (Top) Spectra collected at pH 8.5 (when PA is in the prepore conformation). (Bottom) Spectra collected at pH 5.5 (when PA is in the pore conformation). Spectra were collected at room temperature and normalized to the intensity of the central peak. (B) Black—additive EPR spectrum of LFN  N180R1 or PA N180R1 F427H. Red—EPR spectrum of 1∶1 molar ratio LFN M40R1:PA N180R1 or LFN M40R1:PA N180R1 F427H complex. Spectra were collected at 233 K either at pH 8.5 (when PA is in the prepore state) or pH 5.5 (when PA is in the pore state) and normalized to the same number of spins.
N180R1 or PA N180R1 F427H. Red—EPR spectrum of 1∶1 molar ratio LFN M40R1:PA N180R1 or LFN M40R1:PA N180R1 F427H complex. Spectra were collected at 233 K either at pH 8.5 (when PA is in the prepore state) or pH 5.5 (when PA is in the pore state) and normalized to the same number of spins.
Evidence for a Direct Interaction in Solution Between LFN Residue 40 and the PA α-Clamp.
We sought to determine if the interaction identified between PA and LFN residue 40 (Fig. 5A) was with the PA α-clamp, as predicted by the LFN-PA prepore crystal structure (24). As such, we constructed a PA [N180R1]1[R178A/K214E]6 heteroheptamer (termed PA N180R1 hereafter, Fig. 1 C and D), and tested for spin–spin interaction between this PA variant and LFN M40R1. Heteroheptamers were constructed such that the spin label was on the PAC face of the functional LF binding site [Fig. 1D, nomenclature defined by Feld et al. (24)]; PA residue N180 was chosen for our studies, as it is on the PAC face of the putative LFN-PA α-clamp interaction (24). Our results showed that LFN residue 40 interacted with PA residue 180 in the prepore, with a spin–spin interaction distance of ∼16 Å (Fig. 5B, Upper Left, and Table S2). These results are in good agreement with the PA prepore-LFN crystal structure, in which these residues are ∼14 Å apart (24). Consistent with our mobility shift studies, this interaction was not affected by mutation of the PA Φ-clamp to F427H (Fig. 5B, Upper Right). Interestingly, however, the LFN M40R1-PA N180R1 interaction was abolished upon formation of the PA pore (Fig. 5B, Lower Left). This was not entirely unexpected, as LFN M40R1 was shown to interact only weakly with the pore in our mobility shift assays (Fig. 5A, Lower). It seems likely that the conformational rearrangement associated with the prepore to pore conversion (5) causes a shift in the α-clamp interaction, such that LFN residue 40 and PA residue 180 are no longer within 25 Å [the distance limitation of the technique (34)]. Interestingly, mutation of the PA Φ-clamp to F427H restored a small amount (20–30%) of the LFN M40R1-PA N180R1 interaction in the pore (Fig. 5B, Lower Right, and Table S2), indicating that there is a small but measurable correlation between Φ-clamp functionality and the conformation of the α-clamp binding site in the pore.
LFN Helix α1 Pulls away from Helix α2 upon Binding PA Prepore.
In the crystal structure of LF alone, helix α1 (magenta in Fig. 1B), which contains residue 40, packs tightly against helices α2 and α3 (16) (orange and yellow, respectively, in Fig. 1B). In order for LFN residue 40 to interact with PA prepore or pore, as we observed (Fig. 5), helix α1 must pull away from helices α2 and α3 and adopt a new binding site within PA. Such a conformational rearrangement is observed in the PA prepore-LFN crystal structure (24). We constructed an LFN variant (LFN M40R1/E60R1) with a spin label in helix α1 (at residue 40) and a second one in helix α2 (at residue 60) and measured the distance between these spin-labeled residues in solution. Consistent with the LF crystal structure, LFN residues M40R1 and E60R1 interacted strongly, with ∼80% of the population having a spin–spin distance of < 10 Å (Fig. 6, Left, and Table S2). However, upon binding PA prepore, the M40R1-E60R1 spin–spin interaction within LFN decreased significantly, to < 25% (Fig. 6, Right, and Table S2). These results are consistent with the PA prepore-LFN crystal structure (24) and indicate that LFN helix α1 separates from helix α2 upon binding to the prepore.
Fig. 6.

LFN helix α1 separates from helix α2 and binds in PA α-clamp. (Left) Black—additive spectra of LFN 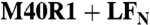 E60R1. Red—Spectrum of LFN M40R1/E60R1 alone. (Right) Black—additive spectrum of LFN M40R1 with WT
E60R1. Red—Spectrum of LFN M40R1/E60R1 alone. (Right) Black—additive spectrum of LFN M40R1 with WT  E60R1 with WT PA. Red—Spectrum of LFN M40R1/E60R1 with WT PA. Spectra were collected at pH 8.5 (when PA is in the prepore state) and normalized to the total number of spins.
E60R1 with WT PA. Red—Spectrum of LFN M40R1/E60R1 with WT PA. Spectra were collected at pH 8.5 (when PA is in the prepore state) and normalized to the total number of spins.
The Globular Portion of LFN Interacts with PA Domain 1′ of Both Prepore and Pore Independently of Φ-Clamp Functionality.
Interaction between the globular domain of LFN and PA domain 1′ has been well characterized in the prepore state of PA. However, it has been unclear how the prepore to pore conversion and mutations in the Φ-clamp affect this interaction. We measured the mobility shift of a spin label attached to LFN residue Y108 (located within the known PA domain 1′ binding site) and found that the spectrum was broadened upon binding either PA prepore or pore (Fig. 7, Left). The Φ-clamp mutation, PA F427H, did not affect broadening with either form of PA (Fig. 7, Right). These results indicate that the LFN-PA domain 1′ binding interaction exists in both the prepore state and the pore state of PA and is independent of a functional Φ-clamp.
Fig. 7.

Interaction between LFN globular domain and PA domain 1′ is independent of PA conformation and Φ-clamp functionality. Shown are EPR spectra of LFN Y108R1 with and without WT PA or PA F427H. Black line—LFN Y108R1 alone. Red line—with WT PA or PA F427H (1∶1 molar ratio LFN∶PA prepore). (Top) Spectra collected at pH 8.5 (when PA is in the prepore conformation). (Bottom) Spectra collected at pH 5.5 (when PA is in the pore conformation). Spectra were collected at room temperature and normalized to the intensity of the central peak.
In conclusion, our results show that the PA-LFN binding interaction is more complex than originally described. Three distinct LFN binding sites within PA can now be defined—the Φ-clamp, the α-clamp, and domain 1′, which interact with the extreme N terminus, helix α1, and the globular domain of LFN, respectively (Fig. 1E). The interactions of LFN with PA domain 1′ and the α-clamp are present in both the prepore and the pore, but that between the LFN N terminus and the PA Φ-clamp is present only in the pore conformation of PA, suggesting that the convergence of the Phe427 residues as the prepore converts to the pore is necessary for efficient binding at this site. Our experiments were performed in solution, in the absence of an applied voltage, indicating that interactions between LFN and the PA pore lumen are an inherent feature of the PA-LFN binding site. Greater understanding of the role each of the subsites plays will provide insights into the translocation mechanism of anthrax toxin and other members of the binary toxin family. Our results exemplify the power of SDSL-EPR as an adjunct to crystallography for defining interactions between oligomeric membrane-spanning proteins and their proteinaceous ligands.
Methods
Protein Expression and Purification.
QuikChange Site-Directed Mutagenesis (Stratagene) was used to introduce mutations into plasmids [pETSUMO (Invitrogen) or pET22b (EMD Biosciences)] encoding either recombinant LFN or PA, respectively. LFN mutants were expressed as His6-SUMO-LFN variants, which were later cleaved by SUMO (small ubiquitin-related modifier) protease, revealing the native LFN N terminus. PA variants that would be subsequently spin-labeled were expressed with a C-terminal His6 tag. All other PA variants were expressed without a purification tag. All LFN and PA mutants were purified as described (5, 21, 35).
Site-Directed Spin Labeling.
Spin labels were introduced into LFN or C-terminally His-tagged PA83 variants by reaction of an introduced cysteine with a nitroxide spin label (MTSL, Toronto Research Chemicals Inc.), resulting in the attachment of the R1 side chain (Fig. 1A). Immediately before reaction with MTSL, the LFN or PA83 variants were incubated with 10 mM DTT for 30 min at room temperature. Buffer exchange into PBS, pH 7.2, was then performed on a G25 Sephadex column, and MTSL was added at a 10× molar ratio. The reaction was allowed to proceed overnight at 4 °C, after which time buffer exchange (into 20 mM Tris, pH 8.5, 150 mM NaCl) was performed to eliminate unreacted MTSL reagent. Spin-labeling efficiency was assessed and found to be at least 70% for each LFN variant and > 95% for each PA variant.
Formation of Heteroheptamers with a Single LFN Binding Site.
His-tagged PA83 monomers (either WT or spin-labeled) were mixed in a 1∶20 ratio with PA83 R178A/K214E monomers. For Φ-clamp mutant heteroheptamers, the F427 mutation was introduced into both the His-tagged PA83 monomers and the R178A/K214E PA83 monomers; thus, the F427 residue was homogeneously mutated in the resulting heteroheptamers. Heteroheptamers were formed and isolated essentially as described, except a Ni column was used in place of an avidin column (30). Briefly, trypsin was added to the PA83 mixtures, and PA heptamers were isolated by anion exchange chromatography. His-tag-containing heteroheptamers were then separated from nontagged homoheptamers by two rounds of Ni affinity chromatography (Fig. S3).
Functionality Test of PA and LFN Variants in Planar Lipid Bilayers.
Single-LFN-binding PA heteroheptamers and all spin-labeled PA and LFN variants were tested for functionality in planar lipid bilayers. Experiments were performed as described (31) except ΔΨ (where ΔΨ = Ψcis - Ψtrans) was held at +20 mV. PA with a single LFN binding site (PA [WT]1[R178A/K214E]6) formed channels and was ∼90% blocked by LFN, although a small amount (∼25%) of LFN dissociated during perfusion (10× volume). Spin-labeled PA heteroheptamers behaved essentially as PA [WT]1[R178A/K214E]6, except for the Φ-clamp mutant heteroheptamers, which behaved as their homoheptameric counterparts. All LFN spin-labeled variants blocked ion conductance through PA channels at near WT levels (≥90%). Results are shown in Table S1.
Room Temperature Mobility Shift EPR Studies.
Spin-labeled LFN variants in 20 mM Tris, pH 8.5, 150 mM NaCl buffer, were either assayed alone or mixed in a 1∶1 molar ratio with non-spin-labeled PA heptamer. The final concentrations of both LFN and PA heptamer were ∼50–100 μM in a volume of 10 μL. For samples in which pore binding was assayed, the pH was dropped to pH 5.5 by addition of 1 μL 1 M sodium acetate buffer, pH 5.0. The EPR spectra were then recorded at room temperature on a Bruker EMX spectrometer at a microwave power of 2 mW, sweeping the magnetic field from 3434–3534 G at a frequency of 9.45 GHz. Spectra were recorded a minimum of two times; little to no variation was observed between trials.
Low Temperature Spin–Spin Interaction EPR Studies.
Spectra were collected of LFN spin-labeled variants alone, spin-labeled PA heteroheptamer variants alone, and a 1∶1 molar ratio mixture of spin-labeled LFN:spin-labeled PA heteroheptamer. The samples were in buffer containing 20 mM Tris, pH 8.5, 150 mM NaCl. The final concentrations of both LFN and PA heptamer were ∼80–200 μM in a volume of 10 μL. The pH was either held at 8.5 or dropped to 5.5 by the addition of 1.6 μL 100 mM HCl or 2 μL 1 M sodium acetate buffer, pH 5.0. As a cryoprotectant, 10 μL of 80% glycerol was then added to the samples, to yield a final concentration of 40%. The EPR spectra were then recorded at 233 K on a Bruker EMX spectrometer at a microwave power of 2 mW sweeping the magnetic field from 3,260 to 3,460 G at a frequency of 9.45 GHz. Spectra were recorded a minimum of two times; little to no variation was observed between trials. To assess spin–spin distances, the EPR spectrum of each spin-labeled LFN:spin-labeled PA heteroheptamer mixture was compared to the spectrum formed by adding that of spin-labeled LFN alone and that of spin-labeled PA heteroheptamer alone. For studies with LFN M40R1/E60R1, spin–spin distances were assessed by comparing the spectrum of doubly labeled LFN to the additive spectrum of LFN M40R1 + LFN E60R1. The spin–spin distances were calculated using a Monte Carlo/simplex Gaussian convolution method (36). The results are shown in Table S2. To ensure that the spin–spin interactions observed were not due to background LFN-LFN spin–spin interaction (which might arise in the unlikely event that two LFN molecules bound to the same PA heteroheptamer), we recorded the spectra of LFN G2R1, LFN G5R1, and LFN M40R1 bound to WT PA. No spin–spin interaction was observed in any of these controls (Fig. S5).
Supplementary Material
Acknowledgments.
We thank Dr. Robin Ross and Lauren Perry of the Biomolecule Production Core of New England Regional Center of Excellence, supported by National Institutes of Health (NIH) Grant AI057159, for helping with bacterial growth and expression of proteins. We also thank Prof. Blythe Janowiak (St. Louis University, St. Louis, MO) for supplying plasmids, Prof. Bryan Krantz, Geoffrey Feld, and colleagues for allowing access to the PA prepore-LFN crystal structure coordinates ahead of print, and Prof. Ellis Reinherz for providing time on the Bruker EMX spectrometer. This work was supported by NIH Grant AI022021 (to R.J.C.).
Footnotes
Conflict of interest statement: R.J.C. holds equity in PharmAthene, Inc.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1018965108/-/DCSupplemental.
References
- 1.Leppla SH. Anthrax toxin edema factor: A bacterial adenylate cyclase that increases cyclic AMP concentrations of eukaryotic cells. Proc Natl Acad Sci USA. 1982;79:3162–3166. doi: 10.1073/pnas.79.10.3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klimpel KR, Arora N, Leppla SH. Anthrax toxin lethal factor contains a zinc metalloprotease consensus sequence which is required for lethal toxin activity. Mol Microbiol. 1994;13:1093–1100. doi: 10.1111/j.1365-2958.1994.tb00500.x. [DOI] [PubMed] [Google Scholar]
- 3.Bradley KA, Mogridge J, Mourez M, Collier RJ, Young JA. Identification of the cellular receptor for Anthrax toxin. Nature. 2001;414:225–229. doi: 10.1038/n35101999. [DOI] [PubMed] [Google Scholar]
- 4.Scobie HM, Rainey GJ, Bradley KA, Young JA. Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc Natl Acad Sci USA. 2003;100:5170–5174. doi: 10.1073/pnas.0431098100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miller CJ, Elliott JL, Collier RJ. Anthrax protective antigen: Prepore-to-pore conversion. Biochemistry. 1999;38:10432–10441. doi: 10.1021/bi990792d. [DOI] [PubMed] [Google Scholar]
- 6.Klimpel KR, Molloy SS, Thomas G, Leppla SH. Anthrax toxin protective antigen is activated by a cell surface protease with the sequence specificity and catalytic properties of furin. Proc Natl Acad Sci USA. 1992;89:10277–10281. doi: 10.1073/pnas.89.21.10277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petosa C, Collier RJ, Klimpel KR, Leppla SH, Liddington RC. Crystal structure of the Anthrax toxin protective antigen. Nature. 1997;385:833–838. doi: 10.1038/385833a0. [DOI] [PubMed] [Google Scholar]
- 8.Kintzer AF, et al. The protective antigen component of anthrax toxin forms functional octameric complexes. J Mol Biol. 2009;392:614–629. doi: 10.1016/j.jmb.2009.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mogridge J, Cunningham K, Collier RJ. Stoichiometry of anthrax toxin complexes. Biochemistry. 2002;41:1079–1082. doi: 10.1021/bi015860m. [DOI] [PubMed] [Google Scholar]
- 10.Abrami L, Liu S, Cosson P, Leppla SH, vander Goot FG. Anthrax toxin triggers endocytosis of its receptor via a lipid raft-mediated clathrin-dependent process. J Cell Biol. 2003;160:321–328. doi: 10.1083/jcb.200211018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Collier RJ. Mechanism of membrane translocation by anthrax toxin: Insertion and pore formation by protective antigen. J Appl Microbiol. 1999;87:283. doi: 10.1046/j.1365-2672.1999.00889.x. [DOI] [PubMed] [Google Scholar]
- 12.Young JA, Collier RJ. Anthrax toxin: Receptor binding, internalization, pore formation, and translocation. Annu Rev Biochem. 2007;76:243–265. doi: 10.1146/annurev.biochem.75.103004.142728. [DOI] [PubMed] [Google Scholar]
- 13.Collier RJ. Membrane translocation by anthrax toxin. Mol Aspects Med. 2009;30:413–422. doi: 10.1016/j.mam.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cunningham K, Lacy DB, Mogridge J, Collier RJ. Mapping the lethal factor and edema factor binding sites on oligomeric anthrax protective antigen. Proc Natl Acad Sci USA. 2002;99:7049–7053. doi: 10.1073/pnas.062160399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lacy DB, Mourez M, Fouassier A, Collier RJ. Mapping the anthrax protective antigen binding site on the lethal and edema factors. J Biol Chem. 2002;277:3006–3010. doi: 10.1074/jbc.M109997200. [DOI] [PubMed] [Google Scholar]
- 16.Pannifer AD, et al. Crystal structure of the anthrax lethal factor. Nature. 2001;414:229–233. doi: 10.1038/n35101998. [DOI] [PubMed] [Google Scholar]
- 17.Shen Y, Zhukovskaya NL, Guo Q, Florian J, Tang WJ. Calcium-independent calmodulin binding and two-metal-ion catalytic mechanism of anthrax edema factor. EMBO J. 2005;24:929–941. doi: 10.1038/sj.emboj.7600574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lacy DB, et al. A model of anthrax toxin lethal factor bound to protective antigen. Proc Natl Acad Sci USA. 2005;102:16409–16414. doi: 10.1073/pnas.0508259102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blaustein RO, Koehler TM, Collier RJ, Finkelstein A. Anthrax toxin: Channel-forming activity of protective antigen in planar phospholipid bilayers. Proc Natl Acad Sci USA. 1989;86:2209–2213. doi: 10.1073/pnas.86.7.2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang S, Udho E, Wu Z, Collier RJ, Finkelstein A. Protein translocation through anthrax toxin channels formed in planar lipid bilayers. Biophys J. 2004;87:3842–3849. doi: 10.1529/biophysj.104.050864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang S, Finkelstein A, Collier RJ. Evidence that translocation of anthrax toxin’s lethal factor is initiated by entry of its N terminus into the protective antigen channel. Proc Natl Acad Sci USA. 2004;101:16756–16761. doi: 10.1073/pnas.0405754101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krantz BA, et al. A phenylalanine clamp catalyzes protein translocation through the anthrax toxin pore. Science. 2005;309:777–781. doi: 10.1126/science.1113380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun J, Lang AE, Aktories K, Collier RJ. Phenylalanine-427 of anthrax protective antigen functions in both pore formation and protein translocation. Proc Natl Acad Sci USA. 2008;105:4346–4351. doi: 10.1073/pnas.0800701105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feld GK, et al. Structural basis for the unfolding of anthrax lethal factor by protective antigen oligomers. Nat Struct Mol Biol. 2010;17:1383–1390. doi: 10.1038/nsmb.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Katayama H, et al. GroEL as a molecular scaffold for structural analysis of the anthrax toxin pore. Nat Struct Mol Biol. 2008;15:754–760. doi: 10.1038/nsmb.1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Katayama H, et al. Three-dimensional structure of the anthrax toxin pore inserted into lipid nanodiscs and lipid vesicles. Proc Natl Acad Sci USA. 2010;107:3453–3457. doi: 10.1073/pnas.1000100107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vernier G, et al. Solubilization and characterization of the anthrax toxin pore in detergent micelles. Protein Sci. 2009;18:1882–1895. doi: 10.1002/pro.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hubbell WL, Cafiso DS, Altenbach C. Identifying conformational changes with site-directed spin labeling. Nat Struct Biol. 2000;7:735–739. doi: 10.1038/78956. [DOI] [PubMed] [Google Scholar]
- 29.Fanucci GE, Cafiso DS. Recent advances and applications of site-directed spin labeling. Curr Opin Struct Biol. 2006;16:644–653. doi: 10.1016/j.sbi.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 30.Janowiak BE, Finkelstein A, Collier RJ. An approach to characterizing single-subunit mutations in multimeric prepores and pores of anthrax protective antigen. Protein Sci. 2009;18:348–358. doi: 10.1002/pro.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Janowiak BE, Fischer A, Collier RJ. Effects of introducing a single charged residue into the phenylalanine clamp of multimeric anthrax protective antigen. J Biol Chem. 2010;285:8130–8137. doi: 10.1074/jbc.M109.093195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mourez M, et al. Mapping dominant-negative mutations of anthrax protective antigen by scanning mutagenesis. Proc Natl Acad Sci USA. 2003;100:13803–13808. doi: 10.1073/pnas.2436299100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krantz BA, Trivedi AD, Cunningham K, Christensen KA, Collier RJ. Acid-induced unfolding of the amino-terminal domains of the lethal and edema factors of anthrax toxin. J Mol Biol. 2004;344:739–756. doi: 10.1016/j.jmb.2004.09.067. [DOI] [PubMed] [Google Scholar]
- 34.Rabenstein MD, Shin YK. Determination of the distance between two spin labels attached to a macromolecule. Proc Natl Acad Sci USA. 1995;92:8239–8243. doi: 10.1073/pnas.92.18.8239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pentelute BL, Barker AP, Janowiak BE, Kent SB, Collier RJ. A semisynthesis platform for investigating structure-function relationships in the N-terminal domain of the anthrax lethal factor. ACS Chem Biol. 2010;5(4):359–364. doi: 10.1021/cb100003r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fajer PG, Brown LJ, Song L. In: ESR Spectroscopy in Membrane Biophysics. Hemminga M, Berliner L, editors. New York: Springer; 2007. pp. 95–128. [Google Scholar]
- 37.Lacy DB, Wigelsworth DJ, Melnyk RA, Harrison SC, Collier RJ. Structure of heptameric protective antigen bound to an anthrax toxin receptor: A role for receptor in pH-dependent pore formation. Proc Natl Acad Sci USA. 2004;101:13147–13151. doi: 10.1073/pnas.0405405101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Melnyk RA, et al. Structural determinants for the binding of anthrax lethal factor to oligomeric protective antigen. J Biol Chem. 2006;281:1630–1635. doi: 10.1074/jbc.M511164200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.