

Abstract
We report a new method for the formal [3+2] reaction of aryl cyclopropyl ketones with olefins to generate highly substituted cyclopentane ring systems. The key initiation step in this process is the one-electron reduction of the ketone to the corresponding radical anion, which is accomplished using a photocatalytic system comprising Ru(bpy)32+, La(OTf)3−, and TMEDA.
Cyclopentanes are important structural elements of a wide variety of organic compounds. One particularly attractive strategy for their synthesis is the [3+2] cycloaddition reaction of activated cyclopropanes with olefins.1 Several straightforward and robust methods for the synthesis of cyclopropyl ketones are available,2 and the strain associated with the three-membered ring provides a substantial driving force for the synthesis of larger carbocycles. However, most [3+2] cycloadditions of cyclopropanes reported to date have utilized “donor-acceptor” cyclopropanes,3 methylene cyclopropanes,4 or similar highly activated cyclopropanes that bear substituents that predispose them towards ring-opening.5 Recently, Montgomery and Ogoshi independently reported cycloadditions of simple cyclopropyl ketones with olefins that are catalyzed by Ni0 complexes.6 Herein, we report a mechanistically distinct method for the activation and [3+2] cycloaddition of simple aryl cyclpropyl ketones based upon our strategy for formation of anion radicals by visible light photocatalysis.7,8,9
The chemistry of cyclopropyl ketyl radicals has been studied in a variety of contexts.10 Synthetically, they have most commonly been exploited for their propensity to undergo reductive fragmentations.11,12 To the best of our knowledge, however, they have not been examined as intermediates in [3+2] cycloaddition reactions. We recently showed that a Ru(bpy)32+ photocatalyst can readily promote the one-electron reduction of a variety of aryl enones upon irradiation with visible light, and we have reported [2+2] cycloadditions involving radical anions generated in this manner.7 We wondered if similar photocatalytic conditions might be used to effect the one-electron reduction of aryl cyclopropyl ketone 1. We expected the resulting radical anion 2 to be capable of ring-opening to distonic radical anion 3. Sequential radical cyclizations might then give rise to cyclized ketyl radical 5, which, upon loss of an electron, would produce 6 as the product of formal intramolecular [3+2] cycloaddition of 1.
However, under the conditions we had utilized for [2+2] cycloadditions of enones (Ru(bpy)3Cl2, LiBF4, i-Pr2NEt), we observed no evidence of ring opening of 1 (Table 1, entry 1). In our original studies, we had discovered that the Lewis acidity of the lithium cation was critical to the success of the reaction and speculated that it served to activate the carbonyl compound towards one-electron reduction. We reasoned, therefore, that stronger Lewis acid additives might better activate the cyclopropyl ketone towards one-electron reduction, better stabilize the ketyl radical intermediate, and thus increase the likelihood of productive cycloaddition. Indeed, modest conversion to 6 was observed using a variety of Lewis acidic additives including Zn(OTf)2, Gd(OTf)3, and La(OTf)3 (entries 2–4). A brief screen of amine additives revealed that TMEDA afforded significantly higher yields of the cycloadduct (entries 5 and 6). Finally, reducing the loading of the photocatalyst to 2.5 mol% produced an additional increase in the yield of the reaction, and under these conditions we were able to isolate 83% of cyclopentane 6 (entry 7). We also conducted control experiments to verify the necessity of each component of the reaction mixture and found that the starting cyclopropane remained unchanged when the Ru(bpy)3Cl2, La(OTf)3, or TMEDA were omitted from the reaction (entries 8-10). These experiments indicate the critical role of the photocatalyst, Lewis acid, and reductive quencher, respectively, for the successful generation of the key radical.13 On the other hand, the use of MgSO4 or other drying agents was not necessary, but its omission resulted in somewhat lower yields and poorer reproducibility (entry 12).
Table 1.
Optimization and control studies.a
| entry | mol% Ru | Lewis acid | amine (equiv) | yieldb | d.r.c |
|---|---|---|---|---|---|
| 1 | 5 | LiBF4 | i-Pr2NEt (10) | 0% | -- |
| 2 | 5 | Zn(OTf)2 | i-Pr2NEt (10) | <5% | -- |
| 3 | 5 | Gd(OTf)3 | i-Pr2NEt (10) | 7% | 7:1 |
| 4 | 5 | La(OTf)3 | i-Pr2NEt (10) | 32% | 6:1 |
| 5 | 5 | La(OTf)3 | Et3N (10) | 7% | 6:1 |
| 6 | 5 | La(OTf)3 | TMEDA (5) | 73% | 9:1 |
| 7 | 2.5 | La(OTf)3 | TMEDA (5) | 87% (83%) d | 6:1 |
| 8 | 0 | La(OTf)3 | TMEDA (5) | 0% | -- |
| 9 | 2.5 | none | TMEDA (5) | 0% | -- |
| 10 | 2.5 | La(OTf)3 | none | 0% | -- |
| 11e | 2.5 | La(OTf)3 | TMEDA (5) | 0% | -- |
| 12f | 2.5 | La(OTf)3 | TMEDA (5) | 82% | 7:1 |
Subjected to irradiation with a 23 W compact fluorescent bulb unless otherwise noted.
Determined by 1H NMR unless otherwise noted.
Ratio of major isomer to all other isomers, as determined by GC.
Isolated yield in parentheses.
No light.
No MgSO4.
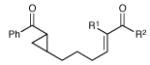
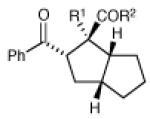
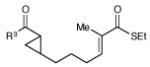
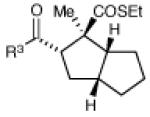
Having developed conditions for this novel [3+2] cycloaddition, we conducted an initial investigation of the scope of this reaction (Table 2). Variation of the enone moiety revealed that a variety of esters, ketones and thioesters participate in this cycloaddition, but that the presence of an α-substituent increased the efficiency and stereoselectivity of the process, and that reactions of α-substituted thioesters were particularly diastereoselective (entries 1–7). We found that aliphatic cyclopropyl ketones were not successful substrates for this reaction, presumably due to the greater difficulty of generating their radical anions. On the other hand, a variety of substituted aryl ketones were easily tolerated (entries 8 and 9), although electron-donating substituents resulted in sluggish reactions. Cycloadditions with various tethers, including three-carbon, four-carbon, and heteroatom linkers, were successful (entries 10–13).14 Finally, in contrast to the [2+2] process of the parent bis(enones), we found that coupling with an enone acceptor was not a requirement; styrenes also resulted in successful [3+2] cycloadditions (entries 12 and 13). Cyclic aliphatic olefins also participate in this reaction, enabling the synthesis of tricyclic structures (entry 14). Finally, both aryl and aliphatic alkynes were excellent reaction partners, (entries 15 and 16), although in the latter case we observed migration of the alkene into conjugation with the ketone.
Table 2.
Scope studies of intramolecular [3+2] cycloadditions.a
| entry | substrate | product | Time | yieldb | d.r.c |
|---|---|---|---|---|---|
|
|
||||
| 1 | R1 = Me, R2 = OEt | 6.5 h | 83% | 6:1 | |
| 2 | R1 = H, R2 = OEt | 5.5 h | 67% | 2:1 | |
| 3 | R1 = Me, R2 = Ot-Bu | 6.5 h | 86% | 5:1 | |
| 4 | R1 = H, R2 = Ot-Bu | 12 h | 84% | 2:1 | |
| 5 | R1 = Me, R2 = t-Bu | 5 h | 82% | 4:1 | |
| 6 | R1 = Me, R2 = SEt | 17 h | 79% | >10:1 | |
| 7 | R1 = Et, R2 = SEt | 16 h | 70% | 10:1 | |
|
|
||||
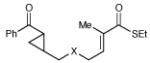
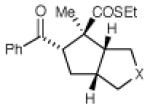
| 8 | R3 = 4-MeO-C6H4 | 48 h | 55% | >10:1 | |
| 9 | R3 = 4-Cl-C6H4 | 12 h | 73% | >10:1 | |
|
|
||||
| 10 | X = C(Me)2 | 19 h | 82% | >10:1 | |
| 11 | X = CH2CH2 | 34 h | 76% | 1:1 | |
|
|
||||
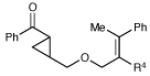
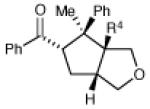
| 12 | R4 = H | 29 h | 63% | >10:1 | |
| 13 | R4 = Me | 48 h | 57% | 3:1 | |
| 14 |
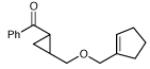
|
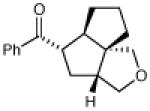
|
18 h | 69% | 2:1 |
| 15 |
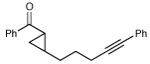
|
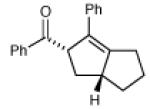
|
16 h | 83% | 9:1 |
| 16 |
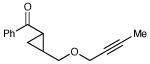
|
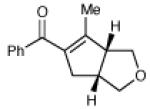
|
13 h | 73% | >10:1 |
Conditions: 2.5 mol% Ru(bpy)3Cl2, 1 equiv of La(OTf)3, 5 equiv of TMEDA, 200 wt% MgSO4, MeCN, 23 °C. Reactions were irradiated with a standard consumer 23 W compact fluorescent light bulb.
Averaged isolated yields from two reproducible experiments.
Ratio of major diastereomer to all other diastereomers as determined by GC.
We also performed the [3+2] cycloaddition of cyclopropane 7 (eq 1). This substrate was prepared as a 1:1 mixture of diastereomers.15 Nevertheless, cycloaddition under our optimized conditions affords the tricyclic cycloadduct 8 as a single diasteromer, which is consistent with the intermediacy of a non-stereogenic ring-opened distonic radical anion in which the stereochemical integrity of the cyclopropane is lost. Thus, the results of this experiment are in good agreement with the design plan outlined in Scheme 1.
Scheme 1.

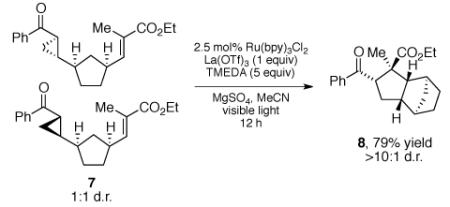 |
(1) |
Finally, we are also interested in developing an intermolecular version of this [3+2] cycloaddition. As an initial exploration of this possibility, we irradiated cyclopropyl phenyl ketone (9) with methacrylonitrile (10) under conditions similar to those we had optimized for the intramolecular cycloaddition and observed successful formation of [3+2] cycloadduct 11 (eq 2). However, the yield and diastereoselectivity of this process are low, and efforts to optimize this promising initial result are continuing in our laboratories.
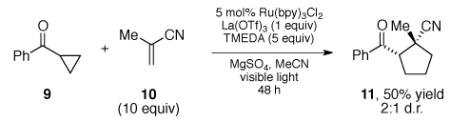 |
(2) |
In conclusion, we have developed a new, mechanistically novel [3+2] cycloaddition of simple aryl cyclopropyl ketones that exploits our recently reported strategy for visible light photocatalysis. A variety of reaction partners are suitable, but α-substituted enoates work particularly well, enabling the rapid diastereoselective construction of quaternary carbon stereocenters within a cyclopentane-containing framework. The further development of this and other synthetically valuable photocatalytic processes is a continuing effort in our labs.
Supplementary Material
Acknowledgment
Financial support from the ACS PRF (49817-ND1), Sloan Foundation, Beckman Foundation, and Research Corporation is gratefully acknowledged. The NMR facilities at UW-Madison are funded by the NSF (CHE-9208463, CHE-9629688) and NIH (RR08389-01).
Footnotes
Supporting Information Available: Experimental procedures and spectral data for all new compounds (PDF format) are provided. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For a recent review, see: De Simone F, Waser J. Synthesis. 2009:3353–3374.
- 2.(a) Corey EJ, Chaykovsky M. J. Am. Chem. Soc. 1965;87:1353–1364. [Google Scholar]; (b) Doyle MP. Chem. Rev. 1986;86:919–939. [Google Scholar]; (c) Lebel H, Marcoux JF, Molinaro C, Charette AB. Chem. Rev. 2003;103:977–1050. doi: 10.1021/cr010007e. [DOI] [PubMed] [Google Scholar]
- 3.For recent reviews of donor-acceptor cyclopropane chemistry, see: Reissig H-U, Zimmer R. Chem. Rev. 2003;103:1151–1196. doi: 10.1021/cr010016n. Yu M, Pagenkopf BL. Tetrahedron. 2005;61:321–347. Campbell MJ, Johnson JS, Parsons AT, Pohlhaus PD, Sanders SD. J. Org. Chem. 2010;75:6317–3290. doi: 10.1021/jo1010735. 2010.
- 4.For recent reviews of methylene cyclopropane chemistry, see: Binger P, Büch HM. Top. Curr. Chem. 1987;135:77–151. Brandi A, Goti A. Chem. Rev. 1998;98:589–635. doi: 10.1021/cr940341t. Brandi A, Cicchi S, Cordero FM, Goti A. Chem. Rev. 2003;103:1213–1269. doi: 10.1021/cr010005u.
- 5.(a) Alper PB, Meyers C, Lerchner A, Siegel DR, Carreira EM. Angew. Chem. Int. Ed. 1999;38:3186–3189. [PubMed] [Google Scholar]; (b) Lautens M, Han W. J. Am. Chem. Soc. 2002;124:6312–6316. doi: 10.1021/ja011110o. [DOI] [PubMed] [Google Scholar]; (c) Carson CA, Kerr MA. Chem. Soc. Rev. 2009;38:3051–3060. doi: 10.1039/b901245c. [DOI] [PubMed] [Google Scholar]; (d) Jiao L, Ye S, Yu Z-X. J. Am. Chem. Soc. 2008;130:7178–7179. doi: 10.1021/ja8008715. [DOI] [PubMed] [Google Scholar]; (e) Jiao L, Lin M, Yu Z-X. Chem. Commun. 2010;46:1059–1061. doi: 10.1039/b922417c. [DOI] [PubMed] [Google Scholar]; (f) Li Q, Jiang G-J, Jiao L, Yu Z-X. Org. Lett. 2010;12:1332–1335. doi: 10.1021/ol100237h. [DOI] [PubMed] [Google Scholar]
- 6.(a) Liu L, Montgomery J. J. Am. Chem. Soc. 2006;128:5348–5349. doi: 10.1021/ja0602187. [DOI] [PubMed] [Google Scholar]; (b) Ogoshi S, Nagata M, Kurosawa H. J. Am. Chem. Soc. 2006;128:5350–5351. doi: 10.1021/ja060220y. [DOI] [PubMed] [Google Scholar]; (c) Liu L, Montgomery J. Org. Lett. 2007;9:3885–3887. doi: 10.1021/ol071376l. [DOI] [PubMed] [Google Scholar]; (d) Tamaki T, Nagata M, Ohashi M, Ogoshi S. Chem. Eur. J. 2009;15:10083–10091. doi: 10.1002/chem.200900929. [DOI] [PubMed] [Google Scholar]
- 7.(a) Ischay MA, Anzovino ME, Du J, Yoon TP. J. Am. Chem. Soc. 2008;130:12886–12887. doi: 10.1021/ja805387f. [DOI] [PubMed] [Google Scholar]; (b) Du J, Yoon TP. J. Am. Chem. Soc. 2009;131:14604–14605. doi: 10.1021/ja903732v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.For reviews on recent developments in transition metal photoredox catalysis in organic synthesis, see: Zeitler K. Angew. Chem. Int. Ed. 2009;48:9785–9789. doi: 10.1002/anie.200904056. Narayanam JMR, Stephenson CRJ. Chem. Soc. Rev. 2010 doi: 10.1039/B913880N. Yoon TP, Ischay MA, Du J. Nature Chem. 2010;2:527–532. doi: 10.1038/nchem.687.
- 9.For recent reports synthetic applications of transition metal photoredox catalysis, see: Nicewicz D, MacMillan DWC. Science. 2008;322:70–80. doi: 10.1126/science.1161976. Nagib DA, Scott ME, MacMillan DWC. J. Am. Chem. Soc. 2009;131:10875–10877. doi: 10.1021/ja9053338. Narayanam JMR, Tucker JW, Stephenson CRJ. J. Am. Chem. Soc. 2009;131:8756–8757. doi: 10.1021/ja9033582. Koike T, Akita M. Chem. Lett. 2009;38:166–167. Tucker JW, Narayanam JMR, Krabbe SW, Stephenson CRJ. Org. Lett. 2010;12:368–371. doi: 10.1021/ol902703k. Condie AG, González-Gómez JC, Stephenson CRJ. J. Am. Chem. Soc. 2010;132:1464–1465. doi: 10.1021/ja909145y. Tucker JW, Nguyen JD, Narayanam JMR, Krabbe SW, Stephenson CRJ. Chem. Commun. 2010;46:4985–4987. doi: 10.1039/c0cc00981d. Furst L, Matsuura BS, Narayanam JMR, Tucker JW, Stephenson CRJ. Org. Lett. 2010;12:3104–3107. doi: 10.1021/ol101146f. Andrews RS, Becker JJ, Gagné MR. Angew. Chem. Int. Ed. 2010:7274–7276. doi: 10.1002/anie.201004311. Shih H-W, Vander Wal MN, Grange RL, MacMillan DWC. J. Am. Chem. Soc. 2010;132:13600 10–13603. doi: 10.1021/ja106593m.
- 10.For seminal studies on the behavior of cyclopropyl ketyl radicals, see: Dauben WG, Deviny EJ. J. Org. Chem. 1966;31:3794–3798. Dauben WG, Wolf RE. J. Org. Chem. 1970;35:2361–2367. Tanko JM, Drumright RE. J. Am. Chem. Soc. 1990;112:5362–5363. Tanko JM, Drumright RE. J. Am. Chem. Soc. 1992;114:1844–1854. Tanko JM, Phillips JP. J. Am. Chem. Soc. 1999;121:6078–6079. Stevenson JP, Jackson WF, Tanko JM. J. Am. Chem. Soc. 2002;124:4271–4281. doi: 10.1021/ja0041831.
- 11.For early examples, see: Packer RA, Whitehurst JS. J. Chem. Soc., Chem. Commun. 1975:757–758. White JD, Torii S, Nogami J. Tetrahedron Lett. 1974;15:2879–2882. Lafontaine J, Mongrain M, Sergent-Guay M, Ruest L, Deslongchamps P. Can. J. Chem. 1980;58:2460–2476. White JD, Ruppert JF, Avery MA, Torii S, Nokami J. J. Am. Chem. Soc. 1981;103:1813–1821.
- 12.For seminal examples of reductive cyclizations initiated by radical fragmentation of cyclopropyl ketones, see: Batey RA, Motherwell WB. Tetrahedron Lett. 1991;32:6211–6214. Cossy J, Furet N, BouzBouz S. Tetrahedron. 1995;51:11751–11764. Kirschberg T, Mattay J. J. Org. Chem. 1996;61:8885–8896. doi: 10.1021/jo961015b. Enholm EJ, Jia ZJ. Chem. Commun. 1996:1567–1568. Molander GA, Alonso-Alija C. Tetrahedron. 1997;53:8067–8084. Enholm EJ, Jia ZJ. J. Org. Chem. 1997;62:174–181. doi: 10.1021/jo961655e. Fagnoni M, Schmoldt P, Kirschberg T, Mattay J. Tetrahedron. 1998;54:6427–6444. Tzvetkov NT, Neumann B, Stammler H-G, Mattay J. Eur J. Org. Chem. 2006:351–370.
- 13.For a more complete discussion of the roles of the various reaction components, see references 7a and 8c.
- 14.We were unable to isolate product, however, from the reactions of substrates with five-carbon tethers.
- 15.See Supporting Information for details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.