

Vibsanine E (1), [now known as vibsanin E (1)] isolated by Kawazu1 from the Japanese fish poison plant Viburnum odoratissimum (Sangoju) in 1978 (Figure 1), was amongst the first vibsane natural products to be isolated. Since this time an entire vibsane family has been isolated from the Viburnum species2 as elucidated by Fukuyama,3 Shen4 and Duh.5 Biological activity in this natural product family is prevalent,3 for example, vibsanin A displayed piscicidal activity, vibsanin B inhibited rice seedling root growth, vibsanins B and C exhibited cytotoxic activities on KB cells, aldolvibsanin B showed lethal brine shrimp activity, vibsanin O demonstrated cytotoxicity against P- 388 cells as did vibsanins P and W, vibsanin K exhibited cytotoxicity against human gastric (NUGC) and oral epidermoid (HONE-1) tumor cells, and vibsanins P and W were cytotoxic against A549 and HT-29 cells. Considering the amount of anti-cancer and cytotoxic activity displayed by the vibsane family of natural products, and our interest in the cancer field,6 we were attracted to vibsanin E (1) as to-date its biological activity had not been reported. We embarked on firstly attempting to confirm the structure of vibsanin E (1) and secondly devise a route that would provide sufficient material for extensive biological evaluation.
Figure 1.
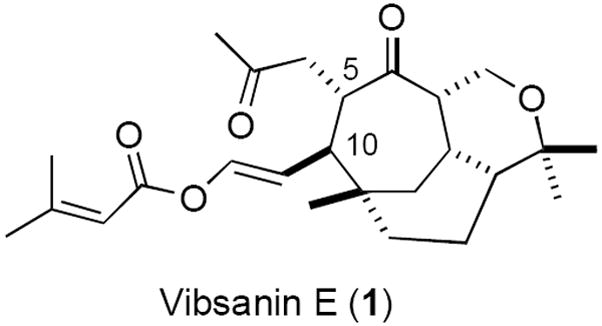
Relative stereochemical representation of vibsanin E (1)
Only very recently have vibsane natural product family members succumbed to total synthesis. So far, the synthesis of (±)-2-O-methylneovibsain H (2),7 (±)-neovibsanin B (3)8 and (−)-5-epi-vibsanin E (4)9 (Figure 2) have been achieved. The stereocontrol in these systems has been challenging, and on many occasions the synthetic efforts have resulted in formation of diastereomers of the natural products [i.e. (+)-6-epi-vibsanin F (5),10 (±)-5,10-bis-epi-vibsanin E (6),11 and (±)-5,14-bis-epi-spirovibsanin A12] or deadend synthetic explorations (Figure 2).13-16 As vibsanin E (1) has a highly functionalized fused seven-membered ring and is one of the original vibsanes, we consider it the pinnacle target in this family of natural products.
Figure 2.
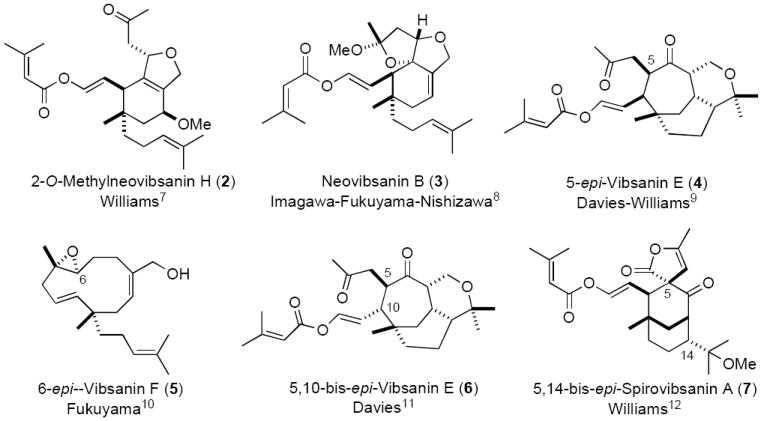
Vibsanin, and epi-vibsanin, family members that have previously been synthesised.
Both Williams and Davies have worked independently towards a total synthesis of vibsanin E (1). The Queensland group pursued a biogenetically modeled approach giving access to bicycle 8,17,18 which, although led to many advanced intermediates (i.e. 9), did not yield the target.19,20 The Davies group utilised three key cycloadditions to achieve a total synthesis of 5-epi-10-epi-vibsanin E 6; a rhodium-catalysed [4+3] and a subsequent heteronuclear [4+2] afforded bicycle 10, followed by a photochemically induced [4+2] (Scheme 1).11
Scheme 1.
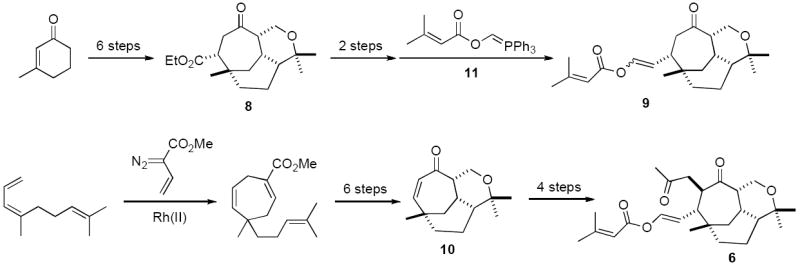
In the view that the Davies strategy readily provided multi-gram quantities of bicycle 1011 and the Queensland group had significant end game experience with molecules of this type,19 including the development of a novel method utilising ylid 11 for introducing the enol ester side chain,20 it made sense to join forces in this common quest. In fact the initial fruits of this collaboration led to the first total synthesis of the natural product (−)-5-epi-vibsanin E (4)9 in which many of the previous difficulties encountered with both introduction and stereocontrol of the side chains were overcome. Highlights of this successful pathway included, conjugate addition of an α-oxa methylene anion (i.e. MOMOCH2Li derived from MOMOCH2SnBu3)21 to bicycle 10, using TMSCl22 activation, affording 12. Metallation then subsequent O-allylation of 12 afforded 13, which gave a mixture of epimers 14 and 15. Ketone 14 was then carried through a series of deprotection oxidation steps to provide aldehyde 16, which afforded (−)-5-epi-vibsanin E (4) on treatment with ylid 11 (Scheme 2).
Scheme 2.
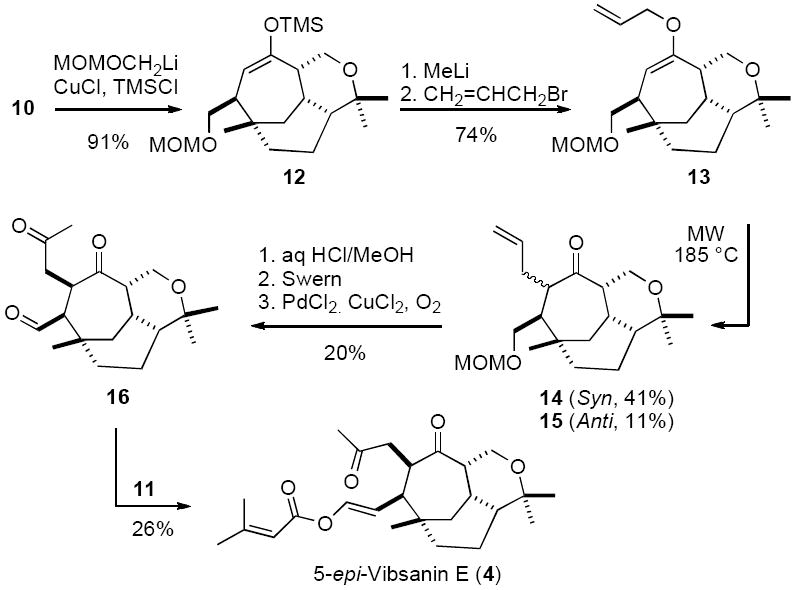
At the time, however, considerable difficulties were being encountered with the Claisen rearrangement in that the anti-isomer 15 was only obtained in low yield preventing a synthesis of vibsanin E (1), but also a considerable amount of by-product 17 was being observed (Scheme 3). If this could be overcome a total synthesis of vibsanin E (1) would be possible.
Scheme 3.
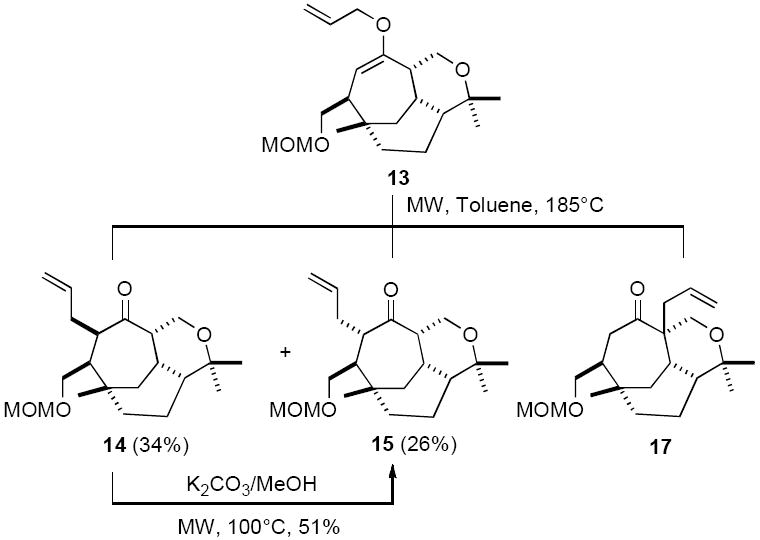
After considerable experimentation it was discovered that C-allylated material 17 could be significantly reduced if the microwave induced Claisen rearrangement was performed in a dilute solution [i.e. toluene (0.02M)]. In addition it was also found that the syn-isomer 14 could be epimerised to the anti-isomer 15 in 51% (K2CO3/MeOH), with further material obtained on recycling. With these developments the anti-isomer 15 could be obtained in 44% yield (56% with recovery) and reasonable quanitity. Subjecting 15 to the same deprotection oxidation sequence used for (−)-5-epi-vibsanin E (4) (see scheme 2) afforded aldehyde 18. Treatment of 18 with ylid 11 produced vibsanin E (1),23 which was an identical 1H and 13C NMR spectroscopic match to the natural material,24 albeit accompanied by trace amounts of the tentatively assigned by-product 19 (Scheme 4).
Scheme 4.
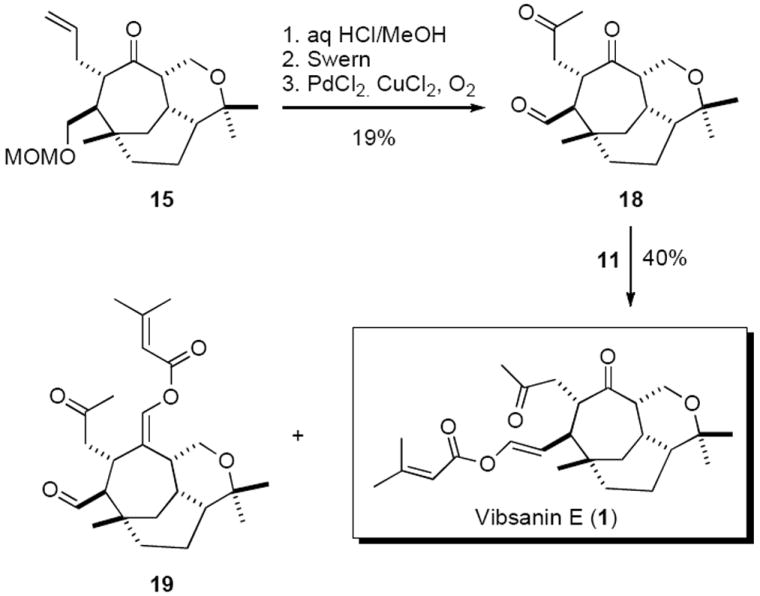
In conclusion, vibsanin E (1), a structurally rare complex diterpene, has been efficiently synthesised in 14 steps confirming the proposed relative stereochemistry. The synthesis combines the rhodium catalyzed [4+3] cycloaddition strategy to rapidly generate the tricyclic core with an effective end game strategy to introduce the remaining side-chains. This represents the first total synthesis of Vibsanin E (1), 31 years since it was isolated, and paves the way for derivative synthesis should structure activity studies be deployed. Vibsanin E (1) is currently undergoing biological evaluation, results of which will be reported in due course.
Acknowledgments
We thank The University of Queensland, Australian Research Council (DP0666855) and the National Institutes of Health (GM080337) for financial support. Prof. Fukuyama from the Tokushima Bunri University (Japan) is gratefully acknowledged for providing NMR spectra of natural vibsanin E. HMLD has financial interests in Dirhodium Technologies, Inc., a company that manufactures chiral dirhodium catalysts.
Footnotes
Accessory Publication
General methods, experimental, scanned copies of 1H and 13C NMR spectra, comparison 1H NMR spectra for vibsanin E and comparison table of 1H and 13C NMR shifts for vibsanin E.
References
- a) Kawazu K. IUPAC Int Symp Chem Nat Prod (11th) 1978;2:101. [Google Scholar]; b) Kawazu K. Agric Biol Chem. 1980;44:1367. [Google Scholar]; c) Fukuyama K, Katsube Y, Kawazu K. J Chem Soc Perkin Trans II. 1980:1701. [Google Scholar]
- 2.Wang L-Q, Chen Y-G, Xu J-J, Liu Y, Li X-M, Zhao Y. Chem Biodiversity. 2008;5:1879. doi: 10.1002/cbdv.200890175. [DOI] [PubMed] [Google Scholar]
- a) Fukuyama Y, Kubo M, Minami H, Yuasa H, Matsuo A, Fujii T, Morisaki M, Harada K. Chem Pharm Bull. 2005;53:72. doi: 10.1248/cpb.53.72. and references therein. [DOI] [PubMed] [Google Scholar]; b) Fukuyama Y, Esumi T. J Org Syn Chem (Japan) 2007;65:585. [Google Scholar]; c) Kubo M, Minoshima Y, Arimoto D, Minami H, Harada K, Hioki H, Fukuyama Y. Heterocycles. 2009;77:539. [Google Scholar]
- a) Shen Y-C, Prakash CVS, Wang L-T, Chien CY, Hung M-C. J Nat Prod. 2002;65:1052. doi: 10.1021/np020007p. [DOI] [PubMed] [Google Scholar]; b) Shen Y-C, Lin C-L, Chien S-C, Khalil AT, Ko C-L, Wang C-H. J Nat Prod. 2004;67:74. doi: 10.1021/np030173c. [DOI] [PubMed] [Google Scholar]
- a) Duh C-Y, El-Gamal AAH, Wang S-K. Tetrahedron Lett. 2003;44:9321. [Google Scholar]; b) El-Gamal AAH, Wang SK, Duh C-Y. J Nat Prod. 2004;67:333. doi: 10.1021/np030447w. [DOI] [PubMed] [Google Scholar]
- 6.Dong L, Gordon VA, Grange RL, Johns J, Parsons PG, Porzelle A, Reddell P, Schill H, Williams CM. J Am Chem Soc. 2008;130:15262–15263. doi: 10.1021/ja807133p. [DOI] [PubMed] [Google Scholar]
- 7.Chen AP-J, Williams CM. Org Lett. 2008;10:3441. doi: 10.1021/ol801117e. [DOI] [PubMed] [Google Scholar]
- 8.Imagawa H, Saijo H, Kurisaki T, Yamamoto H, Kubo M, Fukuyama Y, Nishizawa M. Org Lett. 2009;11:1253. doi: 10.1021/ol802973f. [DOI] [PubMed] [Google Scholar]
- 9.Schwartz BD, Denton JR, Lian Y, Davies HML, Williams CM. J Am Chem Soc. 2009;131 doi: 10.1021/ja9019484. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yuasa H, Makado G, Fukuyama Y. Tetrahedron Lett. 2003;44:6235. [Google Scholar]
- a) Davies HML, Loe Ø, Stafford DG. Org Lett. 2005;7:5561. doi: 10.1021/ol052005c. [DOI] [PubMed] [Google Scholar]; b) Nikolai J, Loe Ø, Dominiak PM, Gerlitz OO, Autschbach J, Davies HML. J Am Chem Soc. 2007;129:10763. doi: 10.1021/ja072090e. [DOI] [PubMed] [Google Scholar]
- a) Gallen MJ, Williams CM. Org Lett. 2008;10:713. doi: 10.1021/ol702827x. [DOI] [PubMed] [Google Scholar]; b) Gallen MJ, Goumont R, Clark T, Terrier F, Williams CM. Angew Chem Int Ed. 2006;45:2929. doi: 10.1002/anie.200504156. In part see. [DOI] [PubMed] [Google Scholar]; c) Gallen MJ, Williams CM. Eur J Org Chem. 2008;4697 In full see. [Google Scholar]
- 13.Tilly DP, Williams CM, Bernhardt PV. Org Lett. 2005;7:5155. doi: 10.1021/ol051897d. [DOI] [PubMed] [Google Scholar]
- 14.Esumi T, Zhao M, Kawakami T, Kukumoto M, Toyota M, Fukuyama Y. Tetrahedron Lett. 2008;49:2692. [Google Scholar]
- 15.Srikrishna A, Pardeshi VH, Satyanarayana G. Tetrahedron: Asymmetry. 2008;19:1984. [Google Scholar]
- 16.Mehta G, Bhat BA. Tetrahedron Lett. 2009;50:2474. [Google Scholar]
- 17.Heim R, Wiedemann S, Williams CM, Bernhardt PV. Org Lett. 2005;7:1327. doi: 10.1021/ol0501222. [DOI] [PubMed] [Google Scholar]
- 18.Schwartz BD, Tilly DP, Heim R, Wiedemann S, Williams CM, Bernhardt PV. Eur J Org Chem. 2006;3181 [Google Scholar]
- 19.Schwartz BD, Williams CM, Bernhardt PV. Beilstein J Org Chem. 2008;4(No. 34) doi: 10.3762/bjoc.4.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schwartz BD, Williams CM, Anders E, Bernhardt PV. Tetrahedron. 2008;64:6482. [Google Scholar]
- 21.Linderman RJ, Godfrey A, Horne K. Tetrahedron. 1989;45:495. [Google Scholar]
- 22.Mander LN, Thomson RJ. J Org Chem. 2005;70:1654. doi: 10.1021/jo048199b. [DOI] [PubMed] [Google Scholar]
- 23.Note: non-stabilized ylids as a general rule give rise to cis (or Z) double bond products, but it has been found20 that steric hindrance dramatically influences the selectivity in the case of 18, hence the observation of an ~10:1 E/Z ratio observed for synthetic vibsanin E (1).
- 24.See supporting information for comparative 1H and 13C NMR spectra of synthetic and natural vibsanin E (1).