

Abstract
The adsorption conditions used to immobilize catalase onto thin-films of carbon nanotubes were investigated to elucidate the conditions that produced films with maximum amounts of active catalase. The adsorption kinetics were monitored by spectroscopic ellipsometry and the immobilized catalase films were then assayed for catalytic activity. The development of a volumetric optical model used to interpret the ellipsometric data is discussed. According to the results herein discussed, not only the adsorbed amount but also the initial adsorption rates determine the final catalytic activity of the adsorbed layer. The results described in the manuscript have direct implications on the rational design and analytical performance of enzymatic biosensors.
Keywords: enzyme adsorption, catalase, carbon nanotubes, biosensors, spectroscopic ellipsometry
Introduction
Exposure of solid surfaces to aqueous protein solutions typically results in the accumulation of protein molecules at the solid/liquid interface. Although it is generally accepted that the adsorption process is mainly driven by a combination of hydrophobic and electrostatic interactions, other processes such as redistribution of charged residues, dehydration of the protein and substrate surface, and structural rearrangements in the protein molecules1 can play a fundamental role. These structural rearrangements can affect the integrity of the active site, thereby affecting the enzymatic activity. From the analytical point of view, the structure of the recognition element in the biosensor is a key element because if it is affected, the performance of the resulting sensor can be significantly impaired. Despite this, only a few reports have approached the design of biosensors by first optimizing the adsorption kinetics and conditions to maximize catalytic activity of the biorecognition element.2-5
Among other relevant enzymes, catalase (hydrogen peroxide oxidoreductase; EC.1.11.1.6) is a convenient probe for protein adsorption studies because it is commercially available and has well-characterized structure and activity. In addition, catalase has one of highest turnover numbers in nature.6 Catalase is an oxidative stress protection enzyme with molecular weight of 240 kDa and an isoelectric point (pI) of 5.4 whose function is to scavenge intracellular H2O2 in aerobic organisms.7 More specifically, this enzyme catalyzes the disproportionation of two molecules of H2O2 to molecular oxygen and water. Besides removal of H2O2 in different industrial processes, the catalytic properties of catalase have been used to facilitate the amperometric detection of hydrogen peroxide8-12 as well as non-native substrates such as dibenzoyl peroxide, 3-chloroperoxibenzoic acid,13 and ethanol.14 Catalase can also be co-immobilized with other enzymes to improve the upper sensing limit in the detection of peroxide15 and to aid in the detection of other analytes such as glucose,16 short chain primary aliphatic alcohols,17 and glutamine.18
Recently, carbon nanotubes (CNTs) have been used as substrates for the immobilization of catalase.19-21 CNTs are conductive, biocompatible,22 stable over a large range of potentials, demonstrate catalytic activities towards many electrochemical reactions,23-25 and provide significant increases in electrode area.25 The increase in electrode area is particularly important because allows the immobilization of higher amounts of enzyme, and therefore significantly improving the limits of detection. Additionally, the nanoenvironment provided by CNTs has been shown to promote greater attachment of protein,26 better retention of catalytic activity,27 and enhancement of the electron transfer rate from catalase to the electrode in a direct and quasi-reversible redox process.19-21, 26, 27
Considering the aforementioned contributions, the aim of this paper is to investigate the adsorption kinetics of catalase (a model enzyme for adsorption processes28) onto CNTs and elucidate the conditions at which maximum amounts of active catalase can be immobilized. For the experiments herein described, spectroscopic ellipsometry (SE) was utilized. SE allows for monitoring the protein adsorption to the CNTs in real time and in addition, has sensitivity down to the nanometer scale. After adsorption, catalytic activity assays were performed to measure the activity of the immobilized enzyme.
Experimental
Reagents and Solutions
All chemicals were analytical reagent grade and used as received. Hydrogen peroxide, sodium phosphate, citric acid, sodium chloride, sodium hydroxide, and sodium dodecyl sulfate were purchased from Fisher Scientific (Fair Lawn, NJ). Citric acid (10 mmol·L−1) was selected as the background electrolyte because it provides high buffer capacity (pKa1 = 3.13, pKa2 = 4.76, pKa3 = 6.40) around the pI of the selected protein (pI = 5.40). All aqueous solutions were prepared using 18 MΩ·cm water (NANOpure Diamond, Barnstead; Dubuque, IA). The pH of the solutions was adjusted using either 1 M NaOH or 1 M HCl and measured using a glass electrode and a digital pH meter (Orion 420A+, Thermo; Waltham, MA). Catalase from bovine liver (Worthington Biochemical Corporation; Lakewood, NJ) was purchased as a crystalline suspension in thymol preservative (≥20,000 U·mg−1) and kept at 2-8°C until used. Before preparing the stock solutions of catalase, the thymol was removed following a procedure recommended by Worthington’s technical notes. Then, a stock solution of catalase was prepared in 10 mmol·L−1 phosphate buffer at pH 7.00, to ensure proper dissolution. The concentration of the stock solution was determined by measuring its absorbance at 405 nm, using 3.24 × 105 M−1 cm−1 as the molar extinction coefficient.29 Other protein solutions were prepared by diluting the corresponding amount of stock in a buffer solution totaling 10% of 10 mmol·L−1 phosphate at pH 7.00 and 90% of 10 mmol·L−1 citrate buffer at the desired pH. The purity of the catalase was evaluated by gel electrophoresis (see Supporting Information). It is important to note that many protein adsorption studies neglect to determine purity of protein prior to experimentation and thus, the adsorption data could be characteristic of a mixture of proteins rather than the one protein intended.
Substrates
CNT-coated silica (Si/SiO2/CNT) surfaces were used for the present studies. As previously described,30, 31 Si/SiO2/CNT surfaces were prepared by Eikos Inc. (Franklin, MA), using <111> silicon wafers (Sumco; Phoenix, AZ) as substrates. According to the provider, a layer of CNT was deposited on the wafers using arc-produced single-wall CNTs having about a 1.3 nm diameter. As described elsewhere,32, 33 the raw material formed in the arc reactor was purified by a process of acid reflux followed by washing and centrifugation (besides removing metal catalyst and non-tubular forms of carbon, the exposure to nitric acid during the purification is known to introduce –COOH groups on the CNTs34). Once purified, the CNTs were dispersed in water and alcohol to form an ink, which was then spray-coated onto the Si/SiO2 wafer heated to 65 °C while monitoring the deposition rate. The formed coating is essentially a layer of pure CNT and contains no residual organic additives or polymeric constituents. Contact angle measurements of the Si/SiO2/CNT substrates were performed using a VCA-Optima surface analysis system (Ast Products, Inc.; Billerica, MA) and analyzed using the software provided by the manufacturer 30 s after dispensing 5 μL of deionized water. The average contact angle for the Si/SiO2/CNT surfaces was 26° ± 1°.
Spectroscopic Ellipsometry and Cell for in Situ Adsorption Monitoring
The substrate characterization as well as the dynamic adsorption experiments were performed at room temperature using a variable angle spectroscopic ellipsometer (VASE, J. A. Woollam Co.; Lincoln, NE). Although other techniques have been used,35-38 SE has proven suitable to study adsorption of proteins in real time and provides useful information about the optical constants and structure of the adsorbed film.39-43 The collected data (amplitude ratio (Ψ) and phase difference (Δ) as function of wavelength or time) was modeled using the WVASE software package (J. A. Woollam Co.; Lincoln, NE) (vide infra). The mean square error (MSE, calculated by a built-in function in WVASE) was used to quantify the difference between the experimental and model-generated data. In all cases, the sample under investigation was mounted on a micrometer-position-controlled translation stage with the gradient direction perpendicular to the plane of incidence.
Dynamic adsorption experiments were performed in a modified44 electrochemical cell (J. A. Woollam Co.; Lincoln, NE), mounted directly on the vertical base of the ellipsometer. In order to control the protein supply to the substrate, the cell was modified by fixing an L-shaped stainless steel tube to the cell. One end of the tube faced the substrate at the same spot where the incident light beam hits the surface. The other end of the tube was connected, using Tygon tubing, to a peristaltic pump (Minipuls3, Gilson; Middleton, WI). A two-way valve (V100D, Upchurch Scientific; Oak Arbor, WA) was also connected in series to enable rapid switching between the buffer and the protein solution. As a difference with respect to batch techniques, this set-up enables combining the advantages of in-situ SE with stagnation point flow conditions. Under the selected experimental conditions, the flux of solute (J) towards the surface can be described by Equation 1,
| Equation 1 |
where ν is the kinematic viscosity of the solvent, R is the inner radius of the tube through which the solution enters the cell (0.381 mm), D is the diffusion coefficient of the studied adsorbate, β is the efficiency factor (2.3406 × Re0.4094), and C is the concentration of the solute (catalase) in the bulk.44
The substrates were initially characterized as a function of wavelength (a spectroscopic scan in the 250-900 nm range with 10 nm steps) in the cell using the selected aqueous buffer as the ambient medium.45 This procedure allowed verification of the thickness of each substrate at the measuring spot, thereby improving the accuracy of the calculation. Then, the dynamic experiment was initiated by pumping background electrolyte through the cell at a rate of 1 mL·min−1 (for 5 min) to measure the baseline. Next, the valve was switched, protein solution was introduced, and the adsorption process initiated. An initial fast process, followed by a slower one, was always observed. In all cases during the dynamic experiments, the variation of Ψ and Δ as a function of time was determined at 500 and 600 nm and at an angle of incidence of 70°, as defined by the inlet/outlet of the UV fused-silica windows. When no significant change in the signal was observed (after ~90 minutes), the dynamic scan was stopped, and the more accurate spectroscopic scan was collected to verify the thickness of the protein layer. Experiments performed in this way provided data for calculating the initial adsorption rate and the saturation amount. The substrates with adsorbed catalase were then removed from the cell and immediately assayed for catalytic activity. In between experiments, the flow cell and tubing were thoroughly rinsed with a solution containing 4 mmol·L−1 sodium dodecyl sulfate and water.
Activity Assays
In order to measure the enzymatic activity of catalase, a solution containing hydrogen peroxide was prepared in 50 mmol·L−1 phosphate buffer at pH 7.0 and 25°C. The solution was then placed in a stirring cell mounted on a Spinette cell stirrer (Starna Cells; Atascadero, CA). The immobilized catalase then immersed in the solution and the rate of decomposition of H2O2 was followed by observing the decrease in absorbance at 240 nm (ε240 nm H2O2 = 43.6 M−1cm−1)46 over time with a GENESYS10 spectrophotometer (Thermo Electron). In agreement with previous reports,47, 48 one unit was defined as the amount of catalase required to decompose 1 μmol of H2O2 per minute at pH 7.0 at 25°C, while the H2O2 concentration falls from 10.3 mmol·L−1 to 9.2 mmol·L−1.
Results and Discussion
Development of the Optical Model
The principle of SE is the measurement of changes in the reflectance and phase difference between the parallel (RP) and perpendicular (RS) components of a polarized light beam upon reflection from a surface. The intensity ratio of RP and RS can be related to the ellipsometric angles (Ψ and Δ) using Equation 2:
| Equation 2 |
Because ellipsometry measures the ratio of two values originated by the same signal, the data collected are highly accurate and reproducible. The changes in polarization measured by ellipsometry are also extremely sensitive to film thickness (down to the monolayer level), optical constants, and film microstructure (such as surface roughness, index grading, and intermixing). This monolayer sensitivity is useful for real-time studies of film deposition, including biological molecules on a variety of substrates.
Interpretation of ellipsometric measurements from the raw data is rather difficult. Hence, construction of an optical model that describes the substrate microstructure in terms of the refractive index (n), extinction coefficient (k), and thickness (d) is required. For this reason, a model containing three uniaxial layers with optical axes parallel to the substrate surface was used to describe the optical properties of the substrates (Si/SiO2/CNT). As previously reported, the dielectric functions of the substrates were described by a layer of Si (bulk, d = 1 mm), a layer of native SiO2 (d = 2.5 ± 0.5 nm), and a two-media Maxwell-Garnett effective medium approximation (EMA)49 layer (d = 25 ± 5 nm) consisting of 98% void and 2% arc-evaporated carbon.50 The thickness value of the CNT layer calculated by the optical model was verified by performing atomic force microscopy and scanning electron microscopy on the selected substrates.51
In order to describe the adsorption of catalase to the silica-supported CNTs, a previously described optical model51 that considers the adsorption of the protein to the surface of the CNT film was initially applied. However, when dynamic experiments were analyzed, significant increases in the MSE value were obtained as the adsorption process progressed (see Supplementary Information), indicating that the selected model cannot accurately describe the optical properties of the sample, and therefore may not represent the physical meaning of the measurement. Therefore, considering that catalase may penetrate into the CNT film rather than adsorbing on top of the CNT layer, the EMA layer was modified to include an additional component, representing the protein (see Figure 1A). Because catalase is transparent at 500 and 600 nm (wavelengths used for the dynamic adsorption experiments), the optical constants of the third component were described using a Cauchy parameterization model according to Equation 3,
| Equation 3 |
where A = 1.465, B = 0.01, and C = 0 are computer-calculated fitting parameters and λ is the wavelength of the incident beam. These parameters yielded index of refraction values ranging from 1.527 to 1.477, which are consistent with previously reported values for other adsorbed proteins.45, 52, 53 In this way, the model was allowed to dynamically replace a certain % of void space in the CNT layer by a % of space occupied by the adsorbed proteins. This volumetric interpretation of the adsorption process is in good agreement with previous reports54 and, to the best of our knowledge, represents the first kinetic study of this kind, with a time resolution of 17 sec/data point.
Figure 1.

A) Optical model used for modeling the adsorption kinetics of catalase to silica-supported CNTs, B) Spectroscopic measurement of Ψ (□) and Δ (■) parameters as measured (points) and calculated (lines) after adsorption of 0.003 mg·mL−1 catalase at pH 5.4 onto CNT substrate, and C) Kinetics (■) and MSE of fitting (□) for the adsorption of 0.003 mg·mL−1 catalase at pH 5.4 onto CNT substrate.
To account for slight variations in the thickness of the different CNT substrates, the percentage of volume of catalase (calculated as the % composition of Cauchy in the EMA layer) was multiplied by the thickness of the CNT layer to obtain an “effective thickness” of catalase inside the CNT film. This calculation, which enabled the comparison of experiments performed with different substrates, was verified by adsorbing catalase to different substrates. In all cases, variations of less than 5% were obtained. Consequently, this optical model was used to analyze the results of the adsorption experiments presented in this manuscript.
As shown in Figure 1B and Figure 1C, the proposed model, which considers the possibility of penetration, not only enables the accurate description of the optical properties of the nanocomposites (Si/SiO2/CNT/Catalase/H2O), but also allows following the dynamic adsorption experiment with MSE values lower than 3 (MSE < 15 are typically considered acceptable55, 56). It is worth noting that the optical description of the CNT film is in good agreement with published results31 but, as expected, differs slightly from other CNT films prepared under different conditions.30, 57-59 It is also important to note that related studies (see the Supporting Information of Ref.51), indicate that although the CNT surface is not perfectly flat and smooth (roughness ~5 nm), adsorbed enzymes do not reach the Si/SiO2 substrate underneath the CNT layer.31
Effect of Catalase Concentration
The effect of catalase concentration on the initial adsorption rate and effective thickness in the CNT films was evaluated by performing adsorption experiments with a continuous flow of 0.05, 0.005, and 0.0005 mg·mL−1 catalase in 10 mmol·L−1 buffer, and at pH 5.4 (pI of catalase). As can be observed in Figure 2, the initial adsorption rate was found to be proportional to the concentration of catalase. These results can be explained by considering that in the cell selected for these studies,44 the flux of protein at the protein/CNT interface is proportional to the concentration of protein in the bulk of the impinging solution, which therefore, increases the probability of adsorption. Further analysis of the data in Figure 2 reveals that the maximum amount of adsorbed protein was also proportional to the concentration of catalase, which, at first glance, can be interpreted as an equilibrium between adsorbed and unadsorbed protein in solution. However, desorption experiments performed by washing the adsorbed protein with buffer did not render significant changes in the adsorbed amount (data not shown), indicating irreversible adsorption of catalase to CNTs. The irreversibility of adsorption precludes the interpretation of the data in Figure 2 as an equilibrium. Furthermore, if the adsorption is irreversible, one might expect that over an extended period of time, all concentrations would result in the same saturation level, which was not observed. Consequently, our results can be interpreted using Norde’s proposition that at low initial rates of adsorption, the adsorbed protein molecules have time to relax onto the surface (to maximize favorable contacts with the substrate) before coming into contact with a neighboring molecule. As a result of the larger area occupied per protein, less protein molecules can be adsorbed per unit of surface area. Conversely, at high concentration, the high initial rate of adsorption causes rapid crowding of the substrate. This crowding forces the protein to keep a more compact structure, since the relaxation process is sterically inhibited.60
Figure 2.
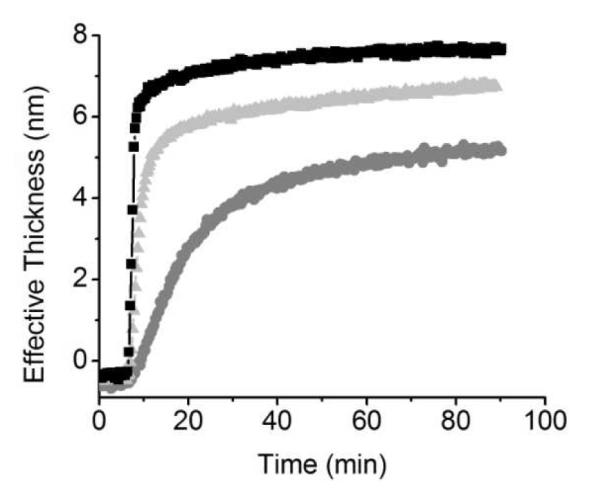
Adsorption kinetics of 0.05 (■), 0.005  , and 0.0005
, and 0.0005  mg·mL−1 catalase onto CNT substrate. Other conditions are as described in the Experimental Section.
mg·mL−1 catalase onto CNT substrate. Other conditions are as described in the Experimental Section.
Effect of pH and Ionic Strength
In order to evaluate the contribution of electrostatic interactions to the adsorption process, the effect of pH was investigated. For these experiments, the pH of the solution containing catalase was adjusted to 4.4, 5.4, and 6.4 to render solutions of catalase that have a net positive, zero, and negative charge, respectively (pI = 5.4). Due to the presence of –COOH groups on the surface of the CNTs (introduced during the purification process), the sorbent surface exposed negatives charges at all the studied pH values. The initial adsorption rates shown in Figure 3 suggest that electrostatic interactions between the surface and the protein influence the initial stages of the adsorption process, slightly favoring the adsorption of catalase at pH = 4.4 and decreasing the adsorption at pH = 6.4. However, the fact that the pH does not produce vastly different initial adsorption rates suggests that the main driving forces for the adsorption are hydrophobic interactions between catalase molecules and the CNT film. Figure 3 also shows that the adsorbed amount is highest when the adsorption pH is equal to the pI of catalase, in accordance with previous studies.61 At the pI, a compact structure is favored owing to an equal number of positive and negative charges within a protein allowing more protein to adsorb per unit area. Adjusting the pH to above or below the pI will increase the number of negative or positive charges, respectively, and due to intra-protein electrostatic repulsion, the structure of each protein will be expanded, occupying a larger footprint, resulting in a decreased amount of total protein adsorbed.
Figure 3.
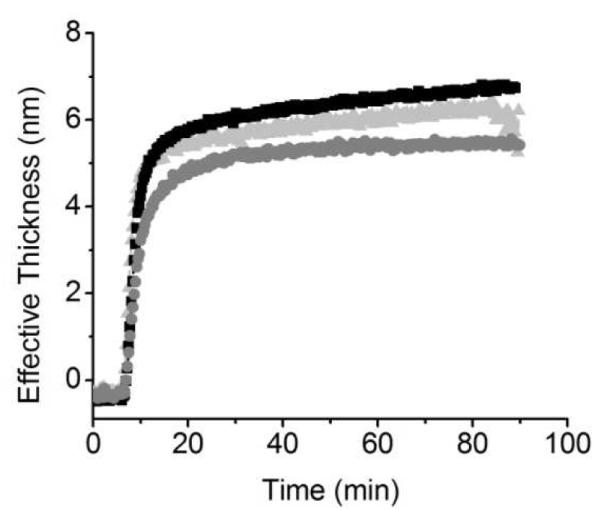
Adsorption kinetics 0.005 mg·mL−1 catalase onto CNT substrate at pH = 5.4 (■), pH = 4.4  , and pH = 6.4
, and pH = 6.4  . Other conditions are as described in the Experimental Section.
. Other conditions are as described in the Experimental Section.
Changing the ionic strength of the background electrolyte can reveal the degree of contribution of electrostatic interactions. For example, if electrostatic interactions play a significant role in the adsorption process, then increasing the concentration of salt should decrease the adsorption of a positively charged protein molecule to a negatively charged surface and increase the adsorption of a negatively charged protein molecule to a negatively charged surface due to shielding effects.62 Therefore, to further explore the role of electrostatic interactions on the adsorption of catalase to CNTs, the effect of the ionic strength was investigated by adding 100 mmol·L−1 NaCl to the buffer solutions at each pH value. According to our results (see Table 1 and Supporting Information), increases in the ionic strength showed little effect in the initial adsorption rate and only led to slightly lower maximum adsorbed amounts at the three selected pH values. Thus, the ionic strength experiments are in agreement with the pH experiments in that they suggest that hydrophobic interactions rather than electrostatic interactions play the dominant role in the adsorption process. Hydrophobic interactions between a protein and surface are favorable due to the increase in entropy as non-polar amino acid residues are dehydrated (most non-polar residues are buried within a globular protein, however the exterior can still have a significant non-polar fraction63). The slight decrease in the saturation adsorbed amount when salt is present is consistent with previous studies64 and may be caused by the increase in area that a protein molecule occupies (increased spreading) as the ionic strength of the solution increases.65
Table 1.
Kinetic Data and Enzymatic Assay Data of Catalase Adsorbed onto CNTs Under Varying Conditions
| Adsorption Conditions |
Initial Adsorption Rate (nm·min−1) |
Maximum Effective Thickness (nm) |
Activity (U·cm−2) |
|||
|---|---|---|---|---|---|---|
| Exp # | [Catalase] (mg·mL−1) |
pH | [NaCl] (mmol·L−1) |
|||
| 1 | 0.0005 | 5.4 | 0 | 0.27 ± 0.01 | 5.2 ± 0.1 | 0.8 ± 0.2 |
| 2 | 0.005 | 5.4 | 0 | 1.58 ± 0.08 | 6.7 ± 0.5 | 1.5 ± 0.1 |
| 3 | 0.05 | 5.4 | 0 | 3.6 ± 0.3 | 7.74 ± 0.02 | 3.5 ± 0.3 |
| 4 | 0.005 | 4.4 | 0 | 2.12 ± 0.09 | 6.20 ± 0.02 | 2.02 ± 0.02 |
| 5 | 0.005 | 6.4 | 0 | 1.20 ± 0.03 | 5.4 ± 0.3 | 1.44 ± 0.03 |
| 6 | 0.005 | 4.4 | 100 | 1.8 ± 0.1 | 5.2 ± 0.3 | 0.923 ± 0.002 |
| 7 | 0.005 | 5.4 | 100 | 1.4 ± 0.1 | 5.6 ± 0.1 | 1.8 ± 0.2 |
| 8 | 0.005 | 6.4 | 100 | 0.71 ± 0.07 | 5.0 ± 0.5 | 1.4 ± 0.3 |
Enzymatic Activity of Catalase Adsorbed to CNTs
In order to investigate the relationship between the adsorption kinetics and the catalytic activity of the resulting film, the activity of each nanocomposite was assayed. The goal of the project was to find the conditions that give the highest enzymatic activities (not necessarily just the highest adsorbed amount) in order to later produce biosensors that have the highest sensitivity possible. Figure 4, which shows activity as a function of effective thickness, shows that in general, the more catalase that is adsorbed to the CNT layer (at saturation), the higher the activity of the layer.
Figure 4.

A Activity as a function of effective thickness for pH = 5.4 (■), pH = 4.4 (▲), pH = 6.4 (•), pH = 4.4 + 100 mmol·L−1 NaCl (Δ), pH = 5.4 + 100 mmol·L−1 NaCl (□), and pH 6.4 + 100 mmol·L−1 NaCl (○). Dashed lines are drawn to guide the eye.
B Activity as a function of initial adsorption rate for pH = 5.4 (■), pH = 4.4 (▲), pH = 6.4 (•), pH = 4.4 + 100 mmol·L−1 NaCl (Δ), pH = 5.4 + 100 mmol·L−1 NaCl (□), and pH 6.4 + 100 mmol·L−1 NaCl (○). Dashed lines are drawn to guide the eye.
A linear analysis of the data included in Figure 4A shows that about 0.77 U can be obtained per nm of catalase (effective thickness) adsorbed to CNT and that when the experimental conditions selected for the adsorption render an effective thickness of catalase ≤ 3.7 nm, the resulting sensor would display no enzymatic activity. The results summarized in Table 1 suggest that in order to inhibit structural relaxation and thus better preserve the native structure of the protein, it is also necessary to maximize the initial adsorption rate.
In order to support this hypothesis, the activity of adsorbed catalase was analyzed as a function of the initial adsorption rate (Figure 4B). As can be observed, a positive correlation (R = 0.87) was obtained. This relationship is highly relevant to the fabrication of not only biosensors, but also other nanocomposites, as it shows that the adsorption conditions should be tailored to increase the initial rate of adsorption and not only the amount of protein adsorbed.
Conclusions
SE was used to investigate the kinetics of the interaction of catalase to CNTs in real-time scales. In order to interpret the adsorption process and describe the optical properties of the system, an improved optical model was developed. This so-called ‘volumetric interpretation’ of adsorption suggests that the enzyme molecules penetrate into the CNT layer enabling the adsorption of catalase throughout the film rather than simply adsorbing onto the surface of the layer. This arrangement is particularly important because it results in a greater amount of enzyme adsorbed to the CNT films, and can potentially render significant improvements in performance in CNT-based biosensors. The adsorption conditions studies showed that high concentrations of catalase, a solution pH at the pI of the protein, and low ionic strength produce films with the highest enzymatic activity. Results also indicate that not only the adsorbed amount, but also a high initial adsorption rate is important to preserve the structure of the adsorbing enzyme, and therefore its catalytic activity. These results also highlight the importance of understanding the kinetic aspects of the adsorption process for the rational design of a biosensor. We envision that a deeper understanding of the interaction of enzymes and CNT films will soon enable combining nanomaterials with various enzymes for biosensor applications.
Supplementary Material
Acknowledgements
The authors thank Dr. H. Soetedjo for performing preliminary experiments in this project and Dr. D. Kurtz for the helpful discussions. This work was financially supported by The University of Texas at San Antonio and the National Institutes of Health / National Institute of General Medical Sciences (NIGMS) 1SC3GM081085. J. Felhofer and J. Caranto also acknowledge the financial support received through the MBRS-RISE Program (Grant # GM60655).
Footnotes
Supporting Information Available SDS-PAGE gel of bovine liver catalase. Optical model previously used for modeling the adsorption kinetics of enzyme to silica-supported CNTs. Kinetics and MSE of fitting for a dynamic scan using the optical model herein discussed. Spectroscopic measurement and fitting with the previously used model. Kinetics of adsorption under various ionic strength conditions. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Karajanagi SS, Vertegel AA, Kane RS, Dordick JS. Langmuir. 2004;20(26):11594–11599. doi: 10.1021/la047994h. [DOI] [PubMed] [Google Scholar]
- 2.He H, Xie Q, Zhang Y, Yao S. J. Biochem. Bioph. Methods. 2005;62(3):191–205. doi: 10.1016/j.jbbm.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 3.Isailovic S, Li H-W, Yeung ES. J. Chromatogr. A. 2007;1150(1-2):259–266. doi: 10.1016/j.chroma.2006.09.093. [DOI] [PubMed] [Google Scholar]
- 4.Massafera MP, Torresi S. I. C. d. Sens. Actuators B. 2009;137(2):476–482. [Google Scholar]
- 5.Zhdanov VP, Kasemo B. Colloids Surf., B. 2010;76(1):28–31. doi: 10.1016/j.colsurfb.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 6.Brigelius-Flohé R, Kipp A. BBA. 2009;1790(11):1555–1568. doi: 10.1016/j.bbagen.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 7.Alptekin Ö, Tükel SS, YildIrIm D, Alagöz D. J. Mol. Catal. B: Enzym. 2009;64(3-4):177–183. [Google Scholar]
- 8.Di J, Zhang M, Yao K, Bi S. Biosens. Bioelectron. 2006;22(2):247–252. doi: 10.1016/j.bios.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 9.O’Brien KB, Killoran SJ, O’Neill RD, Lowry JP. Biosens. Bioelectron. 2007;22(12):2994–3000. doi: 10.1016/j.bios.2006.12.020. [DOI] [PubMed] [Google Scholar]
- 10.Varma S, Mattiasson B. J. Biotechnol. 2005;119(2):172–180. doi: 10.1016/j.jbiotec.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 11.Varma S, Mitra CK. Electrochem. Commun. 2002;4(2):151–157. [Google Scholar]
- 12.Zhang Z, Chouchane S, Magliozzo RS, Rusling JF. Anal. Chem. 2002;74(1):163–170. doi: 10.1021/ac010701u. [DOI] [PubMed] [Google Scholar]
- 13.Horozova E, Dimcheva N, Jordanova Z. Bioelectrochemistry. 2002;58(2):181–187. doi: 10.1016/s1567-5394(02)00153-6. [DOI] [PubMed] [Google Scholar]
- 14.Akyilmaz E, Dinçkaya E. Talanta. 2003;61(2):113–118. doi: 10.1016/S0039-9140(03)00245-5. [DOI] [PubMed] [Google Scholar]
- 15.Tatsuma T, Watanabe T, Tatsuma S, Watanabe T. Anal. Chem. 1994;66(2):290–294. doi: 10.1021/ac00074a017. [DOI] [PubMed] [Google Scholar]
- 16.Santoni T, Santianni D, Manzoni A, Zanardi S, Mascini M. Talanta. 1997;44(9):1573–1580. doi: 10.1016/s0039-9140(96)02181-9. [DOI] [PubMed] [Google Scholar]
- 17.Hnaien M, Lagarde F, Jaffrezic-Renault N. Talanta. 2010;81(1-2):222–227. doi: 10.1016/j.talanta.2009.11.061. [DOI] [PubMed] [Google Scholar]
- 18.Madaras MB, Spokane RB, Johnson JM, Woodward JR. Anal. Chem. 1997;69(18):3674–3678. doi: 10.1021/ac970173f. [DOI] [PubMed] [Google Scholar]
- 19.Wang L, Wang J, Zhou F. Electroanalysis. 2004;16(8):627–632. [Google Scholar]
- 20.Zhou H, Lu T-H, Shi H-X, Dai Z-H, Huang X-H. J. Electroanal. Chem. 2008;612(2):173–178. [Google Scholar]
- 21.Salimi A, Noorbakhsh A, Ghadermarz M. Anal. Biochem. 2005;344(1):16–24. doi: 10.1016/j.ab.2005.05.035. [DOI] [PubMed] [Google Scholar]
- 22.Liu Y, Qu X, Guo H, Chen H, Liu B, Dong S. Biosens. Bioelectron. 2006;21(12):2195–2201. doi: 10.1016/j.bios.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 23.Qi H, Zhang C, Li X. Sens. Actuators B. 2006;114(1):364–370. [Google Scholar]
- 24.Weber J, Kumar A, Kumar A, Bhansali S. Sens. Actuators B. 2006;117(1):308–313. [Google Scholar]
- 25.Zhang M, Gorski W. Anal. Chem. 2005;77(13):3960–3965. doi: 10.1021/ac050059u. [DOI] [PubMed] [Google Scholar]
- 26.Prakash P. Arun, Yogeswaran U, Chen S-M. Talanta. 2009;78(4-5):1414–1421. doi: 10.1016/j.talanta.2009.02.033. [DOI] [PubMed] [Google Scholar]
- 27.Rahimi P, Rafiee-Pour H-A, Ghourchian H, Norouzi P, Ganjali MR. Biosens. Bioelectron. 2010;25(6):1301–1306. doi: 10.1016/j.bios.2009.10.020. [DOI] [PubMed] [Google Scholar]
- 28.Shentu J, Wu J, Song W, Jia Z. Int. J. Biol. Macromol. 2005;37(1-2):42–46. doi: 10.1016/j.ijbiomac.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 29.Samejima T, Yang JT. J. Biol. Chem. 1963;238(10):3256–3261. [PubMed] [Google Scholar]
- 30.Barnes TM, van de Lagemaat J, Levi D, Rumbles G, Coutts TJ, Weeks CL, Britz DA, Levitsky I, Peltola J, Glatkowski P. Phys. Rev. B. 2007;75(23):23541001–2354110. [Google Scholar]
- 31.Valenti LE, Fiorito PA, Garcia CD, Giacomelli CE. J. Colloid Interface Sci. 2007;307(2):349–356. doi: 10.1016/j.jcis.2006.11.046. [DOI] [PubMed] [Google Scholar]
- 32.Chiang IW, Brinson BE, Smalley RE, Margrave JL, Hauge RH. J. Phys. Chem. B. 2001;105(6):1157–1161. [Google Scholar]
- 33.Hu H, Zhao B, Itkis ME, Haddon RC. J. Phys. Chem. B. 2003;107(50):13838–13842. [Google Scholar]
- 34.Hu H, Bhowmik P, Zhao B, Hamon MA, Itkis ME, Haddon RC. Chem. Phys. Lett. 2001;345(1-2):25–28. [Google Scholar]
- 35.Marxer CG, Coen MC, Schlapbach L. J. Colloid Interface Sci. 2003;261(2):291–298. doi: 10.1016/S0021-9797(03)00089-4. [DOI] [PubMed] [Google Scholar]
- 36.Schmidt DR, Waldeck H, Kao WJ. Protein Adsorption to Biomaterials. In: Puleo DA, Bizios R, editors. Biological Interactions on Materials Surfaces. Springer; New York: 2009. pp. 1–18. [Google Scholar]
- 37.Armstrong FA. Curr. Op. Chem. Biol. 2005;9(2):110–117. doi: 10.1016/j.cbpa.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 38.Sultana N, Schenkman JB, Rusling JF. J. Am. Chem. Soc. 2005;127(39):13460–13461. doi: 10.1021/ja0538334. [DOI] [PubMed] [Google Scholar]
- 39.Arwin H. Thin Solid Films. 1998:313–314. 764–774. [Google Scholar]
- 40.Vinnichenko M, Gago R, Huang N, Leng YX, Sun H, Kreissig U, Kulish MP, Maitz MF. Thin Solid Films. 2004:455–456. 530–534. [Google Scholar]
- 41.Karlsson LM, Schubert M, Ashkenov N, Arwin H. Thin Solid Films. 2004:455–456. 726–730. [Google Scholar]
- 42.Wang X, Wang Y, Xu H, Shan H, Lu JR. J. Colloid Interface Sci. 2008;323(1):18–25. doi: 10.1016/j.jcis.2008.04.024. [DOI] [PubMed] [Google Scholar]
- 43.Lousinian S, Logothetidis S, Laskarakis A, Gioti M. Biomol. Eng. 2007;24(1):107–112. doi: 10.1016/j.bioeng.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 44.Mora MF, Nejadnik M. Reza, Baylon-Cardiel JL, Giacomelli CE, Garcia CD. J. Colloid Interface Sci. 2010;346(1):208–215. doi: 10.1016/j.jcis.2010.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Poksinski M, Arwin H. Thin Solid Films. 2004:455–456. 716–721. [Google Scholar]
- 46.Noble RW, Gibson QH. J. Biol. Chem. 1970;245(9):2409–2413. [PubMed] [Google Scholar]
- 47.Beers RF, Sizer IW. J. Biol. Chem. 1952;195(1):133–140. [PubMed] [Google Scholar]
- 48.Stern KG. J. Biol. Chem. 1937;121(2):561–572. [Google Scholar]
- 49.Fujikawa H. Spectroscopic ellipsometry. Principles and applications. J. Wiley & Sons; West sussex, England: 2007. [Google Scholar]
- 50.Arakawa ET, Dolfini SM, Ashley JC, Williams MW. Phys. Rev. B. 1985;31(12):8097–8101. doi: 10.1103/physrevb.31.8097. [DOI] [PubMed] [Google Scholar]
- 51.Mora MF, Giacomelli CE, Garcia CD. Anal. Chem. 2009;81(3):1016–1022. doi: 10.1021/ac802068n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Logothetidis S, Gioti M, Lousinian S, Fotiadou S. Thin Solid Films. 2005;482(1-2):126–132. [Google Scholar]
- 53.Filippini D, Winquist F, Lundström I. Anal. Chim. Acta. 2008;625(2):207–214. doi: 10.1016/j.aca.2008.07.022. [DOI] [PubMed] [Google Scholar]
- 54.Barnthip N, Parhi P, Golas A, Vogler EA. Biomaterials. 2009;30(33):6495–6513. doi: 10.1016/j.biomaterials.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Murakami Y, Einarsson E, Edamura T, Maruyama S. Carbon. 2005;43(13):2664–2676. doi: 10.1103/PhysRevLett.94.087402. [DOI] [PubMed] [Google Scholar]
- 56.Shrestha RP, Yang D, Irene EA. Thin Solid Films. 2006;500(1-2):252–258. [Google Scholar]
- 57.Elim HI, Ji W, Ma GH, Lim KY, Sow CH, Huan CHA. Appl. Phys. Lett. 2004;85(10):1799–1801. [Google Scholar]
- 58.Fanchini G, Miller S, Parekh BB, Chhowalla M. Nano Lett. 2008;8(8):2176–2179. doi: 10.1021/nl080563p. [DOI] [PubMed] [Google Scholar]
- 59.Wu Z, Chen Z, Du X, Logan JM, Sippel J, Nikolou M, Kamaras K, Reynolds JR, Tanner DB, Hebard AF, Rinzler AG. Science. 2004;305(5688):1273–6. doi: 10.1126/science.1101243. [DOI] [PubMed] [Google Scholar]
- 60.Baszkin A, Norde W. Physical Chemistry of Biological Interfaces. Marcel Dekker, Inc.; New York, NY: 2000. [Google Scholar]
- 61.Norde W. Adv.Colloid Interface Sci. 1986;25:267–340. doi: 10.1016/0001-8686(86)80012-4. [DOI] [PubMed] [Google Scholar]
- 62.Norde W. Colloids Surf., B. 2008;61(1):1–9. doi: 10.1016/j.colsurfb.2007.09.029. [DOI] [PubMed] [Google Scholar]
- 63.Lee B, Richards FM. J. Mol. Biol. 1971;55(3):379–400. IN3–IN4. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
- 64.Wendorf JR, Radke CJ, Blanch HW. Colloids Surf., B. 2010;75(1):100–106. doi: 10.1016/j.colsurfb.2009.08.015. [DOI] [PubMed] [Google Scholar]
- 65.Ramsden JJ, Prenosil JE. J. Phys. Chem. 1994;98(20):5376–5381. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.