

Abstract
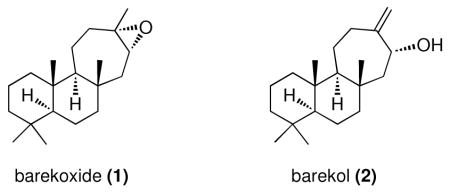
The tandem cyclopropanation/Cope rearrangement between bicyclic dienes and siloxyvinyldiazoacetate, catalyzed by the dirhodium catalyst Rh2(R-PTAD)4 effectively accomplishes enantiodivergent [4 + 3] cycloadditions. The reaction proceeds by a cyclopropanation followed by a Cope rearrangement of the resulting divinylcyclopropane. This methodology was applied to the synthesis (+)-barekoxide (1) and (−)-barekol (2).
Fused seven-membered carbocycles are present in a wide-variety of terpene natural products.1 An effective method for the stereoselective synthesis of seven-membered rings is the formal [4 + 3] cycloaddition between vinylcarbenoids and dienes, which gives predictable diastereocontrol, proceeding through a Cope rearrangement of a cis-divinylcyclopropane intermediate.2,3 Furthermore, chiral dirhodium tetracarboxylate catalysts enable high enantioselectivity to be achieved in these transformations.4 Recently, the Sarpong group applied the formal [4 + 3] cycloaddition in a stereodivergent approach to the core of the cyanthane diterpenes.5 This paper describes a collaborative study between the Davies and Sarpong groups, resulting in a greatly enhanced level of enantiomeric differentiation for this chemistry and its application to the synthesis of (+)-barekoxide (1) and (−)-barekol (2).6
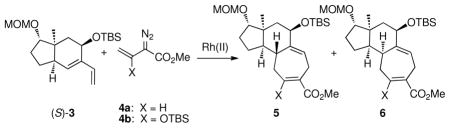
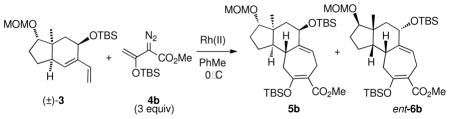
The original studies by Sarpong were conducted on the diene (S)-3 and the unsubstituted vinyldiazoacetate 4a using the enantiomers of Rh2(DOSP)47 as catalysts (Table 1, entries 1 and 2).5 Each enantiomer of the catalyst gave moderate control of which diastereomer of the tricyclic products (5a or 6a) was formed. Davies has recently demonstrated that siloxyvinyldiazoacetate 4b with Rh2(PTAD)48 is a good combination for highly enantioselective [4 + 3] cycloadditions.3b,c Therefore, the reactions of (S)-3 were re-examined using siloxyvinyldiazoacetate 4b as the carbenoid precursor.
Table 1.
Reaction between enantio-pure diene and diazoacetates
 | |||||
|---|---|---|---|---|---|
| entry | substrate | catalyst | diazo | ratio (5/6) | yield (5+6, %) |
| 1a | (R)-3 | Rh2(R-DOSP)4 | 4a | 5:1b | 39c |
| 2a | (R)-3 | Rh2(S-DOSP)4 | 4a | 1:5b | 50c |
| 3d | (S)-3 | Rh2(R-DOSP)4 | 4b | >19:1 | 76 |
| 4d | (S)-3 | Rh2(S-DOSP)4 | 4b | 2:1 | 63 |
| 5d | (S)-3 | Rh2(R-PTAD)4 | 4b | >19:1 | 81 |
| 6d | (S)-3 | Rh2(S-PTAD)4 | 4b | <1:19 | 80 |
Reactions were conducted at 8 °C in pentane with 3.0 equivalents of 4a and 1 mol% catalyst;
Determined by 1H NMR;
Isolated yields of the reduced ester protected as a p-nitrobenzoate;
Reactions were conducted at 0 °C in PhMe with 3.0 equivalents of 4b and 2 mol% catalyst.
The reaction of (S)-3 with 4b catalyzed by Rh2(R-DOSP)4 gave exclusively the tricycle 5b, whereas the reaction catalyzed by Rh2(S-DOSP)4 still gave a slight preference of 5b over 6b (entries 3 and 4). These results indicate that the substrate control in the reaction between (S)-3 and 4b favors the formation of 5b, which is enhanced in the matched reaction using Rh2(R-DOSP)4 as catalyst. The mismatched reaction, however, with Rh2(S-DOSP)4 as catalyst gives a mixture of products.
The diastereocontrol of the [4 + 3] cycloaddition is further enhanced when the enantiomers of Rh2(PTAD)4 are used as catalysts. Once again in the matched reaction with Rh2(R-PTAD)4 as catalyst, 5b is formed with high diastereoselectivity (entry 5). In this case, however, the mismatched reaction with Rh2(S-PTAD)4 as catalyst is also highly diastereoselective, generating 6b exclusively (entry 6).
Having discovered that the siloxyvinyldiazoacetate 4b gives much better stereodifferentiation than 4a, the next series of reactions explored whether a resolution would be possible using (±)−3. The reaction of 4b catalyzed by Rh2(R-DOSP)4 was highly diastereoselective, but the major diastereomer 5b was produced in only 53% ee. In contrast, the reaction catalyzed by Rh2(R-PTAD)4 had excellent reagent control, in which 5b was produced in 90% ee and the other diastereomer (ent-6b) was produced in 99% ee.
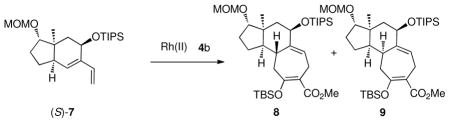
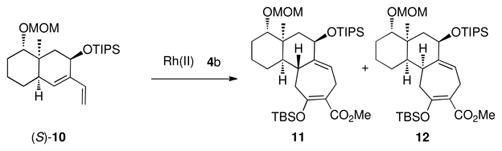
This transformation can also be effectively extended to a more sterically hindered diene (S)-7, using TIPS as the protecting group. The Rh2(R-PTAD)4-catalyzed reaction of (S)-7 and the siloxydiazoacetate 4b generated product 8 as a single diastereomer (entry 1, Table 3), whereas the reaction initiated by Rh2(S-PTAD)4 afforded the other isomer 9 with excellent diastereoselectivity (entry 2, Table 3). Extension of the study from a bicyclo[4.3.0]nonane to a bicyclo[4.4.0]decane system revealed the subtle controlling influences assocciated with this selectivity. The Rh2(R-PTAD)4-catalyzed reaction of diene (S)-10 with siloxydiazoacetate 4b generated two diastereomers of the formal [4 + 3] cycloadducts, 11 and 12 in a 9:1 ratio (entry 3, Table 2). The same reaction catalyzed by Rh2(S-PTAD)4 switched the diastereoselectvity, favoring 12 with a 4: 1 dr. These results indicate that the chiral catalyst has a conrolling influence on the diastereoselectvity in the bicyclo[4.4.0]decane system but the effect is not as overwhelming as it is in the bicyclo[4.3.0]nonane system.
Table 3.
Reaction scope between enantio-pure diene and diazoacetate 4b
 | |||
|---|---|---|---|
| entry | catalyst | ratio (8/9) | yield (major isomer only, %) |
| 1 | Rh2(R-PTAD)4 | >19:1 | 82 |
| 2 | Rh2(S-PTAD)4 | <1:19 | 76 |
 | |||
| entry | catalyst | ratio (11/12) | yield (major isomer only, %) |
| 3 | Rh2(R-PTAD)4 | 9:1 | 77 |
| 4 | Rh2(S-PTAD)4 | 1:5 | 66 |
 | |||
| entry | catalyst | ratio (14/15) | yield (major isomer only, %) |
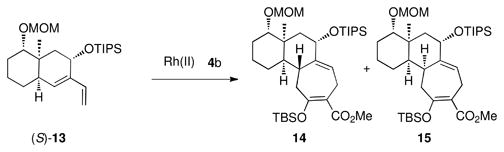
| 5 | Rh2(R-PTAD)4 | 4:1 | 63 |
| 6 | Rh2(S-PTAD)4 | 1:7 | 69 |
 | |||
| entry | catalyst | ratio (17/18) | yield (major isomer only, %) |
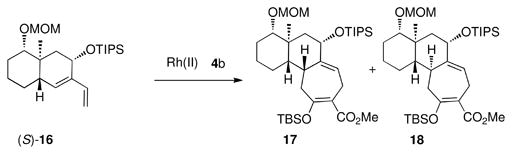
| 7 | Rh2(R-PTAD)4 | 2:1 | 55 |
| 8 | Rh2(S-PTAD)4 | 1:16 | 81 |
Table 2.
Reaction between racemic 3 and diazoacetate 4b
 | |||||
|---|---|---|---|---|---|
| catalyst | ratio (5 b/ent-6b) | yield (5b, %) | yield (ent-6b, %) | ee (5b, %) | ee (ent-6b, %) |
| Rh2(R-DOSP)4 | 10:1 | 48 | NDa | 53 | NDa |
| Rh2(R-PTAD)4 | 1.7:1 | 39 | 25 | 90 | 99 |
Not determined.
With access to a range of diasteromeric bicyclo[4.4.0]decane-derived dienes, studies were then conducted to determine if the catalyst control could be extended to generate different pairs of diastereomeric products. Substrate (S)-13 is epimeric to (S)-10 at the siloxy carbon, which is closely positioned next to the diene. However, the catalyst effect is not greatly influenced by this change. The reaction of (S)-13 with the siloxydiazoacetate 4b catalyzed by Rh2(R-PTAD)4 afforded the formal [4 + 3] cycloadducts, 11 and 12 in a 4:1 dr (entry 5) whereas the Rh2(S-PTAD)4-catalyzed reaction favored 12 by a 7:1 dr (entry 6). The next series of experiments examined the influence of the ring fusion configuration on the selectivity. All the compounds to date have been cis-fused, but diene (S)-16 is trans-fused. The Rh2(R-PTAD)4-catalyzed reaction of (S)-16 with 4b showed poor diastereoselectvity as the formal cycloadducts 17 and 18 were produced in only a 2: 1 dr (entry 7). However, the Rh2(S-PTAD)4-catalyzed reaction was much more diastereoselective, favoring 18 (16: 1 dr, entry 8). Even though these reactions do not display perfect catalyst control, they are still effective for the diastereoselective synthesis of the tricyclic product because six distinct diastereomers wre generated in isolated yields rnaging from 55–81%.
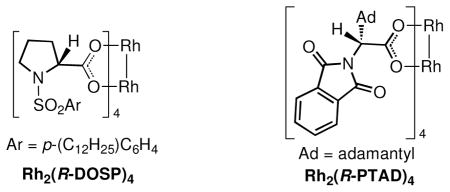
The stereochemistry of the [4 + 3] cycloaddition is controlled in the initial cyclopropanation. Theoretical calculations have shown that the alkene approaches in essentially an end-on mode,9 whereas the Cope rearrangement of the divinylcyclopropane proceeds through a boat transition state.2 Several studies have demonstrated that the same sense of asymmetry is obtained in the reaction catalyzed by either Rh2(R-DOSP)4 or Rh2(R-PTAD)4.3b,c These catalysts will cause the diene to approach from the front face as illustrated in Figure 1.3b,c This results in a matched double stereoselection because the siloxy group of the diene (S)-3 is pointing away from the carbenoid during the cyclopropanation. The Cope rearrangement of the divinylcyclopropane would generate 5b. The reaction of (S)-3 catalyzed by Rh2(S-DOSP)4 or Rh2(S-PTAD)4 are mismatched reactions, but the stereodirecting influence of Rh2(S-PTAD)4 is sufficiently strong to overwhelm the inherent stereodirecting influence of (S)-3, leading to the clean formation of 6b.
Figure 1.

Stereochemical analysis of the matched reactions of (S)-3
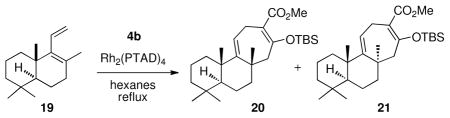
With an understanding of the factors influencing optimal stereocontrol in hand, we then applied it to the synthesis of (+)-barekoxide and (−)-barekol. The requisite diene was prepared in three steps from commercially available scalerolide (see the Supporting Information for details). Due to the more sterically crowded nature of the double bond in the diene 19, a higher temperature (70 °C) and 3–5 equivalents of 4b were required for an efficient reaction. The Rh2(R-PTAD)4 catalyzed reaction gave a 6: 1 mixture of diastereomers favoring 20, whereas the Rh2(S-PTAD)4 catalyzed reaction gave a 9: 1 mixture of diastereomers favoring 21. The mixture of 20 and 21 was only slightly separable by chromatography on silica gel impregnated with 5% AgNO3, but fortunately 20 could be selectively crystallized. In the Rh2(R-PTAD)4 catalyzed reaction, the pure desired diastereomer 20 was isolated in 47% yield after recrystallization. In this way, the configuration of a demanding quaternary stereocenter at the B–C ring fusion was effectively controlled.
The synthesis of (+)-barekoxide (1) and (−)-barekol (2) from the tricycle 20 was readily achieved as illustrated in Scheme 1. Palladium catalyzed hydrogenation of 20 generated 22 in essentially quantitative yield. Reduction of the ester in 22 followed by acidic hydrolysis of the silyl enol ether generated the enone 23 in 64% overall yield for the two steps. DIBAL-H reduction of the enone 23 generated the allylic alcohol 24, an epimer of (−)-barekol (2), in 95% yield. Deoxygenation of 24 with double bond isomerization using the Gevorgyan procedure10 followed by epoxidation with m-CPBA generated (+)-barekoxide (1) in 58% yield over two steps. Acid-catalyzed isomerization of (+)-barekoxide (1) to (−)-barekol (2)11 was achieved in 73% yield using the reported literature procedure.6a
Scheme 1.

In summary, these studies demonstrate that the combination of the siloxyvinyldiazoacetate and Rh2(R-PTAD)4 is very effective in enantiodivergent [4 + 3] cycloadditions. The chiral catalyst controls the diastereoselectivity of the [4 + 3] cycloaddition and can overwhelm the inherent selectivity of the chiral substrate. Even in the system used for the synthesis of (+)-barekoxide (1) and (−)-barekol (2), which required elevated temperatures for an effective reaction, good levels of diastereocontrol was possible, enabling the stereoselective synthesis of an all-carbon quaternary center.
Supplementary Material
Table 4.
Stereodivergent [4 + 3] cycloaddition
 | |||
|---|---|---|---|
| catalyst | ratio (4b/19) | dr (20/21) | yield (20 + 21, %) |
| Rh2(R-PTAD)4 | 5:1 | 6:1 | 65 (47a) |
| Rh2(S-PTAD)4 | 3:1 | 1:9 | 63 |
Isolated yield of pure 20.
Acknowledgments
The research by HMLD was supported by the National Institutes of Health (GM080337). RS is grateful to the ACS (RSG-09-017-01-CDD) and NIH (NIGMS RO1 GM84906-01) for funding. LCM thanks Eli Lilly for a graduate fellowship. We thank Dr. Ken Hardcastle for the X-ray crystallographic structural determination.
Footnotes
Supporting Information Available. Full experimental data for the compounds described in the paper; X-ray crystallographic files in CIF format. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For recent reviews, see: Maimone TJ, Baran PS. Nat Chem Biol. 2007;3:396. doi: 10.1038/nchembio.2007.1.Salem MM, Werbovetz KA. Current Medicinal Chemistry. 2006;13:2571. doi: 10.2174/092986706778201611.Sosa ME, Tonn CE. Phytochem Rev. 2008;7:3.
- 2.For reviews, see: Davies HML, Denton JR. Chem Soc Rev. 2009;38:3061. doi: 10.1039/b901170f.Davies HML. In: Advances in Cycloadditions. Harmata M, editor. Vol. 5. JAI Press; Greenwich, CT: 1998. pp. 119–164.
- 3.For recent examples, see: Olson JP, Davies HML. Org Lett. 2008;10:573. doi: 10.1021/ol702844g.Reddy RP, Davies HML. J Am Chem Soc. 2007;129:10312. doi: 10.1021/ja072936e.Schwartz BD, Denton JR, Lian Y, Davies HML, Williams CM. J Am Chem Soc. 2009;131:8329. doi: 10.1021/ja9019484.
- 4.Davies HML, Stafford DG, Doan BD, Houser JH. J Am Chem Soc. 1998;120:3326. [Google Scholar]
- 5.Miller LC, Ndungu JM, Sarpong R. Angew Chem Int Ed. 2009;48:2398. doi: 10.1002/anie.200806154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Justicia J, Oller-López JL, Campaña AG, Oltra JE, Cuerva JM, Buñuel E, Cárdenas DJ. J Am Chem Soc. 2005;127:14911. doi: 10.1021/ja054316o. [DOI] [PubMed] [Google Scholar]; (b) Rudi A, Shalom H, Schleyer M, Benayahu Y, Kashman Y. J Nat Prod. 2004;67:106. doi: 10.1021/np030261x. [DOI] [PubMed] [Google Scholar]; (c) Kuniyoshi M, Marma MS, Higa T, Bernardinelli G, Jefford CW. Chem Comm. 2000:1155. doi: 10.1021/np000638o. [DOI] [PubMed] [Google Scholar]; (d) Rudi A, Kashman Y. J Nat Prod. 1992;55:1408. [Google Scholar]
- 7.Rh2(S-DOSP)4: Tetrakis[(S)-(−)-N-(p-dodecylphenylsulfonyl)prolinato]-dirhodium (Cas 179162-34-6).
- 8.Rh2(S-PTAD)4: Tetrakis[(S)-(+)-(1-adamantyl)-(N-phthalimido)acetato]-dirhodium(II) (Cas 909393-65-3).
- 9.Hansen J, Autsbach J, Davies HML. J Org Chem. 2009;74:6555. doi: 10.1021/jo9009968. [DOI] [PubMed] [Google Scholar]
- 10.Gevorgyan V, Rubin M, Benson S, Liu J, Yamamoto Y. J Org Chem. 2000;65:6179. doi: 10.1021/jo000726d. [DOI] [PubMed] [Google Scholar]
- 11.The crystal structure of (−)-barekol (2) has been deposited at the Cambridge Crystallographic Data Centre, and the deposition number CCDC-776944 has been allocated. It adopts two conformations in the crystal, which supports the structure analysis by Kashman.6b
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.