

Abstract
Transcription factors that drive complex patterns of gene expression during animal development bind to thousands of genomic regions, with quantitative differences in binding across bound regions mediating their activity. While we now have tools to characterize the DNA affinities of these proteins and to precisely measure their genome-wide distribution in vivo, our understanding of the forces that determine where, when, and to what extent they bind remains primitive. Here we use a thermodynamic model of transcription factor binding to evaluate the contribution of different biophysical forces to the binding of five regulators of early embryonic anterior-posterior patterning in Drosophila melanogaster. Predictions based on DNA sequence and in vitro protein-DNA affinities alone achieve a correlation of ∼0.4 with experimental measurements of in vivo binding. Incorporating cooperativity and competition among the five factors, and accounting for spatial patterning by modeling binding in every nucleus independently, had little effect on prediction accuracy. A major source of error was the prediction of binding events that do not occur in vivo, which we hypothesized reflected reduced accessibility of chromatin. To test this, we incorporated experimental measurements of genome-wide DNA accessibility into our model, effectively restricting predicted binding to regions of open chromatin. This dramatically improved our predictions to a correlation of 0.6–0.9 for various factors across known target genes. Finally, we used our model to quantify the roles of DNA sequence, accessibility, and binding competition and cooperativity. Our results show that, in regions of open chromatin, binding can be predicted almost exclusively by the sequence specificity of individual factors, with a minimal role for protein interactions. We suggest that a combination of experimentally determined chromatin accessibility data and simple computational models of transcription factor binding may be used to predict the binding landscape of any animal transcription factor with significant precision.
Author Summary
During early stages of development, regulatory proteins bind DNA and control the expression of nearby genes, thereby driving spatial and temporal patterns of gene expression during development. But the biochemical forces that determine where these regulatory proteins bind are poorly understood. We gathered experimental data on the activities of several key regulators of early development of the fruit fly (Drosophila melanogaster) and developed a computational method to predict where and how strongly they will bind. We find that competition, cooperativity, and other interactions among individual regulatory proteins have a limited effect on their binding, while the global accessibility of DNA to protein binding has a significant impact on the binding of all factors. Our results suggest a practical method for predicting regulatory binding by combining experimental DNA accessibility assays with computational algorithms to determine where will binding occur among the accessible regions of the genome.
Introduction
In vivo crosslinking studies show that animal transcription factors each bind to many thousands of DNA regions throughout the genome (e.g. [1]–[18]). While many of the strongest binding events are at functional cis regulatory modules and are evolutionary conserved, many thousands of other genomic regions that are bound at lower levels do not appear to be functional targets [2], [13], [14], [16], [18], [19]. In addition, factors with unrelated biochemical or functional properties bind to the same genomic regions with surprisingly high frequency [2], [16], [20], while the biological specificities of factors appear to be determined in part by quantitative differences in occupancy between proteins at these commonly bound sites [2], [16], [21], [22].
It is a fundamental challenge to determine the biochemical mechanisms that direct these complex, quantitative patterns of factor occupancy. Animal transcription factors bind to short (5–12 bp) sequences of DNA that occur with high frequency throughout the genome [23], yet most occurrences of these recognition sequences are not detectably bound in vivo [2], [5], [13], [24], [25]. This discrepancy between predicted and observed factor binding has been attributed to several mechanisms that could alter the simple behavior of a factor, including: (1) competitive inhibition of binding at those DNA regions that overlap sites occupied either by other sequence specific factors [26], [27] or by nucleosomes and chromatin associated proteins [28]–[33]. (2) Direct and indirect cooperative interactions between factors bound at physically proximal sites that increase their affinity at those sites [34]–[42]. The relative influence that each of these biochemical mechanisms on the overall pattern of factor binding in vivo, however, is currently a matter of debate.
The anterior-posterior (A-P) patterning system in early Drosophila melanogaster development offers an excellent system for addressing the question of transcription factor targeting in an animal regulatory network. Prior to gastrulation, the Drosophila embryo is a syncytium of ∼6,000 nuclei distributed around its periphery [43]. Extensive genetic and molecular analyses have established that a simple regulatory cascade involving the morphogens Bicoid (BCD) and Caudal (CAD), and the gap transcription factors Hunchback (HB), Giant (GT), Kruppel (KR) and Knirps (KNI) directs the expression of numerous target genes in complex three dimensional patterns across the embryo, which in turn establish the segmental body plan in the trunk of the developing fly [44]–[46].
To analyze this network at a systems level, we have previously established a complementary set of quantitative datasets describing key aspects of the network. We have measured the widespread, overlapping in vivo binding of these and other early regulatory factors genome wide [13], [16], [18], In addition, we have characterized DNA affinities of these proteins from in vitro selection (SELEX) experiments (Berkeley Drosophila Transcription Network Project, unpublished data), measured the relative concentration of each of these transcription factors in every embryonic nucleus [47], and determined the accessibility of DNA regions throughout the genome.
Here we incorporate this series of datasets into quantitative models of transcription factor binding to measure the impact of different biophysical forces on occupancy levels. Many previous studies have used computational approaches to predict the locations of genomic regions bound by transcription factors [48]–[68]. While these studies have discovered a number of important principles, they have not sought to quantify the contribution of various factor-targeting forces in the context of the highly overlapping, widespread binding seen in animal systems. For example, many have produced qualitative predictions of which genomic regions might be occupied, and thus do not provide information on the levels of expected factor occupancy, which have been shown to be critical for relating binding patterns to biological function [2], [16], [21], [22]. In addition, while some studies have focused on determining the biochemical mechanisms targeting factors to DNA [57], [58], [60], many others have used additional biological information such as microarray expression data or DNA recognition site conservation to predict only those sites that are functional [59], [63], [65], [67], [68], and thus do not, strictly speaking, attempt to predict binding per se. Finally, when predictions of quantitative occupancy levels have been made, these were either part of more complex models of transcriptional output patterns that did not compare the binding models to any experimental in vivo binding data [66], [69]–[71], or were made to coarse in vivo binding information from yeast [58], [62].
Therefore, we have set out to establish a computational framework that predicts the levels of factor occupancy in a way that allows the effect of various proposed biophysical mechanisms influencing factor binding to be quantified. Our results support a long standing model that suggest that animal transcription factors are expressed at sufficiently high concentrations in cells that they can bind to their recognition sites in accessible parts of the genome without the aid of direct cooperative interactions with other proteins [31], [72], [73]. In this view, the dominant force in cells that modifies the intrinsic DNA specificity of transcription factors is the inhibition of DNA binding by chromatin structure. Because of the high frequency of factor recognition sites in most short accessible regions of the genome, this model explains why animal factors show such widespread, overlapping binding.
Results
Quantitative Models of Transcription Factor Binding
We developed a probabilistic framework to infer the occupancies of one or more transcription factors across any DNA sequence given the concentration of these factors and their DNA binding preferences (Figure S1). Our model is based on the formalism of generalized hidden Markov models (gHMMs; [49], [53], [55], [62], [69], ), which allow efficient integration of the different forces that may influence transcription factor binding into the model.
This class of models offers several prominent advantages. gHMMs have very few parameters and are therefore easy to optimize. And, unlike most probabilistic graphical models, they offer exact inference of posterior probabilities in linear time, using the forward-backward dynamic programming algorithm [75]. Finally, gHMMs are related to thermodynamic equilibrium models, with the ensemble of all possible configurations of bound factors viewed as a Boltzmann distribution and each configuration assigned a weight (or probability) depending on its energetic state. The probability that a factor is bound at a given location is assumed to be the fraction of configurations (weighted by their probabilities) in which it is bound [50], [55], [56], [62], [69], [76]–[80].
The Markovian property of gHMMs prevents them from considering the full context in which binding occurs, and thus they offer only an approximation of the full thermodynamic model. We overcome this limitation with a sampling procedure (see below).
In Vitro and In Vivo DNA Binding Data
To model the DNA binding affinities of the five factors considered in this study (HB, BCD, KR, GT and CAD), we used in vitro specificities (expressed as position weight matrices; PWMs) measured using SELEX-Seq by the Berkeley Drosophila Transcription Network Project (bdtnp.lbl.gov).
For in vivo binding data, we used ChIP-seq measurements of formaldehyde crosslinked HB, BCD, KR, GT and CAD from blastoderm embryos of Drosophila melanogaster (Oregon R) [18]. A range of controls establishes that these data provide a quantitative measure of the relative levels of transcription factor directly bound to different genomic DNA regions [2], [13], [16], [18], [31], [81]. In particular, in vitro controls using purified transcription factors and naked DNA, where binding at individual sites is expected to be proportional to the affinity of the factor for that site, show that relative levels of crosslinking closely correlate with relative DNA affinity [81]. “Spike in” experiments using ∼200 kb BAC DNAs show that the ChIP-seq post immunoprecipitation processing steps accurately preserve the relative enrichment levels of different genomic regions, with the enrichments of DNA samples prior to and post processing showing a correlation of 0.997 and a linear fit of slope of 1.14 (Figure S2). In vivo UV crosslinking results show that similar data are obtained when protein/protein crosslinking is absent [2], [16]. And the genomic regions identified as bound by factors in our ChIP-seq experiments are not preferentially enriched in the crosslinked DNA initially used as input to the immunoprecipitations [18].
This latter observation is particularly important, as Auerbach et al have reported that some methods of DNA isolation can introduce a bias that adversely affects ChIP-seq data [82]. In particular, they found that the sonication of intact nuclei leads to the preferential enrichment of regions of open chromatin. Although the crosslinked DNA used in our ChIP-seq experiments is sonicated only after it has been purified away from non-crosslinked proteins by buoyant density centrifugation [13], and thus is unlikely to suffer the above DNA extraction bias, we nonetheless directly compared the input DNA samples from our ChIP-seq experiments to DNase I-seq genome accessibility data for embryos at the same stage of development (Thomas et al, unpublished data). In contrast to Auerbach et al [82], we found no correlation between ChIP-seq input samples and the DNase I-seq data (mean correlation coefficient of -0.0005).
Thus, while we do not believe ChIP-seq represents an absolutely precise measure of binding to each region of the genome (discussed further below), a wide range of evidence indicates that our ChIP-seq data do not appear to suffer large systematic biases that could interfere with our modeling effort and that these data provide a sufficiently quantitative estimate of binding on which to base attempts to predict it.
Comparing Model Predictions and In Vivo Binding Data
We developed a model-based algorithm to compare the binding probabilities predicted by our gHMM model to high-resolution in vivo binding measurements [83]. Using the length distribution of the DNA fragments recovered by ChIP, we simulated the shape of the peak corresponding to a single binding event, as measured by ChIP-seq. We then used that shape to convert the single-nucleotide resolution binding probabilities into an expected ChIP-seq profile (Figure S3, Methods).
To analyze our predictions, we compiled a list of 21 known target loci of the A-P patterning system, primarily known maternal, gap, and pair rule genes [84], expressed during early stage 5 [47]. Each gene was expanded by ∼10 Kb upstream and downstream of the transcription unit to capture its known regulatory sequences. In each of the analyses presented below, we trained the model parameters to optimize the fit between the predicted and observed ChIP-seq landscapes at a set of six loci spanning over 87 Kb (croc, cnc, slp, kni, hkb, D), and evaluated the trained model on a set of 15 loci spanning over 280 Kb (prd, h, eve, cad, oc, opa, ftz, gt, hb, Kr, odd, run, fkh, tll, os). To account for long genomic regions where no binding is observed in vivo, we enhanced the train and test sets by adding three and five control loci, spanning 100 Kb and 221 Kb, respectively (see Table 1).
Table 1. Genes and coordinates for train and test set loci.
| Train | |||
| Symbol | Gene name | Locus coordinates | Length |
| croc | crocodile | chr3L:21,461,001–21,477,000 | 16 Kb |
| cnc | cap-n-collar | chr3R:19,011,001–19,024,000 | 13 Kb |
| slp | sloppy paired | chr2L:3,820,001–3,840,000 | 20 Kb |
| kni | knirps | chr3L:20,683,260–20,695,259 | 12 Kb |
| hkb | huckebein | chr3R:169,001–181,000 | 12 Kb |
| D | Dichaete | chr3L:14,165,001–14,179,000 | 14 Kb |
| control2 | - | chr3L:21,764,501–21,792,500 | 28 Kb |
| control5 | - | chr3R:3,145,001–3,170,000 | 25 Kb |
| control9 | - | chr2L:10,060,001–10,107,000 | 47 Kb |
Genomic coordinates of the six training set loci, spanning a total of 87 Kb, and 15 test set loci, spanning 280 Kb. This list consists of known target genes of the A-P patterning system, that are expressed during early stage 5. Each gene was expanded by ∼10 Kb to include regulatory sequence. In addition, the list includes three control loci that were added to the train set, and five added to the test set.
Quantitative Comparisons of Model Predictions to In Vivo Binding Data
We began with the simplest model – a single transcription factor binding to DNA. This required optimizing only a single parameter, P(t), for each factor, corresponding to its effective concentration in nuclei. After training, the correlation coefficients between the model's predicted binding and the training data range from poor (CAD, 0.11) to reasonable (BCD, 0.58) with a total correlation of 0.37. Correlations for the test set were similar (suggesting that the training data were not over fit), and ranged from 0.15 (GT) to 0.66 (BCD), with a total correlation of 0.36 (Table 2 and Table S1). Figure 1A shows the model's predictions and the in vivo data for several test loci.
Table 2. Factor-specific accuracy at increasing degrees of model complexity.
| Train set | Test set | |||||||||||
| BCD | CAD | GT | HB | KR | Total | BCD | CAD | GT | HB | KR | Total | |
| 1 | 0.58 | 0.1 | 0.18 | 0.49 | 0.52 | 0.38 | 0.66 | 0.28 | 0.15 | 0.37 | 0.35 | 0.36 |
| 2 | 0.54 | 0.07 | 0.12 | 0.44 | 0.51 | 0.33 | 0.65 | 0.24 | 0.08 | 0.35 | 0.34 | 0.33 |
| 3 | 0.55 | 0.07 | 0.12 | 0.46 | 0.53 | 0.35 | 0.65 | 0.24 | 0.09 | 0.36 | 0.36 | 0.34 |
| 4 | 0.49 | 0.07 | 0.18 | 0.43 | 0.55 | 0.35 | 0.56 | 0.16 | 0.10 | 0.38 | 0.37 | 0.31 |
| 5 | 0.58 | 0.11 | 0.21 | 0.50 | 0.55 | 0.39 | 0.65 | 0.29 | 0.15 | 0.39 | 0.38 | 0.37 |
| 6 | 0.87 | 0.67 | 0.66 | 0.75 | 0.71 | 0.73 | 0.78 | 0.79 | 0.53 | 0.58 | 0.60 | 0.65 |
| 7 | 0.90 | 0.67 | 0.72 | 0.79 | 0.71 | 0.76 | 0.79 | 0.78 | 0.59 | 0.58 | 0.62 | 0.67 |
Accuracy of model's predictions at increasing degrees of model complexity. Shown are factor-specific correlations between the predicted binding landscape and measured occupancies for train- (left) and test set loci (right). Variations of the generalized hidden Markov model include (in increasing levels of complexity): (1) independent predictions per factor; (2) joint predictions (allowing for direct binding competition); (3) predictions at single-nucleus resolution; (4) with sequence-specific model of nucleosome binding; (5) with sequence-independent model of nucleosome binding; (6) with non-uniform prior on protein binding, based on DNase I hypersensitivity assay; (7) with cooperative binding interactions.
Figure 1. High-resolution predictions of protein-DNA binding landscape.

(A) The model's binding predictions (red line) are compared to in vivo binding landscape (solid blue). Shown are BCD binding at the 16 Kb eve locus (left), BCD binding at the 15 Kb os locus (middle), and CAD binding at the 24 Kb fkh locus (right). Here, the binding landscape was predicted independently for each transcription factor. (B) Same as (A), except allowing for direct binding competition between the five factors and with nucleosomes, and modeling binding independently in each of 6,078 nuclei of the fly embryo. (C) Same as (B), while incorporating non-uniform DNase I hypersensitivity-based prior on transcription factor binding to account for variations in DNA accessibility (shown in gray). (D) Same as (C), after adding cooperative interactions between adjacently bound factors in a thermodynamic setting.
Minimal Effect of Factor Competition in Predicting In Vivo Binding
Encouraged by results with single factors, we examined the effect of competition between the five factors on our ability to predict in vivo binding. Overlapping binding sites are known to modulate direct competition between factors for some individual cases [26], [27]. Moreover, overlapping sites are often conserved at long evolutionary distances [85], [86], suggesting an important mechanistic role for inter-factor competition. We expanded the gHMM in our model to consider all five transcription factors simultaneously in a probabilistic framework, where the different concentration of each factor t is modeled by an additional probabilistic term P(t). In the single factor model binding of one factor to a site did not affect the binding of a different factor to the same site. In this new model, however, because the total occupancy at a site cannot exceed 1, factors effectively compete for binding to overlapping target sites. While other early regulators bind many of the same regulatory regions as these five factors [16], we have not included them in our model to allow for a focused examination of the factors that are most closely implicated in functioning together and because of a lack of complete DNA binding specificity data.
To our surprise, the five-factor competitive model gave slightly less accurate predictions than its single factor counterpart. On the test data, we observed decrease in the model predictions from a total correlation of 0.36 to 0.33 (see Figure 2 and Table 2 for full data).
Figure 2. Prediction accuracy at increasing degrees of model complexity.

(A) Accuracy of binding predictions at train set, including six known A-P targets and three control loci. Shown are the correlations between the model predictions and the in vivo binding landscape, at various degrees of model complexity. These include, from left to right: (1) independent predictions per transcription factor; (2) allowing binding competition between factors; (3) predictions at a single-nucleus resolution; (4) with sequence-specific model of nucleosome binding; (5) with sequence-independent model of nucleosome binding; (6) adding non-uniform prior on transcription factor binding using DNA accessibility measurements; and (7) adding cooperative binding interactions in a thermodynamic settings. (B) Same as (A), but for test set, including 15 known A-P targets and five control loci.
Expanding the Model to Three Dimensions with Single Nucleus Resolution
One potential explanation for the lack of improvement for the above competition model could have been that, because we were treating the embryo as a homogenous entity, it allowed competition between factors that are expressed together at high levels in few nuclei (e.g. Figure 3A: GT and KR, BCD and CAD, or KR and HB).
Figure 3. Predicting binding in single-nucleus resolution.

(A) Three-dimensional single-cell measurements of protein concentrations [47] were used to estimate the concentration of the five transcription factors across the fly embryo. (B) To model binding competition while considering the differential concentration of factors, we modeled binding in each of the ∼6,000 nuclei of a fly embryo separately. Depicted are the probabilities of binding at the 485 bp-long eve stripe 2 CRM (chr2R:5,865,266-5,865,750) for the five factors, at three example nuclei: one at an the anterior pole, one towards the posterior end, and one at the center of the embryo. (C) Nucleus by nucleus predictions were averaged to predict the binding over the entire embryo. Shown are the predicted occupancies of the five factors along the entire 16 Kb eve locus (below), and along the stripe 2 CRM (inset).
We therefore expanded our algorithm to model the binding of all factors in each of the ∼6000 nuclei separately. We used single nucleus estimates of protein concentration, based on three dimensional fluorescence microscopy of D. melanogaster embryos at early stage 5 [47] to scale the optimized concentration parameters of the five transcription factors. Specifically at each run, we multiplied the concentrations P(t) of every regulator by its protein expression level. We then averaged the predicted binding landscape of all nuclei, to obtain whole-embryo genomic predictions, which were compared to (whole-embryo) in vivo binding measurements (Figure 3). The results were slightly improved relatively to the whole-embryo predictions (3–4% improvement on the training (0.34) and test (0.34) sets), presumably because some inappropriate competition events had been eliminated from the model.
Further analysis suggests why including binding site competition did not have a major affect on the predictive power of the model. The stronger affinity recognition sites for proteins that are co-expressed in the same cells, i.e. those having the potential to significantly affect net occupancy of a protein in vivo, overlap in only a minority of cases in the training or test genomic regions. Thus while binding site competition between the five factors may well play a key regulatory role at a subset of sites in a subset of cells as previously proposed [26], [27], it is unlikely to have a major effect on net occupancy averaged across all cells and many genomic regions.
DNA Accessibility Data Greatly Improve Binding Predictions
Even if direct competition between the five factors does not play a major role in shaping their binding landscape, other mechanisms, including interactions with nucleosomes and covalent modifications to nearby histones [29], [30], [37], [39], [87], [88], could allow transcription factors to affect binding to neighboring sites.
When comparing our computational binding predictions (Figure 1) to experimental maps of in vivo binding, we found that the majority of highly bound regions in vivo were indeed correctly predicted (133 of 240 peaks were correctly predicted, for all factors, with specific percentages varying from 30% (KR, 14/46 peaks) to 86% (BCD, 36/42)). Nonetheless, our model suffered from many false positive predictions (Figure 4), a common problem when attempting to predict in vivo binding from DNA sequence [48], [52]. It is widely accepted that a major source of such over-prediction is that many potential high-affinity binding sites are found in regions of the genome that are densely packed into chromatin states that limit their accessibility to regulatory proteins [28], [31], [33]–[35], [39], [87]–[90] and that information on the state of the chromatin can be used to improve prediction of transcription factor binding [32], [33], [41], [57], [63]–[65], [68], [70]. In some cases, the weak sequence preferences of nucleosomes have been used to predict nucleosomes positions and thereby improve binding site or gene expression predictions [57], [62], [70]. Alternatively, histone modification data has been used to identify genomic regions with putative regulatory function [63], [68]. These previous methods, however, did not make use of direct experimental measurements of DNA accessibility.
Figure 4. Predictions with and without DNA accessibility prior.

(A) Measured (X-axis) vs. predicted (Y-axis) occupancy for all factors along all test loci. Predicted binding is based on a 3D cellular resolution mode, which allows for binding competition between factors and sequence-independent nucleosomes. (B) Same as (A), while coloring each genomic position based on to its DNA accessibility, ranging from pale cyan (lowly accessible) to dark blue (highly accessible). Almost all false binding predictions (dots above the diagonal) are lowly accessible in vivo. (C,D) Same as (A–B), but with DNase I hypersensitivity-based prior on transcription factor binding integrated into the model. This results with more accurate predictions, as measured by the correlation between measured and predicted occupancy, improving from 0.37 to 0.655.
To measure the influence of chromatin state on the accuracy of our model's binding predictions, we turned to two complementary perspectives. First, we integrated the exact position of nucleosomes into our model at single nucleotide resolution to enable the competition between transcription factors and nucleosomes in binding DNA to be modeled [30], [33], [35], [39], [41], [57], [62], [70], [87], [88]. As there are no direct measurements of nucleosome positions from early Drosophila embryos, we modeled these computationally (see below). Secondly, because chromatin accessibility results from the combined effect of nucleosomes, other chromatin binding proteins and the higher-order 3D packaging of the DNA, we used direct genome-wide measurements of DNA accessibility obtained from DNase I digestion of chromatin in isolated blastoderm embryo nuclei [91]. We then quantified the effect of these two ways of assessing chromatin state on predicting the binding landscape in turn.
Predicting Nucleosome Positions Does Not Improve Model Predictions
In analyzing the role of nucleosome positioning, we were particularly interested in the possibility that the binding of one factor would reduce local nucleosome occupancy and therefore increase the occupancy of other factors at adjacent sites, so called indirect cooperative DNA binding [35], [37], [39], [87]. To investigate this, we extended the cellular resolution (3D) model by incorporating an additional state to represent the 141 bp-long sequence bound by a single nucleosome.
Due to uncertainty in the literature about the contribution of sequence specificity to in vivo nucleosome positioning [92], we decided to evaluate two different ways of incorporating positioned nucleosomes into our models. First, we used a sequence-specific model of nucleosome binding, that takes into account presumed preferences for certain DNA sequence features [93] as an additional state of our generalized hidden Markov models. This addition did not improve the predictions of our model, when comparing our predictions to in vivo measurements, obtaining correlations of 0.35 and 0.33, on the training and test sets, respectively (Table 2).
As an alternative, we used a sequence-independent model of nucleosome binding, where nucleosome are viewed as long “space-fillers” that, when present, prevent regulators from binding. This model obtained correlations of 0.37 and 0.36 on the training and test data, respectively, an improvement of 6-7% over the non-nucleosomal 3D model (see Figure 2B and Table 2). Yet, even this model fails to significantly outperform the naïve algorithm of modeling a single-regulator at a time (no competition and no 3D resolution).
Direct Measurements of Chromatin Accessibility
To more directly quantify the effect of chromatin accessibility on in vivo binding, we used genome-wide measurements of DNA accessibility obtained from DNase I digestion of isolated blastoderm embryo nuclei [91], followed by deep sequencing of the short DNA fragments that were released. When comparing these accessibility data to the predictions of our model, we found that genomic loci for which our model correctly predicts DNA binding based on sequence, namely true-positive loci, tend to have higher DNA accessibility (mean DNA accessibility of 0.36, with 25th and 75th percentiles at 0.10 and 0.60, respectively. See Methods), while false-positive loci (for which we predicted stronger binding than actually measured) typically displayed much lower DNA accessibility (mean DNA accessibility of 0.06, with 25th and 75th percentiles at 0.01 and 0.05, respectively).
Based on this observation, we next partitioned the genome based on DNA accessibility (with a threshold of 0.5), and compared the true positive rates (percent of predicted peaks that match measured ones) at high- and low-accessibility regions. Our model obtained high rates among the accessible regions (50–100% true positive rates for the different factors, with a total average of 77%), compared to much lower accuracy (true positive rates of 15–52%, with a total average of 43%) over the low accessibility regions. This differential accessibility is presented in Figure 4B and Figure S4B, where most of the false-positive predictions of the model (top-left corner) indeed show low accessibility.
To better quantify the role of accessibility in factor targeting, we leveraged the statistical framework of generalized hidden Markov models to incorporate DNA accessibility into the model as a non-uniform prior probability of regulatory binding along the genome – with regions of low accessibility having a greatly reduced probability of binding. The incorporation of differential DNA accessibility dramatically boosted the model's accuracy by a twofold for both the training set correlations (from 0.37 using the first, single-factor, model to 0.75) and the test set predictions (to a correlation of 0.70), with factor-specific correlation varying from 0.57 (HB) to 0.81 (BCD) (see Table 2, and Figure 1C and Figure 4C, 4D).
To further control for any possible experimental biases that might be present in the DNase I accessibility data, we also ran our model with DNase I data from a much later developmental stage, after cellular differentiation (embryonic stage 14). The highly bound genomic regions in our training and test sets are mostly comprised of loci that control early developmental patterning at stage 5 and which have been shown to have dramatically lower DNA accessibility at stage 14 (Thomas et al, unpublished data). Consistent with this, when we used DNase I data from the wrong developmental stage to estimate prior probabilities of transcription factor binding, the model's accuracy deteriorated (correlations of 0.36 and 0.28 on the train and test set, respectively), and was similar to the initial runs, where no DNase I data was used at all. Thus, the DNAse I data for stage 5 largely comprises a measurement of developmentally regulated chromatin accessibility, free from any major constitutive experimental bias.
BAC Spike-Ins Results Set the Highest Obtainable Correlation at 0.92
Our scoring system for the accuracy of our model uses the correlation coefficient between predicted and measured binding. We returned to the BAC spike-in data described earlier (Figure S2) to assess the experimental noise introduced into the ChIP-seq experiments by the amplification and sequencing of immunoprecipitated DNA as this would allow us to estimate the maximal correlation possible. We compared the binding landscape measured by the original ChIP data with versions of the same data into which noise was artificially introduced in levels following the BAC data (Methods). This resulted in an average correlation of 0.92 over 100 random perturbations, suggesting that even if our computational model were to perfectly predict the occupancies of proteins in vivo, the maximum obtainable correlation would be ∼0.92. Because additional experimental variation is likely to be introduced into the ChIP-seq data by differences in crosslinking efficiency and immunoprecipitation and because the DNase-seq data must also contain noise, we suspect that the true maximal correlation achievable is probably somewhat lower than this.
Thermodynamic Modeling Via Boltzmann Ensembles Captures Cooperative Binding
Although our predictions with DNA accessibility data were good, especially in light of the above estimates of experimental noise, we sought to further refine our model by considering more complex types of factor-factor interactions than the simple direct competition (via overlapping recognition sites) described above. For example, direct physical interactions between transcription factors bound at neighboring recognition sites has often been found to increase the occupancy of one or both proteins on DNA, both for homomeric and heteromeric interactions [36], [38], [42], and to sharpen the regulatory response to changes in transcription factor concentration [88], [94]–[97].
Generalized hidden Markov models, however, have limited ability to model the broader context of binding events, including interactions between neighboring sites. Therefore we added a second, sampling-based, phase to our computational model. In this phase, a large ensemble of binding configurations is sampled, each with a different set of protein-DNA interactions. The probability of each configuration is then estimated based on all pairs of nearby occupied sites (up to 95 bp apart) and the parameterized energetic gain of each pair. Finally, the overall binding probability at each position is quantified as a weighted sum of all sampled configurations (Figure 5).
Figure 5. Thermodynamic modeling of cooperative interactions.

(A) Cooperative parameters were used to represent the energy gain (or loss) for pairs of factors that bind in proximity (up to 95 bp apart). (B) Binding probabilities for the five factors at the eve stripe 1 locus (chr2R:5873439-5874240), as inferred by the generalized hidden Markov model. (C) Ensemble of configurations sampled from the probabilities in (B). Each row (of the 100 shown) corresponds to one configuration, marking the positions of bound sites. (D) Cooperative parameters for nearby pairs of occupied binding sites, as optimized over the training set.
By adopting a statistical mechanics perspective, the exponential space of protein-DNA binding configurations can be viewed as a canonical ensemble in a thermodynamic equilibrium. Here, the probability of each configuration is directly linked to its energetic state, including direct protein-DNA interactions, steric hindrance constraints and cooperative interactions with neighboring factors [50], [55], [56], [62], [69], [76]–[80].
We extended our model to capture cooperativity using a novel set of 15 parameters (one for each non-redundant pair of the five factors), modeling the energy gain for the nearby binding of every possible pair of the five transcriptional regulators in our model.
To predict binding in this new thermodynamic setting, we first used the generalized hidden Markov model in 3D to analyze the sequence and binding competition, and calculate an approximate map of binding. We then used the predicted binding probabilities to sample likely protein-DNA binding configurations, and re-weighted them to account for additional energetic gain, as modeled via protein-protein cooperative binding interactions [69], [98]. Finally, we averaged over these weighted configurations to predict a 3D map of binding. Using this combination of the gHMM followed by importance-weighted sampling, we were able to approximate the full thermodynamic landscape of binding, using a fast framework with few parameters. These cooperativity parameters, as well as their range of effect, were optimized based on the training set of genomic loci, using multiple random runs of a gradient-based trust-region optimization algorithm [99], [100]. The optimized set of cooperative binding parameters includes predictions of interactions between many homomeric and heteromeric pairs. However, these cooperativity parameters only improved the predictive power of the model by <5%, giving a correlation of 0.79 over the training set, with high accuracy binding predictions for all factors, ranging from 0.70 (CAD) to 0.9 (BCD). The model's accuracy over test set was 0.70, ranging from ∼0.6 (HB) to 0.83 (BCD), a marginal improvement of <1% over the Markovian approach (see Table 2 for full details). To further establish this result, we reran this procedure, this time allowing for both stabilizing and destabilizing interactions. Although we now identified additional interacting protein-protein pairs, the resulting correlations remained similar. Thus, our model suggests that cooperative interactions have a rather limited contribution in shaping the genomic landscape of in vivo binding.
Quantitative Estimations of Various Determinants of Transcriptional Regulation
We have presented here a series of increasingly complex models. The simplest model, using only the DNA sequence and in vitro binding data, obtained a correlation coefficient of 0.36 (on test data). After adding single-nucleus 3D modeling, as well as modeling binding competition with additional transcription factors and nucleosomes, the model's accuracy did not improve. We then incorporated experimental data of the DNA accessibility into the model, and boosted its accuracy to 0.70. Finally, we added thermodynamic parameters to model cooperative binding of neighboring factors, without a significant improvement in the model's accuracy (see Figure 2 and Table 2).
These estimators suggest that while sequence per se is responsible for approximately half of our predictive power, and DNA accessibility contributes the other half, the effect of modeling both binding competition and cooperative interactions is rather minor, and is estimated at the order of 1% of our predictive power.
To quantify the contribution of various transcriptional determinants on the binding landscape, we ran our model in all 16 possible configurations, using or not using: (1) sequence, (2) 3D competition, (3) DNA accessibility data, and (4) cooperativity (see Table S1). We then averaged over particular combinations to quantify the direct contribution of each of the four regulatory determinants. Once again, the relative contribution of each determinant was similar. For example, to quantify the importance of sequence per se, we compared the accuracy of the model in eight combinations (spanning all binary combinations 3D competition, accessibility and cooperativity) to the predictions obtained at the same configuration, only with randomly shuffled DNA sequences. The average difference in correlation over eight configuration pairs was 0.33 (±0.03), suggesting that DNA sequence per se does contribute half of our total predictive power. Similarly, the integration of DNA accessibility resulted in an increase of 0.33 (±0.03) in the model's accuracy (or about a half of its predictive power). The consistency of these estimates over different configurations suggests that the different regulatory determinants are largely independent of each other. Thus, the results suggest that binding within open chromatin is mostly controlled by DNA sequence, with a minimal role for direct cooperative or competitive interactions among factors.
Genome-Wide Prediction of Protein Binding
In the work presented so far, our analysis focused on regions of strong, functional binding, in the proximity of 21 known targets of the A-P patterning system and an additional eight control regions. To quantify the accuracy of the model on a larger genomic scale, we used the DNase I as a proxy, and identified the most accessible 2% of the genome (top 170 DNase I peaks, as called by an iterative peak-fitting algorithm [83], and expanded each peak by ∼10 Kb to each side in a similar manner to the initial set of 21 A-P loci). When applying the model to these open regions, it obtained an accuracy of 0.44, with various factors ranging from 0.38 (CAD) to 0.57 (BCD). Finally, we applied the model to the entirety of chromosome 2R (which was not included in our train set loci). Here, the model's accuracy was measured at 0.33 (or at 0.1 when no DNase I-based prior was used).
While these genomic scale predictions are not as accurate as the ones for the functional train and test set loci, they still offer a crude estimate to the genomic locations where binding occurs. The known target loci are inevitably biased towards genomic regions that are more accessible and more highly bound than the genome average (compare Figure 4 and Figure S4). In that sense, the DNase I peaks offer an unbiased proxy to the regions of the genome where chromatin accessibility plays a lesser role in shaping transcription factor binding in vivo. Moreover, part of the reason for the lower apparent performance of the model at the whole chromosome level could, thus, be technical, due to lower signal-to-noise ratios in the empirical data for the poorly bound, less accessible portions of the genome.
To further understand the decrease in the model's accuracy when shifting from an annotated set of known target loci to chromosome-wide applications, we compared the model's accuracy to the expression level of the underlying gene. We applied the model to predict the genome-wide binding across 4,251 genes for which we had gene expression data from the same developmental stage, taking 10 Kb regions around each gene [101]. We found a strong relation between the model's ability to accurately predict the binding landscape of transcription factors along the gene, and its expression level (Figure S5). These results suggest that while the model is applicable for genome-wide applications, it is mostly useful in predicting binding near actively transcribed genes and highly accessible genomic regions.
Discussion
We have used a thermodynamic computational framework to investigate the biochemical mechanisms that direct the widespread, quantitative patterns of binding by five developmental regulatory factors in the Drosophila blastoderm embryo. Our most striking finding is that a very simple thermodynamic model does a good job of quantitatively predicting the occupancy of transcription factors based on DNA sequence, in vitro DNA binding affinities and DNase I accessibility data.
We find no evidence that either competition between factors for binding sites or direct cooperative interactions between proteins show a significant effect on determining the overall pattern of binding in vivo. In the case of competitive binding, we showed that the reason why these do not dominantly affect net occupancy patterns embryo wide, is because high affinity sites for pairs of factors co-expressed in the same cells rarely overlap in the genome. Because our modeling focused on five early A-P segmentation regulators, it is possible that either competition or direct cooperative interactions with other transcription factors could play some role in shaping binding. The good predictive power of our model, however, sets an upper limit on the degree of this role. Also, it is striking that little evidence was found for positive heteromeric interactions among a set of five proteins that are known to cooperate on common sets of cis regulatory targets.
The relationship between chromatin accessibility and transcription factor binding has been previously described. But there is little agreement on the extent to which transcription factors bind regions of accessible chromatin, or if chromatin becomes accessible as a result of transcription factor binding. Our results are more consistent with the former, and fit well with a long-standing model for transcription factor targeting in which sequence specific transcription factors are expressed in cells at sufficient concentration to occupy their high and moderate affinity sites along accessible parts of the genome, without the aid of heteromeric cooperative interactions with other proteins [1], [2], [72].
Many other studies developed computational algorithms for predicting in vivo binding. Crudely, these studies fall into two categories. Qualitative models that aim at identifying statistically significant appearances of binding sites [49], [50], [53], [55]–[57], [59], [61], [63], [65], [67], [68], [78], [79], [102], and quantitative models that estimate the occupancy (or binding probability) at various sites [58], [60], [62], [66]. While the former group is generally more useful to identify putative cis regulatory modules and to unfold the transcriptional regulatory map, the latter approach is more suitable for modeling the continuous quantitative landscape of binding. An additional advantage of the quantitative approach is in its natural probabilistic settings, which allows for an easy integration of external data. For example, we used high-resolution measurements of DNA accessibility using DNase I hypersensitivity assays as an a-priori estimation of protein binding. Alternatively, previous work used various histone modification data as a proxy for DNA accessibility and to highlight regulatory regions [59], [63], [65], [68]. Nonetheless, our goal was to understand the biophysical forces that shape binding per se. For this, we turned to direct in vivo measurements of DNA accessibility and based our predictions on high-resolution quantitative measurements of in vivo binding data. And, unlike these earlier studies, examined the effects of different biophysical phenomena on our ability to predict binding, thereby revealing aspects of transcription factor biochemistry while providing a means to predict where, how, and to what extent transcription factor bind.
Finally, we wish to refer to a growing body of parallel work that models gene expression. Some of these studies use similar statistical models to ours, yet their modeling of transcription factor binding is intrinsic, limited to short regions of the genome with known regulatory activity, and has not examined the role of chromatin accessibility. Moreover, these models are optimized to recapitulate the patterns of expression for various genes, and do not focus on understanding the mechanisms of protein-DNA targeting [66], [69]–[71], [103].
One could argue that our use of DNA accessibility to eliminate regions where transcription factors are not bound is cheating. And, indeed, to the extent that our ultimate goal is to predict transcription factor binding from first principles it surely is. We have not established what determines the genome-wide landscape of DNA accessibility. But we have taken advantage of these direct in vivo measurements of DNA accessibility, and, in doing so, provide both a practical method for predicting binding, and a platform on which to better understand the forces that shape quantitative variation in the binding of individual factors to regions of open chromatin.
This success suggests a streamlined strategy for estimating of transcription factor binding for large numbers of factors involving in vivo measurement of DNA accessibility and in vitro determination of factor affinities for DNA. While this approach should not be viewed as a substitute for systematic experimental measurement of transcription factor binding in vivo, we believe our predictions are good enough to be useful when such experimental data are unavailable or impractical to obtain. Although our predictions are surprisingly good, they are by no means perfect. We are obviously interested in improving these predictions, primarily by incorporating better data on binding specificities and more realistic models of protein-DNA and protein-protein interactions.
Methods
Transcription Factor Chromatin Immunoprecipitation
We used ChIP-seq data for BCD, CAD, GT, HB and KR, from early stage 5 D. melanogaster embryos [18]. Sequenced reads were mapped to the genome (Apr. 2006 assembly, dm3, BDGP Release 5), extended according to their orientation to a length of 150 bp, and binned (down-sampled) to a 25 bp resolution. Finally, the genomic binding landscape of each factor was smoothed using a running window of 10 bins (or 250 bp), to account for sampling noise.
Generalized Hidden Markov Model
We implemented a generalized hidden Markov model to predict transcription factor binding based on the factor concentration and the underlying DNA sequence. Following a thermodynamic rationale, this statistical framework considers the space of all valid binding configurations as a Boltzmann distribution, where the probability of each configuration Pi, depends on its energetic state Ei
where β equals 1/kBT, with kB being the Boltzmann constant and T is the temperature (25 C). This allows us convert binding energies to probabilities and vice versa.
To model the energetic state of each configuration, we view each unbound nucleotide as generated from a background mononucleotide distribution PB, (0.32 for A/T, 0.18 for G/C). Bound nucleotides are generated using any probabilistic model of transcription factor binding sites [48], [51]. Here we use position weight matrices (PWMs), derived from in vitro SELEX data in which hundreds of bound oligonucleotides are sequenced for 2–4 rounds of SELEX [104] (BDTNP, unpublished data). Using a Maximum-Likelihood estimator with a pseudo-count of 0.01 to prevent zero probabilities, the probability of a subsequence Si to be bound by transcription factor t equals
where P(t) denotes the prior binding probability of transcription factor t (see below), lt denotes the binding site length of factor t, and 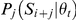 corresponds to the probability of observing the nucleotide Si+j, at the j position of the PWM of factor t.
corresponds to the probability of observing the nucleotide Si+j, at the j position of the PWM of factor t.
The probability of a full binding configuration at the DNA sequence S, with multiple factors T1, …,Tk bound at positions X1…Xk could be written as
with no overlapping binding sites. Moreover, to account for steric hindrance, we artificially extended each binding site model, adding flanking region of 3 bp, modeled by non-specific background distribution PB (0.32 for A/T, 0.18 for G/C), so the minimum distance between two non-overlapping binding sites is 7 (two flanks of 3 bp plus a 1 bp transition through the background state).
In the 3D models, we further scale the prior probability P(t) of each transcription factor t proportionally to its protein expression level (as measured at a single-cell resolution [47]). The prior probability P(t) of the nucleosomal binding state is assumed to be fixed through the embryo.
To account for all possible configurations, and infer the probability of each factor to bind each DNA position, given protein concentrations and DNA sequence, we apply the forward-backward dynamic programming inference algorithm [75]. Specifically, we calculate the probabilities that each factor t binds the DNA starting at each position i, Ut,i = P(t) * Pt(Si). We then calculate the forward potentials Ft,i, and the backward potentials Bt,i, by summing the probabilities of all configurations (paths) that end (or begin) at position i with a binding site of t. By multiplying the forward and backward potentials, we can then directly calculate the exact posterior probability of factor t bound at position i in a linear time.
In Vitro Protein-DNA Affinities
PWMs for the five transcription factors modeled in this study were obtained from the Berkeley Drosophila Transcription Network Project site (http://bdtnp.lbl.gov). PWM counts were then added a pseudo-count of 0.01 and normalized to probabilities. We have also tested other possible sources of PWMs [69], [105], with similar overall results. For example, one-hybrid PWMs yielded correlations of 0.72 for KR or 0.64 for CAD (compared to 0.71 and 0.68 using BDTNP's SELEX PWMs).
Model-Based Simulation of Chromatin Immunoprecipitation
The probabilities of transcription factor binding that were calculated by the generalized hidden Markov model were convolved to predicted ChIP landscape using a customized model-based estimation of a peak shape [83]. Given a distribution of DNA fragment lengths c(l), the estimated shape F of a peak is described as:
where Δx denotes the relative distance from the binding locus (peak center). In general, the probability of sequencing a read Δx bp away from the binding location is proportional to the amount of DNA fragment of length ≥Δx (fragments begin Δx bp away from the binding location and overlap it). We approximate this fraction using a Gamma distribution, with parameters corresponding to mean and standard deviation of fragment length. Finally, we quantify the similarity of the predicted binding landscape for each factor to the in vivo binding measurements using a Pearson correlation coefficient.
Modeling Nucleosome Binding
We used two different probabilistic models to incorporate nucleosome binding into the generalized hidden Markov model. First, we used a 141 bp-long sequence-specific model, based on positional dinucleotide distributions, as described in Segal et al. [93]. Alternatively, we used a 141 bp-long sequence-independent model of nucleosome binding, based on a fixed distribution of nucleotides as in the background state PB of the Markov model (0.32 for A/T, 0.18 for G/C). Similarly to the TF states, the two nucleosomal states were assigned (in turn) a prior probability term P(t) to reflect nucleosomal concentration. P(t) was optimized together with other concentration-related parameters P(t) for all transcription factors.
Non-Uniform Binding Probabilities Using DNA Accessibility
DNase I hypersensitivity data were used to directly compute the in vivo prior probability of transcription factor binding along the genome. We used a logistic sigmoid function to process the genome-wide DNase I read densities DDx into prior probabilities PDx
where α = 6.008 and β = 0.207. These parameters were optimized over the training data, separately from the concentration parameters in an iterative manner (Piecewise Optimization). We then computed these PDx values for every genomic position based on DNase I read densities, and normalized the prior probability of binding by each TF, P(t), by PDx, to calculate the transition probabilities into the bound states of every transcription factor. The transition probability into the nucleosomal state was not affected by this prior. In addition to the sigmoidal prior described here, we also tried a linear transformation from read densities DDx to PDx, resulting with slightly reduced accuracy (0.64 vs. 0.67).
BAC Spike-Ins
Eight BAC were used as spike-ins, including chr2R:8044567-8229187, chr2R:11688866-11882127, chr2R:19255181-19473745, chr3L:5493623-5675833, chr3L:11822927-11997557, chr3L:14593950-14773107, chr3R:12491763-12640405, and chr3R:23311086-23491584 (all given in release 5 coordinates). Three DNA samples were prepared, including: (1) the starting genomic DNA sample; (2) the genomic DNA with the addition of a set of BAC plasmid DNA premixed at different concentrations, and (3) a sample that contains the genomic DNA and the same set of BACs at 2x higher concentrations. These samples were sonicated to an average size of 500 bp, and the concentration of each BAC in the samples was quantified by Q-PCR using BAC specific primer-probe sets, and normalized to the genomic DNA. These samples were then prepared for sequencing following the same procedure we used for preparing sequencing libraries using ChIP samples. DNA fragments in the range from 200–500 bp, including the adapters, were selected for sequencing, and sequenced (4 lanes for genomic DNA, 2 lanes for each of 2 spike-in samples). Reads were mapped using bowtie [106], and the read coverage was normalized to reflect an equal read density on non-BAC background regions. The relative enrichment of each BAC post amplification and sequencing was then calculated, and compared to the Q-PCR enrichments.
Cooperative Binding Modeled Via Importance Sampling
We incorporated cooperative binding in a thermodynamic setting using sampling [69], [98]. This was done by first computing the posterior binding probabilities for every factor/position/nucleus using the generalized hidden Markov model, with sequence-independent nucleosomal state, in 3D resolution. We then sampled 10,000 binding configurations for every setting, calculated the occurrences of neighboring binding events (up to 95 bp apart), and re-weighted each sampled configuration by
where Wi corresponds to the weight of configuration i, xj and xk are the binding positions of transcription factors j and k, and Cj,k corresponds to their protein-protein cooperativity parameter, which we optimized using the train set loci (see below). Finally, the weighted samples were averaged, and the probability of every binding position for every factor estimated.
Optimization of Model Parameters
All parameters were optimized by maximizing the correlation between the model binding predictions over the train loci to their in vivo binding occupancies. The protein concentration parameters were initially optimized using a genetic algorithm [107], with 25 generations across a population size of 15. The optimized concentrations were then further improved using a gradient-based trust-region algorithm [99], [100]. Both phases were implemented in MATLAB. Protein-protein cooperativity parameters were optimized using a gradient-based trust-region algorithm starting from >200 random starting points.
Data Availability
All data, including PWMs, 3D protein concentrations, DNase I hypersensitivity prior, binding probabilities and predicted binding landscapes for all factors (at whole-embryo and single-nucleus resolution) and protein-protein cooperativity parameters are available at http://bdtnp.lbl.gov/gHMM
Supporting Information
The generalized hidden Markov model. Diagram of the model's state machine, including the mononucleotide “no binding” background state (red), five states corresponding to the five transcription factors in the model (blue), and a 141 bp-long nucleosomal binding state (green). The emission probabilities of each TF state are visualized using sequence logos. Transition probabilities depend on the concentrations of transcription factors, and the estimated accessibility of DNA.
(0.65 MB TIF)
BAC spike-ins. Eight long BACs were added to genomic DNA at 16 various concentrations (ranging from ∼2 to ∼40-fold, relative to genomic DNA), and measured before (using Q-PCR, shown along X-axis) and after (using sequencing, shown along Y-axis) amplification and processing for sequencing, resulting with a correlation of 0.997 and a linear fit of y = 1.14x. Vertical error-bars correspond to 1 standard deviation of the enrichment, based on running windows of 250 bp over each BAC.
(0.27 MB TIF)
From binding probabilities to ChIP landscape. (A) Each binding event (left) was transformed to a model-based estimation of peak shape (right, customized from Capaldi et al. [83]), depending on the average length of DNA fragments during the ChIP stage. (B) This model was then used to convolve the model's binding predictions (blue) to the expected landscape of ChIP sequencing assay (green), which was eventually compared to the measured in vivo binding landscape (red).
(0.08 MB TIF)
Chromosomal predictions with and without DNA accessibility prior. Same as Figure 4, but for entire chromosome 2R. (A) Measured (X-axis) vs. predicted (Y-axis) occupancy for all factors. (B) Same as (A), but colored based on DNA accessibility, ranging from pale cyan (lowly accessible) to dark blue (highly accessible). (C–D) Same as (A–C), but with DNase I hypersensitivity-based prior on protein binding integrated into the model.
(2.37 MB TIF)
Higher prediction accuracy for highly expressed genes. Comparison of the expression levels over 4,251 genes [101] vs. the cumulative accuracy of the model's predictions. Genes were binned into 20 groups based on expression levels. Shown are the average expression levels for each group (blue), and the cumulative correlation of the model's predictions vs. measured in vivo data (measured over top K groups) and averaged over all five factors (green).
(0.21 MB TIF)
Model's accuracy at 16 possible combinations of input data. Accuracy of model's predictions at 16 possible binary configurations of input data. These include (1) Sequence, which was either used (Sq = +) or randomly shuffled (Sq = –); (2) Three-dimensional predictions at a single-nucleus resolution, with binding competition among factors and nucleosomes (3D = +) vs. whole embryo factor-independent predictions (3D = –); (3) DNA accessibility prior on protein binding, based on DNase I hypersensitivity (DN = 1) vs. uniform prior (DN = –); and (4) Thermodynamic cooperativity parameters for adjacently bound factors (Co = +) vs. pure Markovian model (Co = –).
(0.10 MB DOC)
Acknowledgments
The authors thank Nir Friedman, Ariel Jaimovich, Richard Lusk, Steven Maere, Mathilde Paris, Devin Scannell, and members of the Eisen lab and the BDTNP for comments and discussions. We would like to thank our anonymous reviewers for useful comments.
Footnotes
MBE is a co-founder and member of the Board of Directors of PLoS.
Experimental work described here was supported by a Howard Hughes Medical Institute Investigator award to MBE and by National Institutes of Health (NIH) grant GM704403 to MBE and MDB. Computational analyses were supported in by NIH grant HG002779 to MBE. Work at Lawrence Berkeley National Laboratory was conducted under Department of Energy contract DE-AC02-05CH11231. TK was supported by a European Molecular Biology Organization (EMBO) long-term post-doctoral fellowship. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Walter J, Dever CA, Biggin MD. Two homeo domain proteins bind with similar specificity to a wide range of DNA sites in Drosophila embryos. Genes Dev. 1994;8:1678–1692. doi: 10.1101/gad.8.14.1678. [DOI] [PubMed] [Google Scholar]
- 2.Carr A, Biggin MD. A comparison of in vivo and in vitro DNA-binding specificities suggests a new model for homeoprotein DNA binding in Drosophila embryos. EMBO Journal. 1999;18:1598–1608. doi: 10.1093/emboj/18.6.1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bieda M, Xu X, Singer MA, Green R, Farnham PJ. Unbiased location analysis of E2F1-binding sites suggests a widespread role for E2F1 in the human genome. Genome Res. 2006;16:595–605. doi: 10.1101/gr.4887606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang A, Zhu Z, Kapranov P, McKeon F, Church GM, et al. Relationships between p63 binding, DNA sequence, transcription activity, and biological function in human cells. Mol Cell. 2006;24:593–602. doi: 10.1016/j.molcel.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 6.Sandmann T, Girardot C, Brehme M, Tongprasit W, Stolc V, et al. A core transcriptional network for early mesoderm development in Drosophila melanogaster. Genes Dev. 2007;21:436–449. doi: 10.1101/gad.1509007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zeitlinger J, Zinzen RP, Stark A, Kellis M, Zhang H, et al. Whole-genome ChIP-chip analysis of Dorsal, Twist, and Snail suggests integration of diverse patterning processes in the Drosophila embryo. Genes Dev. 2007;21:385–390. doi: 10.1101/gad.1509607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- 9.Robertson G, Hirst M, Bainbridge M, Bilenky M, Zhao Y, et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat Methods. 2007;4:651–657. doi: 10.1038/nmeth1068. [DOI] [PubMed] [Google Scholar]
- 10.Chen X, Xu H, Yuan P, Fang F, Huss M, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- 11.Consortium TEP. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Georlette D, Ahn S, MacAlpine DM, Cheung E, Lewis PW, et al. Genomic profiling and expression studies reveal both positive and negative activities for the Drosophila Myb MuvB/dREAM complex in proliferating cells. Genes Dev. 2007;21:2880–2896. doi: 10.1101/gad.1600107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li X-Y, Macarthur S, Bourgon R, Nix D, Pollard DA, et al. Transcription Factors Bind Thousands of Active and Inactive Regions in the Drosophila Blastoderm. PLoS Biol. 2008;6:e27. doi: 10.1371/journal.pbio.0060027. doi: 10.1371/journal.pbio.0060027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fullwood MJ, Liu MH, Pan YF, Liu J, Xu H, et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature. 2009;462:58–64. doi: 10.1038/nature08497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boj SF, Servitja JM, Martin D, Rios M, Talianidis I, et al. Functional targets of the monogenic diabetes transcription factors HNF-1alpha and HNF-4alpha are highly conserved between mice and humans. Diabetes. 2009;58:1245–1253. doi: 10.2337/db08-0812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Macarthur S, Li X-Y, Li J, Brown JB, Chu HC, et al. Developmental roles of 21 Drosophila transcription factors are determined by quantitative differences in binding to an overlapping set of thousands of genomic regions. Genome Biol. 2009;10:R80. doi: 10.1186/gb-2009-10-7-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cao Y, Yao Z, Sarkar D, Lawrence M, Sanchez GJ, et al. Genome-wide MyoD binding in skeletal muscle cells: a potential for broad cellular reprogramming. Dev Cell. 2010;18:662–674. doi: 10.1016/j.devcel.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bradley RK, Li X-Y, Trapnell C, Davidson S, Pachter L, et al. Binding site turnover produces pervasive quantitative changes in transcription factor binding between closely related Drosophila species. PLoS Biol. 2010;8:e1000343. doi: 10.1371/journal.pbio.1000343. doi: 10.1371/journal.pbio.1000343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liang Z, Biggin MD. Eve and ftz regulate a wide array of genes in blastoderm embryos: the selector homeoproteins directly or indirectly regulate most genes in Drosophila. Development. 1998;125:4471–4482. doi: 10.1242/dev.125.22.4471. [DOI] [PubMed] [Google Scholar]
- 20.Moorman C, Sun LV, Wang J, de Wit E, Talhout W, et al. Hotspots of transcription factor colocalization in the genome of Drosophila melanogaster. Proc Natl Acad Sci U S A. 2006;103:12027–12032. doi: 10.1073/pnas.0605003103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ouyang Z, Zhou Q, Wong WH. ChIP-Seq of transcription factors predicts absolute and differential gene expression in embryonic stem cells. Proc Natl Acad Sci U S A. 2009;106:21521–21526. doi: 10.1073/pnas.0904863106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zinzen RP, Girardot C, Gagneur J, Braun M, Furlong EEM. Combinatorial binding predicts spatio-temporal cis-regulatory activity. Nature. 2009;461:65–70. doi: 10.1038/nature08531. [DOI] [PubMed] [Google Scholar]
- 23.Wunderlich Z, Mirny LA. Different gene regulation strategies revealed by analysis of binding motifs. Trends Genet. 2009;25:434–440. doi: 10.1016/j.tig.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iyer VR, Horak CE, Scafe CS, Botstein D, Snyder M, et al. Genomic binding sites of the yeast cell-cycle transcription factors SBF and MBF. Nature. 2001;409:533–538. doi: 10.1038/35054095. [DOI] [PubMed] [Google Scholar]
- 25.Liu X, Lee CK, Granek JA, Clarke ND, Lieb JD. Whole-genome comparison of Leu3 binding in vitro and in vivo reveals the importance of nucleosome occupancy in target site selection. Genome Res. 2006;16:1517–1528. doi: 10.1101/gr.5655606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stanojevic D, Small S, Levine M. Regulation of a segmentation stripe by overlapping activators and repressors in the Drosophila embryo. Science (New York, NY) 1991;254:1385–1387. doi: 10.1126/science.1683715. [DOI] [PubMed] [Google Scholar]
- 27.Makeev VJ, Lifanov AP, Nazina AG, Papatsenko DA. Distance preferences in the arrangement of binding motifs and hierarchical levels in organization of transcription regulatory information. Nucleic Acids Research. 2003;31:6016–6026. doi: 10.1093/nar/gkg799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wolffe AP. Nucleosome positioning and modification: chromatin structures that potentiate transcription. Trends Biochem Sci. 1994;19:240–244. doi: 10.1016/0968-0004(94)90148-1. [DOI] [PubMed] [Google Scholar]
- 29.Cosma MP, Tanaka T, Nasmyth K. Ordered recruitment of transcription and chromatin remodeling factors to a cell cycle- and developmentally regulated promoter. Cell. 1999;97:299–311. doi: 10.1016/s0092-8674(00)80740-0. [DOI] [PubMed] [Google Scholar]
- 30.Agalioti T, Lomvardas S, Parekh B, Yie J, Maniatis T, et al. Ordered recruitment of chromatin modifying and general transcription factors to the IFN-beta promoter. Cell. 2000;103:667–678. doi: 10.1016/s0092-8674(00)00169-0. [DOI] [PubMed] [Google Scholar]
- 31.Carr A, Biggin MD. Accessibility of transcriptionally inactive genes is specifically reduced at homeoprotein-DNA binding sites in Drosophila. Nucleic Acids Res. 2000;28:2839–2846. doi: 10.1093/nar/28.14.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Narlikar GJ, Fan H-Y, Kingston RE. Cooperation between complexes that regulate chromatin structure and transcription. Cell. 2002;108:475–487. doi: 10.1016/s0092-8674(02)00654-2. [DOI] [PubMed] [Google Scholar]
- 33.Morse RH. Transcription factor access to promoter elements. J Cell Biochem. 2007;102:560–570. doi: 10.1002/jcb.21493. [DOI] [PubMed] [Google Scholar]
- 34.Taylor IC, Workman JL, Schuetz TJ, Kingston RE. Facilitated binding of GAL4 and heat shock factor to nucleosomal templates: differential function of DNA-binding domains. Genes Dev. 1991;5:1285–1298. doi: 10.1101/gad.5.7.1285. [DOI] [PubMed] [Google Scholar]
- 35.Adams CC, Workman JL. Binding of disparate transcriptional activators to nucleosomal DNA is inherently cooperative. Mol Cell Biol. 1995;15:1405–1421. doi: 10.1128/mcb.15.3.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnson AD. Molecular mechanisms of cell-type determination in budding yeast. Curr Opin Genet Dev. 1995;5:552–558. doi: 10.1016/0959-437x(95)80022-0. [DOI] [PubMed] [Google Scholar]
- 37.Vashee S, Melcher K, Ding WV, Johnston SA, Kodadek T. Evidence for two modes of cooperative DNA binding in vivo that do not involve direct protein-protein interactions. Curr Biol. 1998;8:452–458. doi: 10.1016/s0960-9822(98)70179-4. [DOI] [PubMed] [Google Scholar]
- 38.Bolouri H, Davidson EH. Transcriptional regulatory cascades in development: initial rates, not steady state, determine network kinetics. Proc Natl Acad Sci U S A. 2003;100:9371–9376. doi: 10.1073/pnas.1533293100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miller JA, Widom J. Collaborative competition mechanism for gene activation in vivo. Mol Cell Biol. 2003;23:1623–1632. doi: 10.1128/MCB.23.5.1623-1632.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zeitlinger J, Simon I, Harbison CT, Hannett NM, Volkert TL, et al. Program-specific distribution of a transcription factor dependent on partner transcription factor and MAPK signaling. Cell. 2003;113:395–404. doi: 10.1016/s0092-8674(03)00301-5. [DOI] [PubMed] [Google Scholar]
- 41.Buck MJ, Lieb JD. A chromatin-mediated mechanism for specification of conditional transcription factor targets. Nat Genet. 2006;38:1446–1451. doi: 10.1038/ng1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mann RS, Lelli KM, Joshi R. Hox specificity unique roles for cofactors and collaborators. Curr Top Dev Biol. 2009;88:63–101. doi: 10.1016/S0070-2153(09)88003-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Campos-Ortega JA, Hartenstein V. Berlin: Springer-Verlag; 1997. The Embryonic Development of Drosophila melanogaster. [Google Scholar]
- 44.Nusslein-Volhard C, Wieschaus E. Mutations affecting segment number and polarity in Drosophila. Nature. 1980;287:795–801. doi: 10.1038/287795a0. [DOI] [PubMed] [Google Scholar]
- 45.St Johnston D, Nusslein-Volhard C. The origin of pattern and polarity in the Drosophila embryo. Cell. 1992;68:201–219. doi: 10.1016/0092-8674(92)90466-p. [DOI] [PubMed] [Google Scholar]
- 46.Rivera-Pomar R, Jäckle H. From gradients to stripes in Drosophila embryogenesis: filling in the gaps. Trends Genet. 1996;12:478–483. doi: 10.1016/0168-9525(96)10044-5. [DOI] [PubMed] [Google Scholar]
- 47.Fowlkes CC, Hendriks CLL, Keränen SVE, Weber GH, Rübel O, et al. A quantitative spatiotemporal atlas of gene expression in the Drosophila blastoderm. Cell. 2008;133:364–374. doi: 10.1016/j.cell.2008.01.053. [DOI] [PubMed] [Google Scholar]
- 48.Stormo GD. DNA binding sites: representation and discovery. Bioinformatics. 2000;16:16–23. doi: 10.1093/bioinformatics/16.1.16. [DOI] [PubMed] [Google Scholar]
- 49.Frith MC, Hansen U, Weng Z. Detection of cis-element clusters in higher eukaryotic DNA. Bioinformatics. 2001;17:878–889. doi: 10.1093/bioinformatics/17.10.878. [DOI] [PubMed] [Google Scholar]
- 50.Rajewsky N, Vergassola M, Gaul U, Siggia ED. Computational detection of genomic cis-regulatory modules applied to body patterning in the early Drosophila embryo. BMC Bioinformatics. 2002;3:30. doi: 10.1186/1471-2105-3-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barash Y, Elidan G, Friedman N, Kaplan T. Proceedings of the seventh annual international conference on Research in computational molecular biology. Berlin, Germany: ACM; 2003. Modeling dependencies in protein-DNA binding sites. pp. 28–37. [Google Scholar]
- 52.Bulyk ML. Computational prediction of transcription-factor binding site locations. Genome Biol. 2003;5:201. doi: 10.1186/gb-2003-5-1-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sinha S, van Nimwegen E, Siggia ED. A probabilistic method to detect regulatory modules. Bioinformatics. 2003;19(Suppl 1):i292–301. doi: 10.1093/bioinformatics/btg1040. [DOI] [PubMed] [Google Scholar]
- 54.Barash Y, Elidan G, Kaplan T, Friedman N. CIS: compound importance sampling method for protein-DNA binding site p-value estimation. Bioinformatics. 2005;21:596–600. doi: 10.1093/bioinformatics/bti041. [DOI] [PubMed] [Google Scholar]
- 55.Granek JA, Clarke ND. Explicit equilibrium modeling of transcription-factor binding and gene regulation. Genome Biol. 2005;6:R87. doi: 10.1186/gb-2005-6-10-r87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sinha S. On counting position weight matrix matches in a sequence, with application to discriminative motif finding. Bioinformatics. 2006;22:e454–463. doi: 10.1093/bioinformatics/btl227. [DOI] [PubMed] [Google Scholar]
- 57.Narlikar L, Gordan R, Hartemink AJ. A nucleosome-guided map of transcription factor binding sites in yeast. PLoS Comput Biol. 2007;3:e215. doi: 10.1371/journal.pcbi.0030215. doi: 10.1371/journal.pcbi.0030215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Roider HG, Kanhere A, Manke T, Vingron M. Predicting transcription factor affinities to DNA from a biophysical model. Bioinformatics. 2007;23:134–141. doi: 10.1093/bioinformatics/btl565. [DOI] [PubMed] [Google Scholar]
- 59.Ward LD, Bussemaker HJ. Predicting functional transcription factor binding through alignment-free and affinity-based analysis of orthologous promoter sequences. Bioinformatics. 2008;24:i165–171. doi: 10.1093/bioinformatics/btn154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.He X, Chen CC, Hong F, Fang F, Sinha S, et al. A biophysical model for analysis of transcription factor interaction and binding site arrangement from genome-wide binding data. PLoS ONE. 2009;4:e8155. doi: 10.1371/journal.pone.0008155. doi: 10.1371/journal.pone.0008155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Narlikar L, Ovcharenko I. Identifying regulatory elements in eukaryotic genomes. Brief Funct Genomic Proteomic. 2009;8:215–230. doi: 10.1093/bfgp/elp014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wasson T, Hartemink AJ. An ensemble model of competitive multi-factor binding of the genome. Genome Res. 2009;19:2101–2112. doi: 10.1101/gr.093450.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Whitington T, Perkins AC, Bailey TL. High-throughput chromatin information enables accurate tissue-specific prediction of transcription factor binding sites. Nucleic Acids Research. 2009;37:14–25. doi: 10.1093/nar/gkn866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Won KJ, Agarwal S, Shen L, Shoemaker R, Ren B, et al. An integrated approach to identifying cis-regulatory modules in the human genome. PLoS ONE. 2009;4:e5501. doi: 10.1371/journal.pone.0005501. doi: 10.1371/journal.pone.0005501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ernst J, Plasterer HL, Simon I, Bar-Joseph Z. Integrating multiple evidence sources to predict transcription factor binding in the human genome. Genome Res. 2010;20:526–536. doi: 10.1101/gr.096305.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.He X, Samee MA, Blatti C, Sinha S. Thermodynamics-based models of transcriptional regulation by enhancers: the roles of synergistic activation, cooperative binding and short-range repression. PLoS Comput Biol. 2010;6:e1000935. doi: 10.1371/journal.pcbi.1000935. doi: 10.1371/journal.pcbi.1000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ramsey SA, Knijnenburg TA, Kennedy KA, Zak DE, Gilchrist M, et al. Genome-wide histone acetylation data improve prediction of mammalian transcription factor binding sites. Bioinformatics. 2010;26:2071–2075. doi: 10.1093/bioinformatics/btq405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Won K-J, Ren B, Wang W. Genome-wide prediction of transcription factor binding sites using an integrated model. Genome Biol. 2010;11:R7. doi: 10.1186/gb-2010-11-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Segal E, Raveh-Sadka T, Schroeder M, Unnerstall U, Gaul U. Predicting expression patterns from regulatory sequence in Drosophila segmentation. Nature. 2008;451:535–540. doi: 10.1038/nature06496. [DOI] [PubMed] [Google Scholar]
- 70.Raveh-Sadka T, Levo M, Segal E. Incorporating nucleosomes into thermodynamic models of transcription regulation. Genome Res. 2009;19:1480–1496. doi: 10.1101/gr.088260.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kazemian M, Blatti C, Richards A, McCutchan M, Wakabayashi-Ito N, et al. Quantitative analysis of the Drosophila segmentation regulatory network using pattern generating potentials. PLoS Biol. 2010;8:e1000456. doi: 10.1371/journal.pbio.1000456. doi: 10.1371/journal.pbio.1000456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lin S, Riggs AD. The general affinity of lac repressor for E. coli DNA: implications for gene regulation in procaryotes and eucaryotes. Cell. 1975;4:107–111. doi: 10.1016/0092-8674(75)90116-6. [DOI] [PubMed] [Google Scholar]
- 73.von Hippel PH, Revzin A, Gross CA, Wang AC. Nonspecific DNA binding of genome regulating proteins as a biological control mechanism: 1. The lac operon: Equilibrium aspects. Proc Natl Acad Sci USA. 1974;71:4808–4812. doi: 10.1073/pnas.71.12.4808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kulp D, Haussler D, Reese MG, Eeckman FH. A generalized hidden Markov model for the recognition of human genes in DNA. Proc Int Conf Intell Syst Mol Biol. 1996;4:134–142. [PubMed] [Google Scholar]
- 75.Rabiner L. A Tutorial on Hidden Markov Moldes and Selected Applications in Speech Recognition. P Ieee. 1989;77:257–286. [Google Scholar]
- 76.Ackers GK, Johnson AD, Shea MA. Quantitative model for gene regulation by lambda phage repressor. Proc Natl Acad Sci USA. 1982;79:1129–1133. doi: 10.1073/pnas.79.4.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Buchler NE, Gerland U, Hwa T. On schemes of combinatorial transcription logic. Proc Natl Acad Sci USA. 2003;100:5136–5141. doi: 10.1073/pnas.0930314100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schroeder MD, Pearce M, Fak J, Fan H, Unnerstall U, et al. Transcriptional control in the segmentation gene network of Drosophila. PLoS Biol. 2004;2:e271. doi: 10.1371/journal.pbio.0020271. doi: 10.1371/journal.pbio.0020271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bintu L, Buchler NE, Garcia HG, Gerland U, Hwa T, et al. Transcriptional regulation by the numbers: models. Current Opinion in Genetics & Development. 2005;15:116–124. doi: 10.1016/j.gde.2005.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gertz J, Cohen BA. Environment-specific combinatorial cis-regulation in synthetic promoters. Mol Syst Biol. 2009;5:1–9. doi: 10.1038/msb.2009.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Toth J, Biggin MD. The specificity of protein-DNA crosslinking by formaldehyde: in vitro and in drosophila embryos. Nucleic Acids Res. 2000;28:e4. doi: 10.1093/nar/28.2.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Auerbach RK, Euskirchen G, Rozowsky J, Lamarre-Vincent N, Moqtaderi Z, et al. Mapping accessible chromatin regions using Sono-Seq. Proc Natl Acad Sci USA. 2009;106:14926–14931. doi: 10.1073/pnas.0905443106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Capaldi A, Kaplan T, Liu Y, Habib N, Regev A, et al. Structure and function of a transcriptional network activated by the MAPK Hog1. Nat Genet. 2008;40:1300–1306. doi: 10.1038/ng.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Brody T. The Interactive Fly: gene networks, development and the Internet. Trends Genet. 1999;15:333–334. doi: 10.1016/s0168-9525(99)01775-8. [DOI] [PubMed] [Google Scholar]
- 85.Hare EE, Peterson BK, Iyer VN, Meier R, Eisen MB. Sepsid even-skipped enhancers are functionally conserved in Drosophila despite lack of sequence conservation. PLoS Genet. 2008;4:e1000106. doi: 10.1371/journal.pgen.1000106. doi: 10.1371/journal.pgen.1000106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kim J, He X, Sinha S. Evolution of regulatory sequences in 12 Drosophila species. PLoS Genet. 2009;5:e1000330. doi: 10.1371/journal.pgen.1000330. doi: 10.1371/journal.pgen.1000330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Polach KJ, Widom J. A model for the cooperative binding of eukaryotic regulatory proteins to nucleosomal target sites. J Mol Biol. 1996;258:800–812. doi: 10.1006/jmbi.1996.0288. [DOI] [PubMed] [Google Scholar]
- 88.Mirny L. Nucleosome-mediated cooperativity between transcription factors. Arxiv preprint arXiv. 2009:09012905. doi: 10.1073/pnas.0913805107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gross DS, Garrard WT. Nuclease hypersensitive sites in chromatin. Annu Rev Biochem. 1988;57:159–197. doi: 10.1146/annurev.bi.57.070188.001111. [DOI] [PubMed] [Google Scholar]
- 90.Hesselberth JR, Chen X, Zhang Z, Sabo PJ, Sandstrom R, et al. Global mapping of protein-DNA interactions in vivo by digital genomic footprinting. Nat Meth. 2009;6:283–289. doi: 10.1038/nmeth.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sabo PJ, Kuehn MS, Thurman R, Johnson BE, Johnson EM, et al. Genome-scale mapping of DNase I sensitivity in vivo using tiling DNA microarrays. Nat Meth. 2006;3:511–518. doi: 10.1038/nmeth890. [DOI] [PubMed] [Google Scholar]
- 92.Zhang Y, Moqtaderi Z, Rattner BP, Euskirchen G, Snyder M, et al. Intrinsic histone-DNA interactions are not the major determinant of nucleosome positions in vivo. Nat Struct Mol Biol. 2009;16:847–852. doi: 10.1038/nsmb.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Segal E, Fondufe-Mittendorf Y, Chen L, Thåström A, Field Y, et al. A genomic code for nucleosome positioning. Nature. 2006;442:772–778. doi: 10.1038/nature04979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Small S, Blair A, Levine M. Regulation of even-skipped stripe 2 in the Drosophila embryo. EMBO J. 1992;11:4047–4057. doi: 10.1002/j.1460-2075.1992.tb05498.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Arnosti DN, Barolo S, Levine M, Small S. The eve stripe 2 enhancer employs multiple modes of transcriptional synergy. Development. 1996;122:205–214. doi: 10.1242/dev.122.1.205. [DOI] [PubMed] [Google Scholar]
- 96.Kulkarni MM, Arnosti DN. cis-regulatory logic of short-range transcriptional repression in Drosophila melanogaster. Molecular and Cellular Biology. 2005;25:3411–3420. doi: 10.1128/MCB.25.9.3411-3420.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fakhouri WD, Ay A, Sayal R, Dresch J, Dayringer E, et al. Deciphering a transcriptional regulatory code: modeling short-range repression in the Drosophila embryo. Molecular Systems Biology. 2010;6:1–14. doi: 10.1038/msb.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hammersley JM, Handscomb DC. London, New York: Methuen; Wiley; 1964. Monte Carlo methods. p. vii, 178. [Google Scholar]
- 99.Steihaug T. The Conjugate Gradient Method and Trust Regions in Large Scale Optimization. SIAM Journal on Numerical Analysis. 1983;20:626–637. [Google Scholar]
- 100.Coleman TF, Li Y. An Interior Trust Region Approach for Nonlinear Minimization Subject to Bounds. SIAM J Optim. 1996;6:418–445. [Google Scholar]
- 101.Arbeitman M, Furlong E, Imam F, Johnson E, Null B, et al. Gene expression during the life cycle of Drosophila melanogaster. Science. 2002;297:2270–2275. doi: 10.1126/science.1072152. [DOI] [PubMed] [Google Scholar]
- 102.Agius P, Arvey A, Chang W, Noble WS, Leslie C. High Resolution Models of Transcription Factor-DNA Affinities Improve In Vitro and In Vivo Binding Predictions. PLoS Comput Biol. 2010;6:e1000916. doi: 10.1371/journal.pcbi.1000916. doi: 10.1371/journal.pcbi.1000916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zinzen RP, Senger K, Levine M, Papatsenko D. Computational models for neurogenic gene expression in the Drosophila embryo. Curr Biol. 2006;16:1358–1365. doi: 10.1016/j.cub.2006.05.044. [DOI] [PubMed] [Google Scholar]
- 104.Roulet E, Busso S, Camargo AA, Simpson AJ, Mermod N, et al. High-throughput SELEX SAGE method for quantitative modeling of transcription-factor binding sites. Nat Biotechnol. 2002;20:831–835. doi: 10.1038/nbt718. [DOI] [PubMed] [Google Scholar]
- 105.Noyes MB, Meng X, Wakabayashi A, Sinha S, Brodsky MH, et al. A systematic characterization of factors that regulate Drosophila segmentation via a bacterial one-hybrid system. Nucleic Acids Research. 2008;36:2547–2560. doi: 10.1093/nar/gkn048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Goldberg D, Holland J. Genetic Algorithms and Machine Learning. Machine learning. 1988;3:95–99. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The generalized hidden Markov model. Diagram of the model's state machine, including the mononucleotide “no binding” background state (red), five states corresponding to the five transcription factors in the model (blue), and a 141 bp-long nucleosomal binding state (green). The emission probabilities of each TF state are visualized using sequence logos. Transition probabilities depend on the concentrations of transcription factors, and the estimated accessibility of DNA.
(0.65 MB TIF)
BAC spike-ins. Eight long BACs were added to genomic DNA at 16 various concentrations (ranging from ∼2 to ∼40-fold, relative to genomic DNA), and measured before (using Q-PCR, shown along X-axis) and after (using sequencing, shown along Y-axis) amplification and processing for sequencing, resulting with a correlation of 0.997 and a linear fit of y = 1.14x. Vertical error-bars correspond to 1 standard deviation of the enrichment, based on running windows of 250 bp over each BAC.
(0.27 MB TIF)
From binding probabilities to ChIP landscape. (A) Each binding event (left) was transformed to a model-based estimation of peak shape (right, customized from Capaldi et al. [83]), depending on the average length of DNA fragments during the ChIP stage. (B) This model was then used to convolve the model's binding predictions (blue) to the expected landscape of ChIP sequencing assay (green), which was eventually compared to the measured in vivo binding landscape (red).
(0.08 MB TIF)
Chromosomal predictions with and without DNA accessibility prior. Same as Figure 4, but for entire chromosome 2R. (A) Measured (X-axis) vs. predicted (Y-axis) occupancy for all factors. (B) Same as (A), but colored based on DNA accessibility, ranging from pale cyan (lowly accessible) to dark blue (highly accessible). (C–D) Same as (A–C), but with DNase I hypersensitivity-based prior on protein binding integrated into the model.
(2.37 MB TIF)
Higher prediction accuracy for highly expressed genes. Comparison of the expression levels over 4,251 genes [101] vs. the cumulative accuracy of the model's predictions. Genes were binned into 20 groups based on expression levels. Shown are the average expression levels for each group (blue), and the cumulative correlation of the model's predictions vs. measured in vivo data (measured over top K groups) and averaged over all five factors (green).
(0.21 MB TIF)
Model's accuracy at 16 possible combinations of input data. Accuracy of model's predictions at 16 possible binary configurations of input data. These include (1) Sequence, which was either used (Sq = +) or randomly shuffled (Sq = –); (2) Three-dimensional predictions at a single-nucleus resolution, with binding competition among factors and nucleosomes (3D = +) vs. whole embryo factor-independent predictions (3D = –); (3) DNA accessibility prior on protein binding, based on DNase I hypersensitivity (DN = 1) vs. uniform prior (DN = –); and (4) Thermodynamic cooperativity parameters for adjacently bound factors (Co = +) vs. pure Markovian model (Co = –).
(0.10 MB DOC)
Data Availability Statement
All data, including PWMs, 3D protein concentrations, DNase I hypersensitivity prior, binding probabilities and predicted binding landscapes for all factors (at whole-embryo and single-nucleus resolution) and protein-protein cooperativity parameters are available at http://bdtnp.lbl.gov/gHMM