

Abstract
Catalytic mechanism for butyrylcholineserase (BChE)-catalyzed hydrolysis of acetylcholine (ACh) has been studied by performing pseudobond first-principles quantum mechanical/molecular mechanical-free energy (QM/MM-FE) calculations on both acylation and deacylation of BChE. It has been shown that the acylation with ACh includes two reaction steps including the nucleophilic attack on the carbonyl carbon of ACh and the dissociation of choline ester. The deacylation stage includes nucleophilic attack of a water molecule on the carboxyl carbon of substrate and dissociation between the carboxyl carbon of substrate and hydroxyl oxygen of Ser198 side chain. Notably, despite of the fact that acetylcholinesterase (AChE) and BChE are very similar enzymes, the acylation of BChE with ACh is rate-determining, which is remarkably different from AChE-catalyzed hydrolysis of ACh in which the deacylation is rate-determining. The computational prediction is consistent with available experimental kinetic data. The overall free energy barrier calculated for BChE-catalyzed hydrolysis of ACh is 13.8 kcal/mol, which is in good agreement with experimentally-derived activation free energy of 13.3 kcal/mol.
Introduction
Cholinesterases are a family of enzymes that catalyze the hydrolysis of neurotransmitter acetylcholine (ACh), an essential reaction necessary to allow a cholinergic neuron to return to the resting state after impulse transmission. There are two types of cholinesterases, i.e. acetylcholinesterase (AChE) and butyrylcholinesterase (BChE), that differ in their distribution in the body. AChE mainly exists in neuromuscular junctions and cholinergic synapses, and hydrolyzes ACh with extremely high efficiency1. BChE is known as plasma cholinesterase, which is widely distributed in tissues and plasma. Although the physiological roles of BChE are still not completely clear, it has been found that BChE can catalyze the hydrolyses of various acyl choline, acyl thiocholine2, cocaine3, and acetanilides4. It has been noted that BChE can rapidly hydrolyze ACh in the nerves and brain5-6 and, thus, BChE can apparently substitute for AChE in maintaining the structural and functional integrity of central cholinergic pathways.
In addition, BChE has been proven a therapeutically important protein. First of all, BChE has been used as a bioscavenger in clinic for detoxification of organophosphorus (OP) nerve agents7-8. Our recently reported studies3, 9-22 have led to discovery of high-activity mutants of BChE with a considerably improved catalytic efficiency against naturally-occurring, widely-abused cocaine. The high-activity mutants of BChE have been recognized as promising candidates for therapeutic treatment of cocaine overdose and addiction23. In addition, BChE is an important target for cholinergic drugs (that include reversible inhibitors of BChE and/or AChE) in treatment of Alzheimer disease (AD) etc.24.
According to X-ray crystal structures of AChE and BChE reported so far, the overall architecture of BChE is quite similar to that of AChE25. The active site of BChE is located at the bottom of deep “aromatic gorge” lined by side chains of several aromatic residues. The channel leading from the surface to the active site is much less confining than that of AChE26. Hence the active site of BChE can accommodate larger ligands such as cocaine10. Similar to that in AChE, the active site of BChE possess a catalytic triad (consisting of Ser198, Glu325, and His438) and an oxyanion hole (consisting of Gly116, Gly117, and Ala199) that are essential for the catalytic function. It is commonly accepted that, when a substrate reaches the active site of BChE, the hydroxyl group of Ser198 acts as a nucleophile to attack the carbonyl carbon of the substrate, which initiates the acylation of BChE. The acylation gives an acyl-enzyme intermediate. Subsequently, a water molecule initiates the deacylation to break the ester linkage (Scheme 1). Throughout the hydrolysis process, the Glu325-His438 pair is crucial in activating the nucleophile and in transferring a proton to the leaving group and, thus, significantly accelerates the hydrolysis reaction.
Scheme 1.

Proposed catalytic reaction pathway for BChE-catalyzed hydrolysis of acetylcholine. Atoms colored in blue are treated by QM method in the pseudobond first-principles QM/MM calculations. Three boundary carbon atoms (Cα or Cβ) are treated with the improved pseudobond parameters 39. All other atoms belong to the MM subsystem.
Cholinesterase-catalyzed hydrolysis of ACh has been the topic for extensive experimental and computational studies1,27-38. Kinetic study37 revealed that the reaction rate constant for the deacylation is smaller than that for the acylation, indicating that the deacylation is rate-determining for AChE-catalyzed hydrolysis of ACh. Reported QM/MM studies36,38 on AChE-catalyzed hydrolysis of ACh further suggested that the first reaction step in deacylation is rate-determining for the catalytic hydrolysis process.
Despite of extensive computational studies on the catalytic mechanisms of AChE1,15,27-30,33-34,36,38, the detailed reaction mechanism for BChE-catalyzed hydrolysis of ACh still remain to be examined. Based on the high similarity between AChE and BChE structures, it seems to be reasonable assuming that the catalytic mechanism for BChE-catalyzed hydrolysis of ACh is the same as that for AChE-catalyzed hydrolysis of ACh. However, to our surprise, the present study has demonstrated that the reaction mechanism for BChE-catalyzed hydrolysis of ACh is remarkably different from the well-known mechanism for AChE-catalyzed hydrolysis of ACh in terms of the rate-determining reaction step.
In the present study, we have carried out pseudobond first-principles quantum mechanical/molecular mechanical-free energy (QM/MM-FE) calculations39-42 to study the detailed reaction pathways for the BChE-catalyzed hydrolysis of ACh. The pseudobond first-principles QM/MM-FE approach39-40,42-43 has been demonstrated to be a powerful tool in simulating a variety of enzymes12,30,41,44-46, and some theoretical predictions30,46 were subsequently confirmed by experimental studies47-49. The computational data clearly reveal the detailed reaction pathway and the corresponding free energy profiles for the BChE-catalyzed hydrolysis of ACh. The rate-determining step is thereby identified, and the roles of essential residues including the catalytic triad and oxyanion hole are discussed on the basis of the QM/MM-optimized geometries of key states in each catalytic hydrolysis reaction process.
Computational and Experimental Methods
Preparation of the Initial Structures
The X-ray crystal structure of BChE (PDB ID: 1P0M)50 and the structure of ACh in its fully extended conformation30 were used to construct the initial Michaelis–Menten complex structure of BChE-ACh complex (ES in Scheme 1). The water molecules in the active site and the choline molecule in the BChE crystal structure were removed from the BChE crystal structure. In the initial complex structure, the carbonyl oxygen of ACh was placed in the oxyanion hole consisting of Gly116, Gly117, and Ala199, and the positively charged –N-(CH3)3 moiety of ACh was placed close to the choline binding site nearby Trp8236,50. The molecular geometry of ACh was optimized by performing ab initio quantum chemical calculation using Gaussian 03 program51 at the HF/6-31G* level. The optimized geometry was used to calculate the electrostatic potential on the molecular surface at the same HF/6-31G* level. The calculated electrostatic potential was used to determine partial atomic charges with the standard restricted electrostatic potential (RESP) fitting procedure52-53. The determined RESP charges were used for the MD simulations. The constructed Michaelis–Menten complex was solvated in a rectangle box of TIP3P water molecules54, with a minimum solute wall distance of 10 Å. One chloride ion was added to neutralize the charge of the reaction system. As seen in Scheme 1, there are two stages in the BChE-catalyzed hydrolysis of ACh. The choline leaves the system after acylation. Consequently, we constructed the structure of INT2′ by removing the choline out of the QM/MM-optimized INT2 structure. The constructed INT2′ structure was then relaxed by performing ∼2 ns MD simulation in which the system was also solvated in a rectangle box with TIP3P water molecules54, with minimum solute wall distance of 10 Å.
For both acylation and deacylation stages, the last snapshots of MD simulations were used to prepare the pseudobond first-principles QM/MM calculations, as the structure of the last snapshot was close to the average structure simulated. Since we are interested in the reaction center, the water molecules beyond 50 Å of the carbonyl carbon of ACh were removed, leaving the QM/MM system with 3,022 water molecules and a total of 17,457 atoms for the acylation stage, and 2,993 water molecules and a total of 17,348 atoms for the deacylation stage. The QM/MM interface was dealt with by using a pseudobond approach39-40, 43. The used boundary of the QM-MM system for both stages is depicted in Scheme 1. Prior to the QM/MM geometry optimizations, each initial reaction system was energy-minimized with the MM method by using the revised AMBER8 program55, where the convergence criterion is a root-mean-square deviation (rmsd) of the energy gradient of less than 0.1 kcal·mol-1·Å-1.
Minimum-Energy Path of the Enzymatic Reaction
With a reaction coordinate driving method and an iterative energy minimization procedure42, the enzyme reaction path was determined by the pseudobond QM/MM calculations at the B3LYP/6-31G*:AMBER level, in which the QM calculations were performed at the B3LYP/6-31G* level of theory by using a modified version of Gaussian03 program51 and the MM calculations were performed by using a modified version of the AMBER8 program55. Normal mode analyses were performed to characterize the reactants, intermediates, transition sates, and final products. In addition, single-point energy calculations were carried out at the QM/MM(MP2/6-31+G*:AMBER) level on the QM/MM-optimized geometries. Throughout the QM/MM calculations, the boundary carbon atoms were treated with improved pseudobond parameters39. No cutoff for non-bonded interactions was used in the QM/MM calculations. For the QM subsystem, the convergence criterion for geometry optimizations follows the original Gaussian03 defaults. For the MM subsystem, the geometry optimization convergence criterion is when the rmsd of energy gradient is less than 0.1 kcal·mol-1·Å-1. Prior to QM/MM calculations, the MM subsystem was relaxed by performing ∼500 steps of energy minimization with the AMBER8 program. Then atoms within 20 Å of the carbonyl carbon (C1) of ACh were allowed to move while all the other atoms outside this range were frozen in all QM/MM calculations.
Free Energy Perturbation
After the minimum-energy path was determined by the QM/MM calculations, the free energy changes associated with the QM-MM interactions were determined by using the free energy perturbation (FEP) method42. In the FEP calculations, sampling of the MM subsystem was carried out with the QM subsystem frozen at different states along the reaction path. The point charges on the frozen QM atoms used in the FEP calculations were those determined by fitting the electrostatic potential (ESP) for the QM part of the QM/MM single-point calculations. The total free energy difference between the transition state and the reactant was calculated with the same procedure used in our previous work on other reaction systems44. The FEP calculations enabled us to more reasonably determine relative free energy changes due to the QM-MM interaction. Technically, the final (relative) free energy determined by the QM/MM-FE calculations is the QM part of the QM/MM energy (excluding the Coulumbic interaction energy between the point charges of the MM atoms and the ESP charges of the QM atoms) plus the relative free energy change determined by the FEP calculations. In the FEP calculations, the time step used was 2 fs, and bond lengths involving hydrogen atoms were constrained. In sampling of the MM subsystem by MD simulations, the temperature was maintained at 298.15 K. Each FEP calculation consisted of 50 ps of equilibration and 300 ps of sampling.
The MD simulations and QM/MM-FE calculations were performed on a supercomputer (e.g. IBM X-series cluster with 340 nodes or 1360 processors) at the University of Kentucky Center for Computational Sciences. The other modeling and computations were carried out on SGI Fuel workstations and a 34-processor IBM ×335 Linux cluster in our own laboratory.
Results and Discussion
BChE-ACh Binding Structure from MD Simulation
We performed ∼2 ns MD simulation on the BChE-ACh complex to study the enzyme-substrate (ES) binding. Collected in Figure 1 are plots of key internuclear distances vs simulation time in the MD-simulated ES complex. Traces D1, D2, and D3 represent the internuclear distances between the carbonyl oxygen O1 of ACh and the NH hydrogen atoms of residues Gly116, Gly117, and Ala199, respectively. Trace D4 is the internuclear distance between hydroxyl oxygen (Oγ) of Ser198 side chain and the carbonyl carbon C1 of ACh. Trace D5 is the internuclear distance between the hydroxyl hydrogen (Hγ) of Ser198 side chain and the Nε atom of the His438 side chain. As seen in Figure 1B, in the MD-simulated ES complex, the average values of D1, D2, and D3 are ∼2.1, ∼2.2, and ∼2.7 Å, respectively, showing that the carbonyl oxygen O1 of ACh forms two hydrogen bonds with the oxyanion hole formed by the backbone NH groups of Gly116, Gly117, and Ala199. The average value of D4 in Figure 1C is ∼3.1 Å, indicating an appropriate distance for the Ser198 hydroxyl to start nucleophilic attack on the carbonyl carbon C1 of ACh. The average value of D5 is ∼2.0 Å, suggesting that His438, the general base in the BChE catalytic triad, has been positioned well and is ready for facilitating the nucleophilic attack process through accepting a proton from the nucleophile.
Figure 1.

Key internuclear distances (D1 to D5) vs simulation time in the MD-simulated BChE-ACh complex.
Fundamental Reaction Pathway for BChE-Catalyzed Hydrolysis of ACh
The MD simulation led to a dynamically stable ES complex. Our QM/MM reaction coordinate calculations at the B3LYP/6-31G*:AMBER level starting from the MD-simulated ES complex revealed that the BChE-catalyzed hydrolysis of ACh consists of four reaction steps. The first step is the nucleophilic attack on the carbonyl carbon C1 of ACh by the Oγ atom in Ser198. The second reaction step is the dissociation between the acetyl group and the choline of ACh. The third reaction step is the nucleophilic attack on the carboxyl carbon of acylated Ser198 by a water molecule. The fourth reaction step is the dissociation between the acetyl group and Ser198 of BChE. The optimized geometries of the reactant, intermediates, transition states, and final product are shown in Figure 2. Hereby we will discuss each of these reaction steps in detail.
Figure 2.
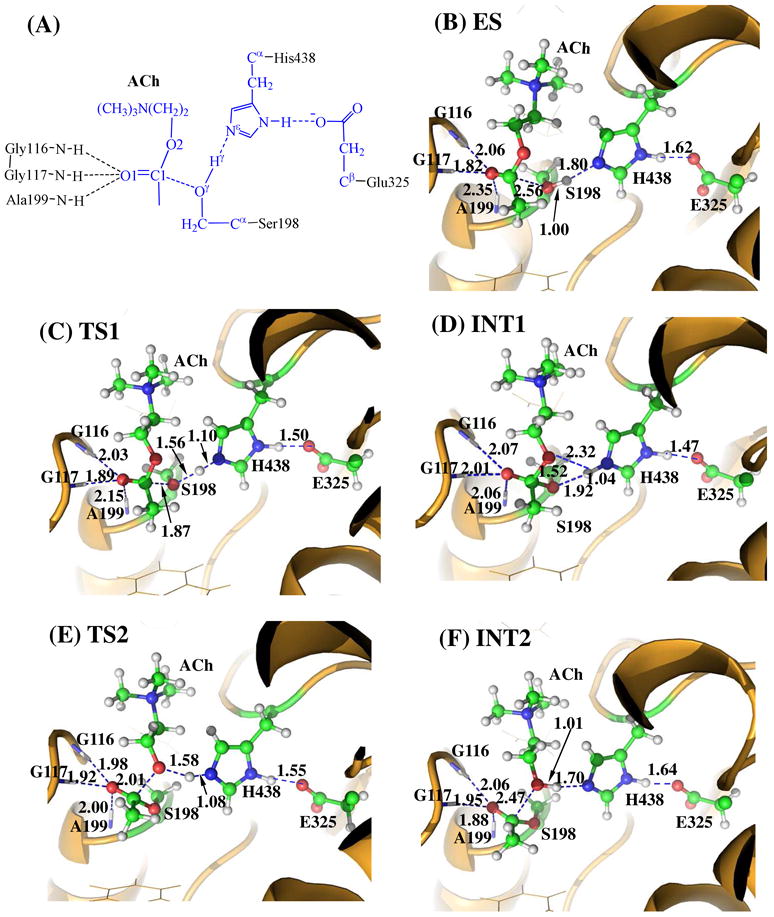
Key states for the acylation reaction stage of BChE-catalyzed ACh hydrolysis. The geometries were optimized at the QM/MM(B3LYP/6-31G*:AMBER) level. The key distances in the figure are in Å. Carbon, oxygen, nitrogen, and hydrogen atoms are colored in greed, red, blue, and white, respectively. The backbone of the protein is rendered in orange. The QM atoms are represented as balls and sticks and the surrounding residues are rendered as sticks or lines. The figures below are represented using the same method.
Step 1: Nucleophilic Attack on the Carbonyl Carbon C1 by the Oγ Atom of Ser198
Before studying the reaction pathway, the ES complex structure was first optimized at the QM/MM(B3LYP/6-31G*:AMBER) level. As shown in Figure 2B, the QM/MM-optimized geometrical parameters D1, D2, D3, and D4 (refer to Figure 1A) are 2.06, 1.82, 2.35, and 2.56 Å, respectively. It has been shown that the corresponding values of D1 to D4 in the calculated the AChE–ACh Michaelis–Menten complex30, 36 were estimated to be 1.86, 1.82, 2.24, and 2.59 Å, respectively. The calculated values of D1 to D3 reflect the binding of ACh with the oxyanion hole of BChE/AChE. Obviously, the binding of ACh with the oxyanion hole of BChE is slightly weaker than that of AChE36.
The nucleophilic attack process then proceeds as the hydroxyl oxygen Oγ of Ser198 gradually approaches the carbonyl carbon C1 of ACh. Meanwhile, the hydroxyl hydrogen Hγ of Ser198 gradually moves toward the nitrogen (Nε) atom of the His438 side chain. Since this reaction step involves the breaking of the Oγ-Hγ bond and the formation of both C1-Oγ and Nε-Hγ bonds as shown in Scheme 1, the distance between Oγ and Hγ (ROγ-Hγ), the distance between C1 and Oγ (RC1-Oγ), and the distance between Nε and Hγ (RNε- Hγ) reflect the nature of chemical reaction step 1. Therefore, the reaction coordinate for the current reaction step was set as ROγ-Hγ − RC-Oγ − RNε-Hγ. As shown in the QM/MM-optimized geometries (Figure 2), when the Oγ atom of Ser198 gradually approaches the C1 atom, the geometry of the reactant (ES) in which the C1 atom is sp2-hybridized and is in a planar geometry with its three attached groups gradually changes into a tetrahedral geometry centered at the sp3-hybridized C1 atom in an intermediate (INT1) through a transition state (TS1).
Step 2: Dissociation of the Choline Ester
In this reaction step, the choline moiety of ACh gradually departs from the acetyl group in which the choline ester bond C1-O2 is broken. Meanwhile, the proton (Hγ) attached to the Nε atom of the His438 side chain transfers to choline oxygen atom (O2) of ACh. The changes of the distances RC1-O2, RO2-Hγ, and RNε-Hγ reflect the nature of the dissociation process. Thus, the reaction coordinate for the current reaction step was chosen to be RC1-O2 + RNε-Hγ − RO2-Hγ.
In the geometry of INT1 where the serine hydroxyl proton (Hγ) has been transferred to the Nε atom of His438 in reaction step 1, the optimized distance (ROγ-Hγ) is 1.92 Å, suggesting the interaction between the Ser198 terminal oxygen and the protonated His438 side chain is weak. At the same time, the distance (RO2-Hγ) between Hγ and the leaving ester oxygen O2 to which Hγ is about to be transferred is 2.32 Å, indicating a weak hydrogen bond between the Hγ atom and the O2 atom. Hence, the Hγ atom is in the appropriate place to protonate the leaving O2 atom of ACh.
In changing from INT1 to TS2, there are two major types of structural changes. One is the gradual breaking of the covalent bond C1-O2 (RC1-O2 is 1.52 Å in INT1 and 2.01 Å in TS2). The other is the formation of a hydrogen bond (Nε-Hγ⋯O2) indicated by the shorter and shorter distance RO2-Hγ in going from INT1 to TS2 (2.32 Å in INT1and 1.58 Å in TS2). In the meantime, the hydrogen bond Nε-Hγ⋯Oγ involving the transferring proton (Hγ) and the Oγ atom of Ser198 becomes progressively weaker (ROγ-Hγ is 1.92 Å in INT1 and 2.52 Å in TS2), which is reasonable as the transferring proton (Hγ) is about to be transferred to the leaving ester oxygen (O2) in the current reaction step.
Step 3: Nucleophilic Attack on the Carbonyl Carbon by a Water Molecule
The choline was removed from the above-discussed QM/MM-optimized geometry of INT2 to construct the structure of INT2′, which was then relaxed by performing MD simulation. A water molecule close to the carbonyl carbon (C1) of the substrate was selected as the nucleophile and was treated by the QM method. Similarly to that in the acylation stage, the carbonyl oxygen of the substrate is also stabilized by the oxyanion hole. As shown in Figure 3B, two strong hydrogen bonds are formed between the carbonyl oxygen (O1) of ACh and the oxyanion hole in INT2′.
Figure 3.
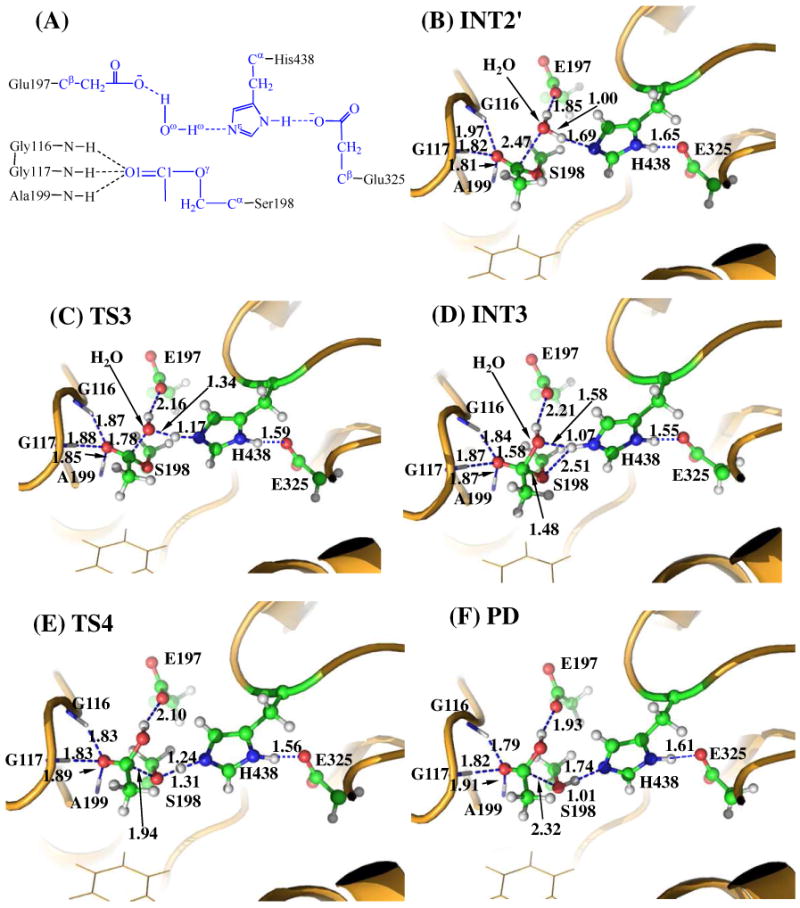
Key states for the deacylation reaction of BChE-catalyzed ACh hydrolysis. The geometries were optimized at QM/MM(B3LYP/6-31G*:AMBER) level. See caption of Figure 2 for the color codes for different types of atoms.
The current nucleophilic attacking process involves the breaking of the Oω-Hω bond and the formation of both C1-Oω and Nω-Hω bonds (see Figure 3A). Thus, the distances ROω-Hω, RC1-oω, and RNε-Hω were chosen to establish the reaction coordinate as ROω-Hω − RC1-Oω + RNε-Hω for the current reaction step. In proceeding from INT2′ to INT3 through the transition state TS3 (Figure 3), the coplanar geometry changes into tetrahedral centering on the sp3-hybridized carbonyl carbon (C1) atom as the nucleophilic water gradually approaches the C1 atom with a spontaneous proton (Hω) transfer from the Oω atom of the nucleophilic water to the Nε atom of the His438 side chain. The QM/MM-optimized geometry of INT3 shows that the nucleophilic attack process is completed with water dissociating into hydroxide ion attaching to the C1 atom and a proton (Hω) attaching to the Nε atom.
Step 4: Dissociation between the Acetyl Group and Ser198 of BChE
The proton transfer between the Nε atom of His438 side chain and the Oγ atom of Ser198 side chain and the breaking of the covalent bond C1-Oγ are involved in the dissociation of acetyl-enzyme. The changes of the distances RC1-Oγ, ROγ- Hω, and RNε-Hω reflect the nature of reaction step 4. Thus, the reaction coordinate for the current reaction step was expressed as RC1-Oγ − RNε-Hω − ROγ-Hω. Reaction step 4 is similar to reaction step 2. In both reaction steps, the C-O covalent bond is broken and one proton is transferred from the Nε atom of His438 to the oxygen atom of the broken C-O covalent bond. As shown in Figure 3, during the breaking of the C1-Oγ covalent bond, the distance between the Oγ and Hω atoms becomes closer and closer, illustrating a spontaneous proton transfer from the Nε atom of His438 side chain to the Oγ atom of Ser198.
Energetics
Depicted in Figure 4 is the free energy profile for BChE-catalyzed hydrolysis of ACh, determined by the QM/MM-FE calculations at the MP2/6-31+G*:AMBER level excluding the zero-point and thermal corrections for the QM subsystem. The values given in parentheses are the corresponding relative free energies including the zero-point and thermal corrections for the QM subsystem.
Figure 4.

Free energy profiles for the acylation and deacylation stages of BChE-catalyzed hydrolysis of ACh. The relative free energies were determined by the QM/MM-FE calculations at the MP2/6-31+G*:AMBER level, excluding the zero-point and thermal corrections for the QM system. Values in the parenthesis are relative free energies including the zero-point and thermal corrections for the QM subsystem.
As shown in Figure 4A, with the zero-point and thermal corrections for the QM subsystem, the free energy barriers calculated for the first and second reaction steps (acylation) of BChE-catalyzed hydrolysis of ACh are 10.3 and 6.1 kcal/mol, respectively. Although the free energy barrier for the first reaction step is higher than that for the second reaction step, the relative free energy of TS2 is higher than that of TS1, indicating that the calculated overall free energy barrier for the acylation is the free energy change from ES to TS2, which is 13.8 kcal/mol. Similarly, in the deacylation stage of BChE-catalyzed hydrolysis of ACh, the free energy barrier (9.1 kcal/mol) calculated for the third reaction step is higher than that (4.5 kcal/mol) calculated for the fourth reaction step, but the relative free energy of TS4 is higher than that of TS3, implying that the calculated overall free energy barrier for the deacylation is the free energy change from INT2′ to TS4, which is 11.9 kcal/mol. Apparently, the acylation stage with the overall free energy barrier of 13.8 kcal/mol, which is the free energy change from ES to TS2, is rate-determining for BChE-catalyzed hydrolysis of ACh.
As mentioned above, the overall architecture of BChE structure is quite similar to that of AChE structure. In particular, the catalytic residues in the active site gorge of both enzymes are identical. Hence, it is surprising to note that the mechanism for BChE-catalyzed hydrolysis of ACh, where the acylation is rate-determining, is remarkably different from the well-known mechanism for AChE-catalyzed hydrolysis of ACh where the deacylation is rate-determining37. The mechanistic difference between BChE and AChE helps to better understand available experimental kinetic data in literature. In particular, it has been known that37 the catalytic rate constant (kcat) for AChE-catalyzed hydrolysis of ACh is identical to that for AChE-catalyzed hydrolysis of acetylthiocholine (ATCh) within the experimental errors, due to the fact that AChE-catalyzed hydrolyses of ACh and ATCh share a common rate-determining deacylation stage. The molecular geometry of ATCh is essentially the same as that of ACh, as the only structural difference between the two substrates is that the ester oxygen (O) in ACh is replaced by a sulfur (S) atom in ATCh. The experimental catalytic rate constants for BChE-catalyzed hydrolyses of ACh and ATCh were also reported in literature: kcat = 6.12 × 104 min-1 for BChE-catalyzed hydrolysis of ACh56 and kcat = 2.02 × 104 min-1 for BChE-catalyzed hydrolysis of ATCh2. The two kcat values are significantly different. The significant difference in kcat between ACh and ATCh hydrolyses catalyzed by BChE indicates that the rate-determining step should not be the common deacylation stage. So, the available experimental kinetic data are consistent with our computational prediction that the acylation is rate-determining for BChE-catalyzed hydrolysis of ACh.
Further, according to the conventional transition state theory (CTST)57, a rate constant of 6.12 × 104 min-1 (reported in ref.56) for BChE-catalyzed hydrolysis of ACh is associated with an activation free energy of 13.3 kcal/mol at room temperature (25°C). The experimentally-derived activation free energy of 13.3 kcal/mol is in good agreement with our predicted free energy barrier of 13.8 kcal/mol.
Conclusion
In this work, we employed pseudobond first-principles QM/MM-FE approach to study the reaction pathway for BChE-catalyzed hydrolysis of ACh and the corresponding free energy profile. The computational results demonstrate that ACh hydrolysis catalyzed by BChE consists of two major reaction stages, i.e. acylation and deacylation of BChE. The acylation of BChE with ACh includes two reaction steps. The first reaction step is the nucleophilic attack on the carbonyl carbon of ACh by the hydroxyl oxygen of Ser198 side chain; the second reaction step is the dissociation of the choline ester. The deacylation stage includes the nucleophilic attack of a water molecule on the carboxyl carbon of substrate, followed by the dissociation between the carbonyl carbon of substrate and hydroxyl oxygen of Ser198 side chain. The computationally determined free energy profile indicates that the acylation is rate-determining for BChE-catalyzed hydrolysis of ACh, which is remarkably different from the well-known mechanism for AChE-catalyzed hydrolysis of ACh where the common deacylation stage is rate-determining. The computational prediction is consistent with available experimental kinetic data showing that the catalytic rate constants for BChE-catalyzed hydrolyses of ACh and ATCh are significantly different. The calculated overall free energy barrier of 13.8 kcal/mol for BChE-catalyzed hydrolysis of ACh is in good agreement with the experimentally-derived activation free energy of 13.3 kcal/mol.
Acknowledgments
This work was supported in part by NIH (grants R01 DA025100 and R01 DA013930 to Zhan). Chen worked in Zhan's laboratory for this project at the University of Kentucky. The entire work was performed at University of Kentucky. The authors also acknowledge the Center for Computational Sciences (CCS) at University of Kentucky for supercomputing time on IBM X-series Cluster with 340 nodes or 1,360 processors.
References
- 1.Fuxreiter M, Warshel A. J Am Chem Soc. 1998;120(1):183–194. [Google Scholar]
- 2.Boeck AT, Schopfer LM, Lockridge O. Biochem Pharmacol. 2002;63(12):2101–2110. doi: 10.1016/s0006-2952(02)01029-8. [DOI] [PubMed] [Google Scholar]
- 3.Zhan CG, Gao DQ. Biophys J. 2005;89(6):3863–3872. doi: 10.1529/biophysj.105.070276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Masson P, Froment MT, Gillon E, Nachon F, Lockridge O, Schopfer LM. FEBS J. 2008;275(10):2617–2631. doi: 10.1111/j.1742-4658.2008.06409.x. [DOI] [PubMed] [Google Scholar]
- 5.Mesulam MM, Guillozet A, Shaw P, Levey A, Duysen EG, Lockridge O. Neuroscience. 2002;110(4):627–39. doi: 10.1016/s0306-4522(01)00613-3. [DOI] [PubMed] [Google Scholar]
- 6.Mesulam M, Guillozet A, Shaw P, Quinn B. Neurobiology of Disease. 2002;9(1):88–93. doi: 10.1006/nbdi.2001.0462. [DOI] [PubMed] [Google Scholar]
- 7.Millard CB, Lockridge O, Broomfield CA. Biochemistry. 1995;34(49):15925–15933. doi: 10.1021/bi00049a007. [DOI] [PubMed] [Google Scholar]
- 8.Lockridge O, Blong RM, Masson P, Froment MT, Millard CB, Broomfield CA. Biochemistry. 1997;36(4):786–795. doi: 10.1021/bi961412g. [DOI] [PubMed] [Google Scholar]
- 9.Pan YM, Gao DQ, Yang WC, Cho H, Yang GF, Tai HH, Zhan CG. P Natl Acad Sci USA. 2005;102(46):16656–16661. doi: 10.1073/pnas.0507332102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhan CG, Zheng F, Landry DW. J Am Chem Soc. 2003;125(9):2462–74. doi: 10.1021/ja020850+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao DQ, Cho H, Yang WC, Pan YM, Yang GF, Tai HH, Zhan CG. Angew Chem Int Edit. 2006;45(4):653–657. doi: 10.1002/anie.200503025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zheng F, Yang WC, Ko MC, Liu JJ, Cho H, Gao DQ, Tong M, Tai HH, Woods JH, Zhan CG. J Am Chem Soc. 2008;130(36):12148–12155. doi: 10.1021/ja803646t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zheng F, Zhan CG. J Comput Aid Mol Des. 2008;22(9):661–671. doi: 10.1007/s10822-007-9144-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zheng F, Zhan CG. Org Biomol Chem. 2008;6(5):836–843. doi: 10.1039/b716268e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao DQ, Zhan CG. The J Phys Chem B. 2005;109(48):23070–23076. doi: 10.1021/jp053736x. [DOI] [PubMed] [Google Scholar]
- 16.Zhan CG. Expert Rev Clin Pharmacol. 2009;2(1):1–4. doi: 10.1586/17512433.2.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang W, Pan Y, Zheng F, Cho H, Tai HH, Zhan CG. Biophys J. 2009;96(5):1931–1938. doi: 10.1016/j.bpj.2008.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pan YM, Gao DQ, Yang WC, Cho H, Zhan CG. J Am Chem Soc. 2007;129(44):13537–13543. doi: 10.1021/ja073724k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hamza A, Cho H, Tai HH, Zhan CG. J Phys Chem B. 2005;109(10):4776–4782. doi: 10.1021/jp0447136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao DQ, Zhan CG. Proteins. 2006;62(1):99–110. doi: 10.1002/prot.20713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang W, Xue L, Fang L, Chen X, Zhan CG. Chem-Biol Interact. 2010;187(1-3):148–152. doi: 10.1016/j.cbi.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang W, Pan Y, Fang L, Gao D, Zheng F, Zhan CG. J Phys Chem B. 2010;114(33):10889–10896. doi: 10.1021/jp104989b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brimijoin S, Gao Y, Anker JJ, Gliddon LA, LaFleur D, Shah R, Zhao QH, Singh M, Carroll ME. Neuropsychopharmacology. 2008;33(11):2715–2725. doi: 10.1038/sj.npp.1301666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Decker M. Eur J Med Chem. 2005;40(3):305–313. doi: 10.1016/j.ejmech.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 25.Sussman JL, Harel M, Frolow F, Oefner C, Goldman A, Toker L, Silman I. Science. 1991;253(5022):872–879. doi: 10.1126/science.1678899. [DOI] [PubMed] [Google Scholar]
- 26.Saxena A, Redman AMG, Jiang XL, Lockridge O, Doctor BP. Biochemistry. 1997;36(48):14642–14651. doi: 10.1021/bi971425+. [DOI] [PubMed] [Google Scholar]
- 27.Wlodek ST, Antosiewicz J, Briggs JM. J Am Chem Soc. 1997;119(35):8159–8165. [Google Scholar]
- 28.Wang QM, Jiang HL, Chen JZ, Chen KX, Ji RY. Int J Quantum Chem. 1998;70(3):515–525. [Google Scholar]
- 29.Vagedes P, Rabenstein B, Aqvist J, Marelius J, Knapp EW. J Am Chem Soc. 2000;122(49):12254–12262. [Google Scholar]
- 30.Zhang YK, Kua J, McCammon JA. J Am Chem Soc. 2002;124(35):10572–10577. doi: 10.1021/ja020243m. [DOI] [PubMed] [Google Scholar]
- 31.Manojkumar TK, Cui CZ, Kim KS. J Comput Chem. 2005;26(6):606–611. doi: 10.1002/jcc.20199. [DOI] [PubMed] [Google Scholar]
- 32.Suarez D, Field MJ. Proteins. 2005;59(1):104–117. doi: 10.1002/prot.20398. [DOI] [PubMed] [Google Scholar]
- 33.Tachikawa H, Igarashi M, Nishihira J, Ishibashi T. J Photochem Photobiol, B. 2005;79(1):11–23. doi: 10.1016/j.jphotobiol.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 34.Sant'Anna CMR, Viana AD, do Nascimento NM. Bioorg Chem. 2006;34(2):77–89. doi: 10.1016/j.bioorg.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 35.Suarez D, Diaz N, Fontecilla-Camps J, Field MJ. Biochemistry. 2006;45(24):7529–7543. doi: 10.1021/bi052176p. [DOI] [PubMed] [Google Scholar]
- 36.Zhou Y, Wang S, Zhang Y. J Phys Chem B. 2010;114(26):8817–8825. doi: 10.1021/jp104258d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Froede HC, Wilson IB. J Biol Chem. 1984;259(17):1010–1013. [PubMed] [Google Scholar]
- 38.Nemukhin AV, Lushchekina SV, Bochenkova AV, Golubeva AA, Varfolomeev SD. J Mol Model. 2008;14(5):409–416. doi: 10.1007/s00894-008-0287-y. [DOI] [PubMed] [Google Scholar]
- 39.Zhang YK. J Chem Phys. 2005;122(2):024114. doi: 10.1063/1.1834899. [DOI] [PubMed] [Google Scholar]
- 40.Zhang YK, Lee TS, Yang WT. J Chem Phys. 1999;110(1):46–54. [Google Scholar]
- 41.Hu P, Zhang YK. J Am Chem Soc. 2006;128(4):1272–1278. doi: 10.1021/ja056153+. [DOI] [PubMed] [Google Scholar]
- 42.Zhang YK, Liu HY, Yang WT. J Chem Phys. 2000;112(8):3483–3492. [Google Scholar]
- 43.Zhang YK. Theor Chem Acc. 2006;116(1-3):43–50. [Google Scholar]
- 44.Liu JJ, Hamza A, Zhan CG. J Am Chem Soc. 2009;131(33):11964–11975. doi: 10.1021/ja903990p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu JJ, Zhang YK, Zhan CG. J Phys Chem B. 2009;113(50):16226–16236. doi: 10.1021/jp9055335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu HY, Zhang YK, Yang WT. J Am Chem Soc. 2000;122(28):6560–6570. [Google Scholar]
- 47.Poyner RR, Larsen TM, Wong SW, Reed GH. Arch Biochem Biophys. 2002;401(2):155–163. doi: 10.1016/S0003-9861(02)00024-3. [DOI] [PubMed] [Google Scholar]
- 48.Cisneros GA, Wang M, Silinski P, Fitzgerald MC, Yang WT. Biochemistry. 2004;43(22):6885–6892. doi: 10.1021/bi049943p. [DOI] [PubMed] [Google Scholar]
- 49.Metanis N, Brik A, Dawson PE, Keinan E. J Am Chem Soc. 2004;126(40):12726–12727. doi: 10.1021/ja0463841. [DOI] [PubMed] [Google Scholar]
- 50.Nicolet Y, Lockridge O, Masson P, Fontecilla-Camps JC, Nachon F. J Biol Chem. 2003;278(42):41141–41147. doi: 10.1074/jbc.M210241200. [DOI] [PubMed] [Google Scholar]
- 51.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, J, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03, Revision C.02. Gaussian, Inc.; Wallingford, CT: 2004. [Google Scholar]
- 52.Cieplak P, Cornell WD, Bayly C, Kollman PA. J Comput Chem. 1995;16(11):1357–1377. [Google Scholar]
- 53.Bayly CI, Cieplak P, Cornell WD, Kollman PA. J Phys Chem. 1993;97(40):10269–10280. [Google Scholar]
- 54.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. J Chem Phys. 1983;79(2):926–935. [Google Scholar]
- 55.Case DA, Darden TA, Cheatham TE, I, Simmerling CL, Wang J, Duke RE, Luo R, Merz KM, Wang B, Pearlman DA, Crowley M, Brozell S, Tsui V, Gohlke H, Mongan J, Hornak V, Cui G, Beroza P, Schafmeister C, Caldwell JW, Ross WS, Kollman PA. AMBER8. University of California; San Francisco: 2004. [Google Scholar]
- 56.Gao Y, LaFleur D, Shah R, Zhao QH, Singh M, Brimijoin S. Chem Biol Interact. 2008;175(1-3):83–87. doi: 10.1016/j.cbi.2008.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Alvarez-Idaboy JR, Galano A, Bravo-Perez G, Ruiz ME. J Am Chem Soc. 2001;123(34):8387–8395. doi: 10.1021/ja010693z. [DOI] [PubMed] [Google Scholar]