

Abstract
Objectives:
To evaluate in vivo brain metabolite differences in control subjects, individuals with premanifest Huntington disease (pre-HD), and individuals with early HD using 1H magnetic resonance spectroscopy (MRS) and to assess their relationship with motor performance.
Methods:
Eighty-five participants (30 controls, 25 pre-HD, and 30 early HD) were recruited as part of the TRACK-HD study. Eighty-four were scanned at 3 T with single-voxel spectroscopy in the left putamen. Disease burden score was >220 among pre-HD individuals. Subjects underwent TRACK-HD motor assessment including Unified Huntington's Disease Rating Scale (UHDRS) motor scoring and a novel quantitative motor battery. Statistical analyses included linear regression and one-way analysis of variance.
Results:
Total N-acetylaspartate (tNAA), a neuronal integrity marker, was lower in early HD (∼15%) vs controls (p < 0.001). N-acetylaspartate (NAA), a constituent of tNAA, was lower in pre-HD (∼8%) and early HD (∼17%) vs controls (p < 0.05). The glial cell marker, myo-inositol (mI), was 50% higher in early HD vs pre-HD (p < 0.01). In early HD, mI correlated with UHDRS motor score (R 2 = 0.23, p < 0.05). Across pre-HD and early HD, tNAA correlated with performance on a tongue pressure task (R 2 = 0.30, p < 0.0001) and with disease burden score (R 2 = 0.17, p < 0.005).
Conclusions:
We demonstrate lower putaminal tNAA in early HD compared to controls in a cross-section of subjects. A novel biomarker role for mI in early HD was also identified. These findings resolve disagreement in the literature about the role of MRS as an HD biomarker. We conclude that putaminal MRS measurements of NAA and mI are promising potential biomarkers of HD onset and progression.
GLOSSARY
- AD
= Alzheimer disease;
- Cr
= creatine;
- DBS
= disease burden score;
- Glu
= glutamate;
- GPC
= glycerophosphocholine;
- HD
= Huntington disease;
- mI
= myo-inositol;
- MR
= magnetic resonance;
- MRS
= 1H magnetic resonance spectroscopy;
- NAA
= N-acetylaspartate;
- NAAG
= N-acetylaspartylglutamate;
- PC
= phosphocholine;
- PCr
= phosphocreatine;
- pre-HD
= premanifest Huntington disease;
- SNR
= signal-to-noise ratio;
- tCho
= total choline;
- tCr
= total creatine;
- TE
= echo time;
- tNAA
= total N-acetylaspartate;
- TR
= repetition time;
- UBC
= University of British Columbia;
- UHDRS
= Unified Huntington's Disease Rating Scale;
- VBM
= voxel-based morphometry.
The eventual development of Huntington disease (HD) symptoms can be predicted through detection of CAG trinucleotide repeat expansion in the HTT gene1 in premanifest gene carriers. However, definitive HD biomarkers, able to objectively identify disease onset and progression, are lacking. Evaluation of potential biomarkers are the focus of longitudinal studies such as TRACK-HD2 and PREDICT-HD.3
1H magnetic resonance spectroscopy (MRS) has previously been evaluated as a biomarker modality in premanifest and early HD.4–11 However, the literature is conflicted with regard to the utility of metabolites such as total N-acetylaspartate (tNAA), a measure of neuronal integrity,12 as potential biomarkers in HD. In an attempt to address these discrepant findings, arising in part from small subject numbers, poorly comparable outcome measures and heterogeneity within patient cohorts, we sought to determine the potential of MRS as an HD biomarker using large numbers of subjects in defined, homogeneous clinical cohorts and examined MRS outcome measures normalized to unsuppressed water rather than metabolites such as total creatine (tCr) which are affected in HD.6,11
TRACK-HD is an observational HD study that is taking place in Europe and 1 location in North America (Vancouver). Eighty-four individuals (30 controls, 25 pre-HD, and 29 early HD individuals) from the University of British Columbia (UBC), Vancouver, Canada, cohort underwent cross-sectional MRS examination of the left putamen and immediately surrounding white matter. We report baseline data from this prospective longitudinal study and correlate findings with selected measures of motor dysfunction.
METHODS
Subjects.
All subjects were recruited and evaluated at UBC, Vancouver, as part of TRACK-HD.2 Eighty-five participants were enrolled between January and August 2008. These comprised early HD (n = 30), pre-HD (n = 25), and control (n = 30) individuals. Pre-HD participants were selected based on 1) CAG repeat expansion within the HTT gene (40 CAG repeats or greater), 2) Unified Huntington's Disease Rating Scale (UHDRS) motor component score of 5 or less, indicating absence of marked motor deficits, and 3) disease burden score (DBS) of 220 or greater. The DBS, an index of mutant huntingtin protein exposure calculated as (age × [CAG − 35.5]),13 was used to select pre-HD individuals closer to predicted disease onset to maximize comparisons with controls. Of 25 pre-HD individuals, 22 had DBS of >250, with scores between 220 and 240 in the remaining 3.
Standard protocol approvals, registrations, and patient consents.
The study was approved by UBC Human Research Ethics Committee. Written informed consent was obtained from each participant.
Procedures.
Research participants were assessed using the UHDRS-99 motor component, performed by a trained rater (A.S.). DBS was calculated for early and pre-HD individuals.
The TRACK-HD battery employs novel quantitative motor assessments including tongue force measurement (glossomotography) and force transducer based self-paced tapping tasks (digitomotography). Glossomotography involves assessment of isometric forces during sustained tongue protrusion using a force transducer. Subjects matched a target force (0.5 N) presented on a monitor for 20 seconds (4 trials). Tongue pressure variability (coefficient of variation) and contact time (percentage of time tongue was contacting transducer) were calculated. During digitomotography, precision of timing for self-paced tapping was recorded. Subjects matched auditory cues (1.8-Hz rate) and continued tapping with the same frequency after cues stopped (5 trials). Variability of deviations of 1) tap initiation from predefined tapping frequency and 2) interval between midpoints of consecutive taps were calculated for the self-paced period (after cues had stopped). These tasks are sensitive to pre-HD motor impairment and can differentiate pre-HD and early HD individuals.2 Since we were interested in left hemisphere MRS changes, right-handed self-paced tapping was used. Tongue pressure tasks involved midline protrusion of the tongue. No lateral tongue movement was required.
Subjects underwent MRI examination at 3 T (Philips Achieva magnetic resonance [MR] scanner). MR spectra were obtained using single-voxel point resolved spectroscopy localization sequence in the left putamen. Scan parameters included 3.5 cm × 1 cm × 1.5 cm (5.25 cm3) voxel size. A voxel of this size inevitably incorporated white matter adjacent to putamen; however, no other gray matter or CSF was included. Repetition time (TR) was 2,000 msec, echo time (TE) = 35 msec, 1,024 samples performed per spectrum. There were 128 signal averages. Spectral bandwidth was 2,000 Hz. Second order shimming was performed. Water concentration was 43,300 mM. The default LCModel basis set for Philips at 3.0 T, TE = 35 msec, was used for analysis. T1-weighted images were collected for volumetric analyses (for TRACK-HD) using 3D T1 fast field echo (TR = 7.7 msec, TE = 3.5 msec, voxel size = 1.1 × 1.1 × 1.1 mm3, 164 slices). Time constraints prevented repeat scanning where spectra were inadequate or affected by movement. If TRACK-HD study images were also poor, subjects were recalled for reexamination and repeat spectra used.
MRS spectra were fit using LCModel.14 Metabolites were normalized to unsuppressed water spectrum. LCModel estimates “concentration” measurement reliability, returning standard deviations (%SD) for each metabolite. Standard deviations below 20% are considered reliable15; higher %SD were excluded. Five metabolites of principal interest were examined: tNAA (summed N-acetylaspartate [NAA] and N-acetylaspartylglutamate [NAAG], neuronal integrity marker), tCr (summed creatine [Cr] and phosphocreatine [PCr], brain energy metabolism and potential gliosis marker), tCho (predominantly glycerophosphocholine [GPC] and phosphocholine [PC], marker of neuronal membrane turnover), Glu ([glutamate], CNS excitatory neurotransmitter), and myo-inositol ([mI], astrocyte marker). NAA measurements, in addition to tNAA, were also performed.
To estimate contribution of white matter (nonputaminal) volumes to metabolite concentrations, blinded, manual voxel segmentation was performed. Percentage putaminal tissue in 3 (3-mm) slices per voxel (top, middle, bottom) was measured using Photoshop (Adobe Systems, Inc.) and averaged, giving percentage putamen volume per voxel (see appendices e-1 and e-2 on the Neurology® Web site at www.neurology.org).
Figure 1 shows putaminal voxel placement and sample spectra.

Figure 1 Samples of manual putamen segmentation and magnetic resonance spectra by subject group
Axial MRI brain slices, at 3 consecutive anatomical levels, used to perform manual segmentation of the voxel. A and B provide an example of the putaminal delineation required for manual segmentation. D–F: Representative MR spectra from the control, pre-HD, and early HD groups. Per our group findings, NAA levels displayed are lower and mI levels are higher in the early HD spectra than in the control spectra. Cho = choline; Cr = creatine; Glu = glutamate; HD = Huntington disease; mI = myo-inositol; NAA = N-acetylaspartate; pre-HD = premanifest Huntington disease.
Statistics.
The TRACK-HD study's lead statistician, Dr. Doug Langbehn, ensured that participants recruited into TRACK-HD were gender- and age-matched across groups. All group comparisons were made using one-way analysis of variance with post hoc Newman-Keuls multiple comparison tests. Scatterplots and linear regression were used to compare metabolites against DBS and motor outcomes. Linear correlations were investigated using the method of least squares.
RESULTS
Subject characteristics.
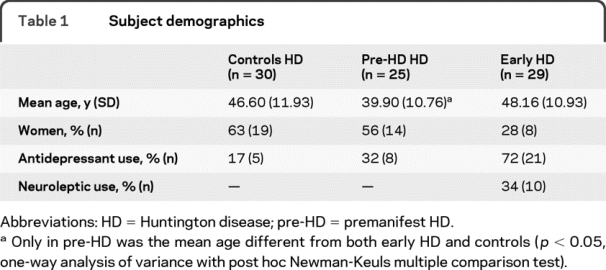
Of 85 individuals who underwent MRS, spectra were obtained from 84 individuals (30 controls, 25 pre-HD, 29 early HD). (Table 1) shows group demographics. Adequate LCModel data fits were obtained for tNAA, tCr, and tCho for all 84 individuals, mI for 74 (28 controls, 21 pre-HD, 25 early HD), Glu for 74 (29 controls, 23 pre-HD, 22 early HD), and NAA for 73 (30 controls, 24 pre-HD, 19 early HD). MR spectra were not included if poor fit quality precluded accurate (%SD >20) measurement. Exclusion was blind to participant group.
Table 1 Subject demographics
Metabolite differences across groups.
Figure 2 shows metabolite differences between groups. In early HD, tNAA and tCr were 15% and 18% lower than in controls (p < 0.001). In contrast, mI was 50% higher in early HD compared to pre-HD (p < 0.01). These findings suggest sensitivity for these metabolites as markers of altered neurochemistry in early HD.

Figure 2 1H magnetic resonance spectroscopy in controls and subjects with premanifest and early Huntington disease (HD)
Error bars denote SEM for each group. Asterisks indicate level of significance: *p < 0.05, **p < 0.01, ***p < 0.001. Group comparisons were made using one-way analysis of variance with post hoc Newman-Keuls Multiple Comparison Tests. Glu = glutamate; mI = myo-inositol; NAA = N-acetylaspartate; pre-HD = premanifest Huntington disease; tCho = total choline; tCr = total creatine; tNAA = total N-acetylaspartate.
mI and mI/tNAA correlate with UHDRS motor scores in early HD.
In an attempt to maximize sensitivity to disease severity, 2 metabolites exhibiting marked differences in early HD were combined as mI/tNAA. None of these metabolite measures correlated with declining motor performance across control, pre-HD, and early HD (data not shown).
Since mI differences occurred in early HD, motor correlations were investigated within this group and across pre-HD and early HD (table 2). In early HD, mI and mI/tNAA correlated with UHDRS motor score.
Table 2 Correlations between metabolite concentration and motor measurements in early HD and across pre-HD and early HD
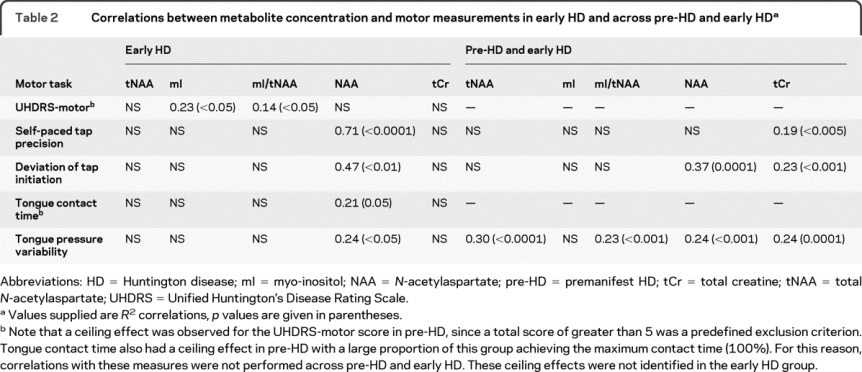
Metabolite correlations with quantitative motor performance across pre-HD and early HD.
Across pre-HD and early HD, tNAA and mI/tNAA correlated with the tongue pressure variability measure. For digitomotography correlations 6 left-handed individuals (4 pre-HD, 2 early HD) were excluded to ensure homogeneity on this right-handed task.
Metabolite correlations with motor performance in pre-HD.
Metabolite correlations with pre-HD motor performance were few (data not shown). Exceptions were tNAA correlation with tongue pressure variability (R 2 = 0.32, p < 0.005), and tCr correlations with self-paced tapping (R 2 = 0.27, p < 0.05), deviation of tap initiation (R 2 = 0.19, p < 0.05), and tongue pressure variability (R 2 = 0.18, p < 0.05).
Metabolite correlations with DBS across pre-HD and early HD.
tNAA correlated with DBS across pre-HD and early HD (figure 3), but not within groups (pre-HD: R 2 = 0.02, p = 0.46; early HD: R 2 = 0.08, p = 0.07). There was no clear trend between mI, mI/tNAA, or tCr and DBS.
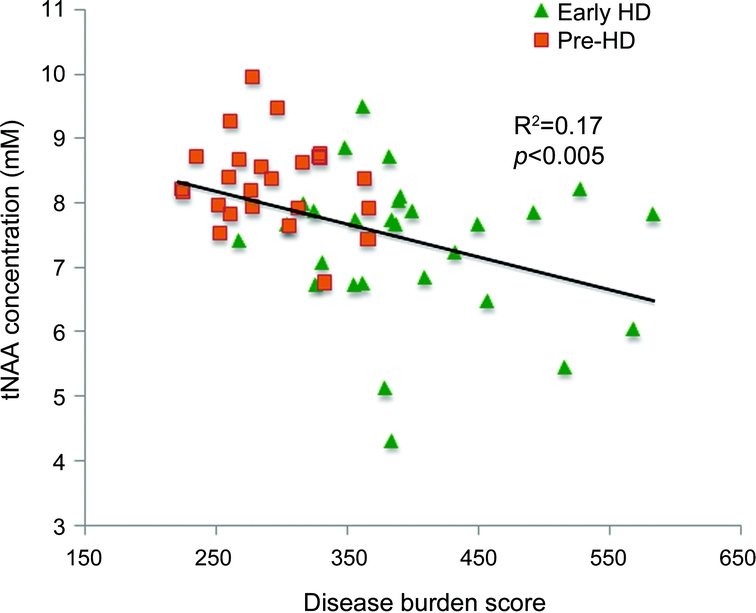
Figure 3 Scatterplot of N-acetylaspartate (NAA) concentration against disease burden score
HD = Huntington disease; pre-HD = premanifest Huntington disease.
DISCUSSION
An objective of this study was to clarify conflicting reports regarding the utility of MRS as an HD biomarker modality. Our focus was mI, tNAA, and their differences between control, pre-HD, and early HD individuals. In early HD, we correlated metabolites with performance on standard and novel quantitative motor tasks sensitive to early deficits in HD. A strength of this study is the normalization of metabolites to unsuppressed water signal, a technique previously reported in HD spectroscopy studies.5,7,8 By avoiding normalization to other metabolites (e.g., tCr), which are also disturbed in the HD brain,8 these measurements more likely reflect true biochemical change and permit direct comparison between studies. Furthermore, unlike most HD MRS studies, which utilize 1.5 T magnets,7,9,10,16–18 3 T MRS provides improved signal-to-noise ratio (SNR). This study also has larger, more homogeneous subject groups than earlier studies and the longitudinal design of TRACK-HD allows future evaluations of the same subjects.
The role of NAA in neuronal function is poorly understood. Synthesized within mature neurons, tNAA concentration within gray matter reflects neuronal number and viability, while in white matter it is a marker of axonal density.19–22 Total NAA was 15% lower in early HD than controls. It also correlated with a glossomotography measure in pre-HD and across pre-HD and early HD. Total NAA correlated with DBS across pre-HD and early HD. In pre-HD we did not identify the recognized association of tNAA with DBS.10 A narrow DBS range in pre-HD, a likely indirect consequence of enforcing a strict UHDRS motor score upper limit, may have contributed to this lack of correlation. In early HD also, tNAA did not correlate with DBS. A possible explanation may be that loss of putaminal tissue in early HD is paralleled by tNAA reduction, theoretically causing absolute tNAA concentration to plateau with resulting lack of correlation with DBS. Our findings are consistent with reported lower tNAA in akinetic HD (−66%) compared to controls, and correlation of tNAA with motor dysfunction.7
Interpretation of findings from other studies is limited by reporting tNAA values normalized to other metabolites, obscuring individual metabolite changes. Lower striatal and thalamic tNAA/tCr has been reported in early HD.6,11 Mean tNAA/tCr was comparable in our groups (mean [SD], controls: 1.01 [0.10] vs pre-HD: 1.04 [0.09] vs early HD: 1.05 [0.17], p > 0.05), suggesting that early HD absolute tNAA levels, in previous reports, may have been lower than our observations. NAA reductions have been reported on MR spectra of transgenic HD mice,23 and in putamen of HD-affected individuals.24
Lack of consistent striatal NAA and tNAA alteration in premanifest and early HD has been reported.8,10 Premanifest cohort heterogeneity, in time to predicted disease onset (range −7 to +29 years vs +7 to +18 years in our study), may have obscured metabolite variation in one study.8 Heterogeneity in our pre-HD subgroup was minimized by ensuring adherence to a maximum UHDRS motor score, a practice not specifically reported in other studies.8,10
mI is an osmolyte and astrocyte marker that is elevated in Alzheimer disease (AD).25,26 Manifest HD in adults and juveniles has been associated with high striatal mI.5,27 We demonstrate mI differences in early HD that correlate with UHDRS measured motor dysfunction. We found no relationship between mI and quantitative motor battery performance in pre-HD or early HD. mI/tNAA correlated less strongly with motor measures in early HD, and across pre-HD and early HD, than the correlations observed separately for mI and tNAA. Similar metabolite changes occur in asymptomatic individuals carrying gene defects associated with familial AD.28 Interestingly, in “premanifest AD,” tNAA was reduced, and tNAA/mI discriminated presymptomatic spectra from controls.28
While tNAA correlated with DBS across premanifest and early HD, it correlated more strongly with glossomotor dysfunction in these individuals. Together with mI, tNAA appears to have a potential role as a marker of HD-related motor decline. Putaminal metabolite changes are pertinent since this is a site of early atrophy, identifiable years prior to clinical diagnosis.29 Furthermore, putaminal atrophy correlates with psychomotor and motor deficits in premanifest HD30 and early HD.31 Our correlations suggest a possible role for MRS in evaluating the ability of an intervention to slow disease progression. It should be borne in mind that these cross-sectional assessments were performed at a single timepoint. Future longitudinal comparison is necessary to appreciate metabolite sensitivity to disease progression.
NAA and tCr were correlated against motor performance. NAA was less robustly measured than tNAA (11 measurement exclusions vs no exclusions) and is difficult to differentiate from NAAG, the other tNAA constituent. The lower NAA in pre-HD compared to controls was not accompanied by lower tNAA. NAA analyses should be treated cautiously, even though potential causes of NAAG artifact—spectral linewidth broadening (mean [SD], pre-HD: 8.00 [2.37] Hz vs controls: 7.55 [1.66] Hz, p > 0.05) and low SNRs (pre-HD: 9.36 [2.12] vs controls: 10.17 [2.18], p > 0.05)—were comparable in pre-HD and controls. NAA was 8% lower in pre-HD than controls. NAA correlated with quantitative motor performance almost consistently and more strongly than any other metabolite in early HD and across pre-HD and early HD.
In all groups tCr values were comparable to tNAA, while normally tNAA is about 30%–40% higher (depending on brain region).32 Our findings are supported by very similar, low tNAA/tCr ratios identified within basal ganglia structures of controls and premanifest and manifest HD individuals at 1.5 T and 7 T MRS.5,33 This possibly reflects increased basal energy metabolism within these structures.5 Since all spectra were analyzed using the same protocol with consistent scaling applied across metabolites, scaling difficulties would not have affected comparisons of estimated tNAA and tCr. Despite lower tCr in early HD, and reports of potential biomarker roles for tCr in HD,7 we were unable to correlate tCr with early HD motor performance. In pre-HD, and across pre-HD and early HD, tCr correlated with a number of quantitative motor measures. While this may represent altered energy metabolism,7 our tCr observations are likely to have been impacted by partial volume effects (discussed below).
Spectral quality was worst in early HD, with lower SNRs (mean [SD], early HD: 6.70 [2.00], controls: 10.17 [2.18], p < 0.001) and broader spectral linewidths (early HD: 10.51 [2.67] Hz, controls: 7.55 [1.66] Hz, p < 0.001) than controls. Movement artifact probably contributed by attenuating SNR.17 However, based on findings from a 4-T MRS study, we would expect the SNR reductions and linewidth increases seen in our early HD group to generate up to 4%–6% variation in tNAA and mI estimations.34 Small SNR or linewidth effects alone would therefore not account for larger mI and tNAA differences in early HD. Furthermore, at 1.5 T, marked SNR reduction and linewidth broadening either did not affect estimated tNAA or generated only 4% increases in tNAA and mI estimations.35,36
While the earliest HD neuropathologic changes occur in caudate and putamen,37 the putamen was selected to avoid averaging CSF into the brain parenchymal compartment. CSF partial volume effects may generate spurious metabolite measurements5 and is a risk with caudate voxel placement. Due to scanning time restrictions and the finding of early metabolic changes within the left striatum in premanifest HD,7 the left putamen was chosen as the structure of interest.
The use of larger voxels to improve SNR generates partial volume white matter effects through inclusion of nonputaminal tissue within the voxel. A 3-T MRS study of brain metabolites reported lower mI in white matter than gray matter.32 This suggests that partial volume white matter effects caused by loss of putaminal volume in transition from premanifest to early HD would diminish the mI difference observed. Hence concentrations presented may underestimate true mI differences. Studies indicate a cortical gray matter/white matter tNAA ratio between 0.8 and 1.2, in adulthood.38,39 We cannot guarantee that white matter components are not, partly, responsible for group tNAA differences, but our findings are consistent with smaller voxel studies that minimized partial volume effects.6,7 Higher tCho, lower Glu and tCr are found in white matter,32 making it impossible to know whether our observations reflect authentic pathophysiologic change. Consequently, tCr analyses presented here should be interpreted with care. Estimations of percentage putamen volumes per voxel were performed solely to ascertain the importance of partial volume effects in interpreting results (see appendices e-1 and e-2).
Subjects participating in TRACK-HD were demographically matched across international sites rather than at individual sites, explaining demographic discrepancies between our groups. Fewer early HD women may have impacted metabolite concentrations for this group (figure 2) but regional brain MRS did not identify relevant gender differences in tNAA or mI.38 Mean age in pre-HD was lower than in early HD and control groups (which were comparable). Consequently, age differences would not explain metabolite differences in early HD.
Use of neurotropic medications was highest in early HD (table 1). Antidepressant use in a total of over 30 depressed individuals was associated with either unchanged or increased cortical tNAA/tCr ratios at 1.5 T and 3 T.40 ,e1 All neuroleptic medicated patients with early HD were taking atypical agents. Atypical antipsychotic medication usage in over 50 patients with schizophrenia did not impact water-normalized thalamic or caudate tNAA levels (1.5 T).e2,e3 At 1.5 T, an atypical neuroleptic caused 27% elevation in thalamic mI/H2O levels.e4 While one-third of patients with early HD could have experienced medication-induced mI elevations, this would not explain the 50% higher mean mI concentration. Although not all studies were water-normalized, the evidence suggests that neurotropic usage would not explain our findings.
MRS has advantages over PET in monitoring disease progression. MRS data collection is less expensive and time-consuming, and does not require IV tracer isotope injection.8 ,e5,e6 Additionally, MRS may be less prone to interoperator variability than some VBM biomarker applications.e7 These advantages support ongoing assessment of MRS as an HD biomarker modality.
The ability of MRS to track pathologic change at the biochemical level makes it potentially responsive to acute therapeutic interventions. In a creatine supplementation study in early HD, MRS identified cortical glutamate reductions within 10 weeks.18 Thus MRS can potentially identify early reversal of pathologic processes, a finding that would be unlikely with structural imaging. The longitudinal assessment of our cohort, and replication of this study in a second large premanifest and early HD cohort, ideally in the setting of a therapeutic trial, will be necessary to fully validate these findings.
AUTHOR CONTRIBUTIONS
A.S. is the UBC Clinical Research Fellow and motor-rater for TRACK-HD. As such, he performed all standard and quantitative motor assessments at UBC. He was also involved in discussion, analysis, and interpretation of data, drafting the manuscript, and participated in patient recruitment. C.L. is a Research Associate at the UBC MRI Research Centre. She was responsible for overseeing MRS protocol development and data collection, analysis of all MRS data, and in the discussion/interpretation of data. She participated in the drafting of the manuscript. J.D. is a Clinical Research Coordinator at the UBC Huntington's Disease Clinic. She was responsible for TRACK-HD recruitment at UBC and also coordinated MRS. She made an intellectual contribution to this manuscript. R.D.S. and A.J.C. are Clinical Research Coordinators at the UBC Huntington's Disease Clinic. They assisted in MRS organization and administration. S.C. is the Genetic Counsellor for the UBC Huntington's Disease Clinic. She assisted in the identification and recruitment of suitable subjects for TRACK-HD and the MRS study. She also made an intellectual contribution to this manuscript. N.B. is the Research Fellow for the quantitative motor study from the HD Clinic at the University of Münster. She was responsible for analysis of quantitative motor assessment raw data and also guided selection of motor tasks for evaluation against metabolite profiles. R.R. is PI of the TRACK-HD quantitative motor substudy and Chair of the EHDN HD Clinic and Biomarker Laboratory at the University of Münster. He developed the quantitative motor equipment for TRACK-HD and did the blinded quality control and analysis of quantitative motor data. He also participated in protocol writing/setting up of and data interpretation for the main TRACK-HD study. M.R.H. was involved in identification and recruitment of suitable subjects and establishing UBC as the North American TRACK-HD site. He also made an intellectual contribution to this manuscript. S.J.T. is global PI for TRACK HD and PI for the London site. She has responsibility for overseeing the entire study. She had a role in obtaining funding, study conception and design, writing the protocol, setting up the study, and data interpretation for the main TRACK-HD study. She made intellectual contributions to both the MRS study and this manuscript. A.L.M. is the Director of the UBC MRI Research Centre. He was involved in protocol development and study design for the UBC MRS arm of the TRACK-HD study. He contributed to data interpretation, discussion of analyzed data, and writing of the final manuscript. B.R.L. is the Vancouver site PI for the main TRACK-HD study. He was responsible for obtaining funding for and setting up the UBC MRS study and for the study's conception and design. He also contributed to the interpretation, analysis, and writing of the final manuscript. Statistical analysis was conducted by Aaron Sturrock and Dr. Cornelia Laule.
ACKNOWLEDGMENT
The authors thank the TRACK-HD study participants, UBC MRI Research Centre MRI technologists, and CHDI, a non-profit organization dedicated to finding treatments for HD. They also thank members of TRACK-HD central coordination: Gail Owen, Saiqah Munir, Azra Hassanali (UCL Institute of Neurology), and Sherry Lifer (CHDI). They also thank Daniel van Kammen (Chief Medical Officer, CHDI), Beth Borowsky (TRACK-HD Scientific Director, CHDI), and Ethan Signer (Senior Scientific Advisor, CHDI) for their advice and assistance in developing the TRACK-HD project, and the data monitoring team at the University of Ulm, Germany: Katja Vitkin, Felix Mudoh Tita, Irina Vainer, and Theresia Kelm. They also thank the following for helping with aspects of data handling/interpretation: Tanka Acharya (Statistics Research Assistant, Psychiatry Research, University of Iowa) and the Laboratory of Neuroimaging UCLA (LONI) and IXICO (London) for their technical imaging assistance. They also thank Ruth Milner (Senior Statistical Advisor, CFRI) for her comprehensive statistical advice. Finally, the authors also thank the members of the TRACK-HD Steering Committee: Sarah J. Tabrizi, Douglas R. Langbehn (University of Iowa), Blair R. Leavitt, Raymund A.C. Roos (Leiden, Netherlands), Alexandra Durr (Hospital de la Salpetriere, France), David Craufurd (University of Manchester), Christopher Kennard and Stephen L. Hicks (Oxford University), Nick C. Fox and Rachael I. Scahill (UCL Institute of Neurology), Beth Borowsky, Allan J. Tobin (CHDI), H. Diana Rosas (Harvard), Hans Johnson (University of Iowa), Ralf Reilmann, Bernhard Landwehrmeyer (University of Ulm), and Julie C. Stout (Monash University, Australia), for their comments and discussion of the manuscript.
DISCLOSURE
Dr. Sturrock receives research support from the CHDI (full-time Clinical Research Fellow). Dr. Laule reports no disclosures. J. Decolongon receives salary support from the CHDI. R. Dar Santos receives salary support from the CHDI. A.J. Coleman receives salary support from the CHDI. S. Creighton serves on the editorial board of the Journal of Genetic Counseling. Dr. Bechtel receives research support from the CHDI/High Q Foundation, Inc. Dr. Reilmann serves on scientific advisory boards for Wyeth, the CHDI, Novartis, Siena Biotech S.p.A., and Neurosearch; holds a patent re: Glossomotography; received a speaker honorarium from Temmler Pharma GmbH & Co. KG; and receives research support from the European Huntington's Disease Network and the High Q Foundation, Inc. Dr. Hayden serves on the editorial board of Clinical Genetics. Prof. Tabrizi receives research support from the CHDI, Wellcome Trust, and MRC. Dr. MacKay serves on a scientific advisory board for the Institute for Biodiagnostics and receives research support from Genzyme Corporation, Teva Pharmaceutical Industries Ltd., the Multiple Sclerosis Society of Canada, and the Natural Sciences and Engineering Research Council. Dr. Leavitt receives research support from CHDI, the Canadian Institutes of Health Research, and the Michael Smith Foundation.
Supplementary Material
Address correspondence and reprint requests to Dr. Blair R. Leavitt, Huntington Disease Medical Clinic, UBC Hospital, 2211 Wesbrook Mall, UBC, Vancouver, V6T 2B5, British Columbia, Canada bleavitt@cmmt.ubc.ca
Supplemental data at www.neurology.org
*These authors contributed equally to this work.
References e1–e7 are available on the Neurology® Web site at www.neurology.org.
Study funding: Supported by the CHDI Foundation, Inc. (CHDI); NIH grant 20R-03189.
Disclosure: Author disclosures are provided at the end of the article.
Received February 24, 2010. Accepted in final form July 9, 2010.
REFERENCES
- 1.Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 1993;72:971–983. [DOI] [PubMed] [Google Scholar]
- 2.Tabrizi SJ, Langbehn DR, Leavitt BR, et al. Biological and clinical manifestations of Huntington's disease in the longitudinal TRACK-HD study: cross-sectional analysis of baseline data. Lancet Neurol 2009;8:791–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paulsen JS, Langbehn DR, Stout JC, et al. Detection of Huntington's disease decades before diagnosis: the Predict-HD study. J Neurol Neurosurg Psychiatry 2008;79:874–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harms L, Meierkord H, Timm G, Pfeiffer L, Ludolph AC. Decreased N-acetyl-aspartate/choline ratio and increased lactate in the frontal lobe of patients with Huntington's disease: a proton magnetic resonance spectroscopy study. J Neurol Neurosurg Psychiatry 1997;62:27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoang TQ, Bluml S, Dubowitz DJ, et al. Quantitative proton-decoupled 31P MRS and 1H MRS in the evaluation of Huntington's and Parkinson's diseases. Neurology 1998;50:1033–1040. [DOI] [PubMed] [Google Scholar]
- 6.Jenkins BG, Rosas HD, Chen YC, et al. 1H NMR spectroscopy studies of Huntington's disease: correlations with CAG repeat numbers. Neurology 1998;50:1357–1365. [DOI] [PubMed] [Google Scholar]
- 7.Sánchez-Pernaute R, García-Segura JM, del Barrio Alba A, Viaño J, de Yébenes JG. Clinical correlation of striatal 1H MRS changes in Huntington's disease. Neurology 1999;53:806–812. [DOI] [PubMed] [Google Scholar]
- 8.Reynolds NC Jr, Prost RW, Mark LP. Heterogeneity in 1H-MRS profiles of presymptomatic and early manifest Huntington's disease. Brain Res 2005;1031:82–89. [DOI] [PubMed] [Google Scholar]
- 9.Gómez-Ansón B, Alegret M, Muñoz E, Sainz A, Monte GC, Tolosa E. Decreased frontal choline and neuropsychological performance in preclinical Huntington disease. Neurology 2007;68:906–910. [DOI] [PubMed] [Google Scholar]
- 10.van Oostrom JC, Sijens PE, Roos RA, Leenders KL. 1H magnetic resonance spectroscopy in preclinical Huntington disease. Brain Res 2007;1168:67–71. [DOI] [PubMed] [Google Scholar]
- 11.Ruocco HH, Lopes-Cendes I, Li LM, Cendes F. Evidence of thalamic dysfunction in Huntington disease by proton magnetic resonance spectroscopy. Mov Disord 2007;22:2052–2056. [DOI] [PubMed] [Google Scholar]
- 12.Duncan JS. Magnetic resonance spectroscopy. Epilepsia 1996;37:598–605. [DOI] [PubMed] [Google Scholar]
- 13.Penney JB Jr, Vonsattel JP, MacDonald ME, Gusella JF, Myers RH. CAG repeat number governs the development rate of pathology in Huntington‘s disease. Ann Neurol 1997;41:689–692. [DOI] [PubMed] [Google Scholar]
- 14.Provencher SW. Estimation of metabolite concentrations from localized in vivo proton NMR spectra. Magn Reson Med 1993;30:672–679. [DOI] [PubMed] [Google Scholar]
- 15.Provencher S. LCModel and LCMGui User's Manual. Stephen W. Provencher; 2005. [Google Scholar]
- 16.Davie CA, Barker GJ, Quinn N, Tofts PS, Miller DH. Proton MRS in Huntington's disease. Lancet 1994;343:1580. [DOI] [PubMed] [Google Scholar]
- 17.Taylor-Robinson SD, Weeks RA, Bryant DJ, et al. Proton magnetic resonance spectroscopy in Huntington's disease: evidence in favour of the glutamate excitotoxic theory. Mov Disord 1996;11:167–173. [DOI] [PubMed] [Google Scholar]
- 18.Bender A, Auer DP, Merl T, et al. Creatine supplementation lowers brain glutamate levels in Huntington's disease. J Neurol 2005;252:36–41. [DOI] [PubMed] [Google Scholar]
- 19.Tallan HH. Studies on the distribution of N-acetyl-L-aspartate acid in brain. J Biol Chem 1956;224:41–45. [PubMed] [Google Scholar]
- 20.Gujar SK, Maheshwari S, Bjorkman-Burtscher I, Sundgren PC. Magnetic resonance spectroscopy. J Neuro-Ophthalmol 2005;25:217–226. [DOI] [PubMed] [Google Scholar]
- 21.Xu V, Chan H, Lin AP, et al. MR spectroscopy in diagnosis and neurological decision-making. Semin Neurol 2008;28:407–422. [DOI] [PubMed] [Google Scholar]
- 22.Pouwels PJ, Frahm J. Differential distribution of NAA and NAAG in human brain as determined by quantitative localized proton MRS. NMR Biomed 1997;10:73–78. [DOI] [PubMed] [Google Scholar]
- 23.van Dellen A, Welch J, Dixon RM, et al. N-Acetylaspartate and DARPP-32 levels decrease in the corpus striatum of Huntington's disease mice. Neuroreport 2000;11:3751–3757. [DOI] [PubMed] [Google Scholar]
- 24.Dunlop DS, Mc Hale DM, Lajtha A. Decreased brain N-acetylaspartate in Huntington's disease. Brain Res 1992;580:44–48. [DOI] [PubMed] [Google Scholar]
- 25.Moats RA, Ernst T, Shonk TK, Ross BD. Abnormal cerebral metabolite concentrations in patients with probable Alzheimer disease. Magn Reson Med 1994;32:110–115. [DOI] [PubMed] [Google Scholar]
- 26.Griffith HR, den Hollander JA, Okonkwo OC, O'Brien T, Watts RL, Marson DC. Brain metabolism differs in Alzheimer's disease and Parkinson's disease dementia. Alzheimers Dement 2008;4:421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reynolds NC, Prost RW, Mark LP, Joseph SA. MR-spectroscopic findings in juvenile-onset Huntington's disease. Mov Disord 2008;23:1931–1935. [DOI] [PubMed] [Google Scholar]
- 28.Godbolt AK, Waldman AD, MacManus DG, et al. MRS shows abnormalities before symptoms in familial Alzheimer disease. Neurology 2006;66:718–722. [DOI] [PubMed] [Google Scholar]
- 29.Aylward EH, Codori AM, Barta PE, Pearlson GD, Harris GJ, Brandt J. Basal ganglia volume and proximity to onset in presymptomatic Huntington disease. Arch Neurol 1996;53:1293–1296. [DOI] [PubMed] [Google Scholar]
- 30.Jurgens CK, van de Wiel L, van Es AC, et al. Basal ganglia volume and clinical correlates in ‘preclinical’ Huntington's disease. J Neurol 2008;255:1785–1791. [DOI] [PubMed] [Google Scholar]
- 31.Harris GJ, Aylward EH, Peyser CE, et al. Single photon emission computed tomographic blood flow and magnetic resonance volume imaging of basal ganglia in Huntington's disease. Arch Neurol 1996;53:316–324. [DOI] [PubMed] [Google Scholar]
- 32.Baker EH, Basso G, Barker PB, Smith MA, Bonekamp D, Horská A. Regional apparent metabolite concentrations in young adult brain measured by 1H MR spectroscopy at 3 Tesla. J Mag Res Imaging 2008;27:489–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van den Bogaard SJA, Dumas EM, Teeuwisse WM, van Buchem MA, van der Grond J, Roos RAC. Exploratory 7T magnetic resonance spectroscopy in Huntington's disease. Clin Genet 2009;76(suppl 1):55. [Google Scholar]
- 34.Bartha R. Effect of signal-to-noise ratio and spectral linewidth on metabolite quantification at 4 T. NMR Biomed 2007;20:512–521. [DOI] [PubMed] [Google Scholar]
- 35.Kanowski M, Kaufmann J, Braun J, Bernarding J, Tempelmann C. Quantitation of simulated short echo time 1H human brain spectra by LCModel and AMARES. Magn Reson Med 2004;51:904–912. [DOI] [PubMed] [Google Scholar]
- 36.Macrì MA, Garreffa G, Giove F, et al. In vivo quantitative 1H MRS of cerebellum and evaluation of quantitation reproducibility by simulation of different levels of noise and spectral resolution. Magn Reson Imaging 2004;22:1385–1393. [DOI] [PubMed] [Google Scholar]
- 37.Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP Jr. Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol 1985;44:559–577. [DOI] [PubMed] [Google Scholar]
- 38.Pouwels PJ, Frahm J. Regional metabolite concentrations in human brain as determined by quantitative localized proton MRS. Magn Reson Med 1998;39:53–60. [DOI] [PubMed] [Google Scholar]
- 39.Lundbom N, Barnett A, Bonavita S, et al. MR Image segmentation and tissue metabolite contrast in 1H spectroscopic imaging of normal and aging brain. Magn Reson Med 1999;41:841–845. [DOI] [PubMed] [Google Scholar]
- 40.Gonul AS, Kitis O, Ozan E, et al. The effect of antidepressant treatment on N-acetyl aspartate levels of medial frontal cortex in drug-free depressed patients. Prog Neuropsychopharmacol Biol Psychiatry 2006;30:120–125. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.