

Abstract
Proteomics is gradually complementing large shotgun qualitative studies with hypothesis-driven quantitative experiments. Targeted analyses performed on triple quadrupole instruments in selected reaction monitoring mode are characterized by a high degree of selectivity and low limit of detection; however, the concurrent analysis of multiple analytes occurs at the expense of sensitivity because of reduced dwell time and/or selectivity due to limitation to a few transitions. A new data acquisition paradigm is presented in which selected reaction monitoring is performed in two ways to simultaneously quantify and confirm the identity of the targeted peptides. A first set of primary transitions is continuously monitored during a predetermined elution time window to precisely quantify each peptide. In addition, a set of six to eight transitions is acquired in a data-dependent event, triggered when all the primary transitions exceed a preset threshold. These additional transitions are used to generate composite tandem mass spectra to formally confirm the identity of the targeted peptides. This technique was applied to analyze the tryptic digest of a yeast lysate to demonstrate the performance of the technique. We showed a limit of detection down to tens of attomoles injected and a throughput exceeding 6000 transitions in one 60-min experiment. The technique was integrated into a linear work flow, including experimental design, data acquisition, and data evaluation, enabling large scale proteomic studies.
Proteomics is gradually complementing qualitative studies focused on protein identification relying on shotgun strategies (1, 2) with large scale quantitative experiments. This change was prompted by the growing demand for qualification and verification of putative protein biomarkers through analysis of larger cohorts of clinical samples on one hand and the need for consistent quantitative data sets to facilitate modeling in systems biology studies on the other. In either case, the number of proteins under target is quite large (tens to hundreds), and traditional immunoassay approaches are not suited for use because of the cost, time, and difficulty of developing multiplexed assays. In this context, the selected reaction monitoring (SRM)1 technique performed on a triple quadrupole mass spectrometer is increasingly applied to quantitative proteomics because of its sensitivity, selectivity, and wide dynamic range (3–6). Mass spectrometry assays can be developed very rapidly via the use of commodity synthetic reference peptides libraries (7), and large resources of peptide MS/MS data (MRMAtlas) (8) are available to design the initial assays. However, developing a robust and precise SRM-based multiple assay remains demanding for proteomics experiments.
One of the main challenges is that most proteomics samples are highly complex, and several interfering signals are detected within a given time window that require systematic verification of the target peptide identity first to ensure its accurate quantification. Using isotopically labeled counterparts of the targeted analytes is a common way to confirm the target peptide identity (9, 10). As this may not always be practical for large scale quantitative proteomics studies, an alternative way to verify the peptide identity is to use SRM-initiated full MS/MS scans (11). However, the disadvantage of this method is a lower sensitivity and selectivity compared with SRM as it uses a broader mass selection window, which results in MS/MS spectra often containing signals from multiple components co-eluting in the case of biological samples with a complex background. Furthermore, SRM-initiated MS/MS scans require much longer duty cycle times that will disrupt the predefined SRM sequence of events in the case of complex multiplexed assays even when performed on a triple quadrupole instrument equipped with a linear ion trap. Recently, instead of using MS/MS spectra, a composite MS/MS spectrum that is generated by measuring multiple fragments ions (8–10 ions) from one specific peptide has been proven to provide sufficient information for peptide identification.2 The peptide is verified both by the overlay of the chromatographic elution profiles of the fragment ions and by matching the composite MS/MS spectrum that comprises multiple SRM transitions to the MS/MS spectral library entry (12, 13). Because it is based on SRM acquisition, this method provides a rapid, sensitive, and selective way to perform peptide verification, which is desirable for large scale screening experiments. The drawback of this approach is that only a limited number of compounds can be practically analyzed in one HPLC-MS run because the instrument is continuously monitoring 8–10 transitions for each peptide no matter whether the peptide is detected from the sample or not, resulting in the waste of a significant portion of the instrument time. To overcome this issue and thus increase the throughput, we propose that the instrument constantly monitor only a small subset of SRM transitions for each peptide for the actual quantification and in addition confirm the peak identity using the full set of fragment ions, which are acquired in a data-dependent mode.
To provide this instrument capability, we developed an innovative instrument control software, called intelligent selected reaction monitoring (iSRM), that can use the specificity of a small subset of SRM transitions to quantify and intelligently trigger the full list for confirmation of target peptides, thereby allowing the simultaneous qualitative and quantitative analysis of up to 1000 peptides in a single LC-MS experiment. Here we describe the concept of iSRM and the work flow associated to it, and we demonstrate the increased throughput facilitating the development of SRM assays and its ability to perform large scale screens targeting a respectable number of proteins.
EXPERIMENTAL PROCEDURES
Sample Preparation
A whole yeast cell lysate was solvated in 0.1 m ammonium bicarbonate buffer containing 8 m urea at a final concentration of 1 mg/ml. The proteins were reduced with 12 mm dithiothreitol for 30 min at 35 °C and alkylated with 40 mm iodoacetamide for 45 min at 25 °C in the dark. Samples were diluted with 0.1 m NH4HCO3 to a final concentration of 1.5 m urea, and sequencing grade porcine trypsin (Promega) was added to a final enzyme:substrate ratio of 1:100 and incubated for 8 h at 37 °C. Peptide mixtures were cleaned by Sep-Pak tC18 cartridges (Waters, Milford, MA) and eluted with 60% acetonitrile. The resulting peptide samples were evaporated on a vacuum centrifuge to dryness and resolubilized in 0.1% formic acid to 1 μg/μl concentration. The same yeast digest was used through out all experiments described in this study and was prepared as described earlier (14).
HPLC
A nano-LC pump from Eksigent was used. HPLC separations were performed on a Picofrit column (C18, 75 μm × 100 mm, 15-μm tip; New Objective) using a gradient starting at 5% B and increasing to 45% B in 40 min at a flow rate of 300 nl/min. The gradient was generated by mixing water containing 0.1% formic acid (A) and acetonitrile containing 0.1% formic acid (B). One microliter of each sample was injected.
Mass Spectrometry
LC-MS experiments were performed on a triple quadrupole mass spectrometer (TSQ Vantage, Thermo Scientific) operated in the nanoelectrospray mode. For ionization, a 1800-V spray voltage and 200 °C capillary temperature were used. The selectivity for both Q1 and Q3 were set to 0.7 Da (full-width half-maximum). The collision gas pressure of Q2 was set at 1.2 millitorrs of argon. The collision energy (CE) was calculated using the following formula: CE = 0.034 × m/z of precursor ion + 2.
Analysis in SRM Mode (iSRM)
For the selected reaction monitoring experiments using the iSRM instrument control software (part of the TSQ driver 2.2.0), two primary and an additional six secondary fragment ions were used for each targeted peptide. The instrument acquisition parameters, including elution time window, collision energy, and intensity threshold for triggering the data-dependent SRM scan, were generated by using Pinpoint 1.0 software (Thermo Scientific). To predict retention times, first we trained Pinpoint on the liquid chromatographic settings. A mixture of 10 isotopically labeled peptides (ELASGLSFPVGFK*, GAIAAAHYIR*, GILFVGSGVSGGEEGAR*, ELGQSGVDTYLQTK*, LTILEELR*, AIPGEYITYALSGYVR*, TTFINTLFQTVLK*, LVEDPQVIAPFLGK*, GISNEGQNASIK*, and LGNDDEVILFR* with the asterisk indicating 13C isotopically labeled amino acids) was run on the TSQ Vantage where we monitored all y-ions for those peptides. Using these transitions, we identified the elution time for each peptide. In addition, for each peptide, we also calculated its hydrophobicity using the SSRCalc algorithm presented in Krokhin et al. (15). Then a least square linear curve was generated between the observed retention time and the hydrophobicity factor for these 10 peptides, and the goodness of fit was evaluated using the r2 value. Using this linear curve, we also established our prediction error (±2 min). Next for any novel peptide sequence, we first calculated its hydrophobicity using the SSRCalc algorithm, and then using the linear curve, we predicted its elution time. Then using ±2-min error around the predicted elution time, we established a 4-min window for the transitions of this peptide. The generated iSRM method with the above acquisition parameters was transferred directly to the TSQ Vantage instrument method setup program directly. Additional instrument parameters used for the iSRM experiments include cycle time, scan time, and dynamic exclusion set up for triggering the data-dependent SRM scan. The overall cycle time of 2 s was used as default. The scan times used for the data-dependent SRM scans were 0.1 s. Dynamic exclusion was enabled with 300 counts of intensity threshold. The dynamic exclusion was set up to trigger the dependent SRM scan events only when the intensities of all primary SRM transitions exceeded 300 counts for five successive cycles. The dynamic exclusion time was 20 s. Three iSRM experiments were performed based on the resources of peptide MS/MS data from MRMAtlas. For each targeted peptide, the two most intense fragment ions were selected as primary ions, and an additional six fragments were selected as secondary ions.
Experiment 1
A total of 128 transitions (32 primary and 96 secondary) were used for targeting eight endogenous/heavy peptide pairs. The isotopically labeled peptides were spiked into the yeast digest mixture at different concentrations (0.01, 0.1, 1, 10, and 100 fmol/μl). The analyses were performed by injecting 1 μl of sample. The completed transition list can be found in supplemental Table 1.
Experiment 2
A total of 6056 transitions (1514 primary and 4542 secondary) were used for large scale screening, targeting 757 literature peptides from MRMAtlas. The completed transition list can be found in supplemental Table 2.
Experiment 3
A total of 544 transitions (136 primary and 408 secondary for 68 peptides) were used for targeting 34 pairs of endogenous and 15N-labeled peptides in the TCA cycle of Saccharomyces cerevisiae. The completed transition list can be found in supplemental Table 3.
Data Processing Using Pinpoint
All raw files were processed by Pinpoint 1.0. The targeted peptides were verified by both overlaid chromatographic profiles of multiple fragment ions and the correlation between the composite MS/MS spectrum that comprises multiple SRM transitions with the MS/MS spectral library entry. The MS/MS spectral library was built by reading in fragment ion intensity data from the database of MRMAtlas directly. To compare the targeted SRM data generated from a triple quadrupole mass spectrometer with MS/MS data of a peptide stored in a spectrum library, a novel spectrum library searching method was used (12). The peak area intensities for all SRM transitions (primary and secondary) that belonged to the same precursor ion m/z were ranked in decreasing order of intensity. Then the same product ions from the MS/MS library spectrum were also ranked in decreasing order of intensity. A Costa-Soares rank correlation (16) was computed as a metric of similarity between the SRM data and the MS/MS spectrum stored in the library for the target peptide,
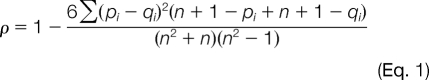 |
where ρ is the correlation coefficient (expected range of values are 1 >= ρ>= −1), pi is the rank of the product ion i in the library spectrum and qi is the rank of the product ion i in the signal intensities from the SRM data, and n is the total number of product ions under consideration. To provide an estimate of the null distribution, the correlation coefficient was compared with library spectra whose rank intensities were shuffled and then rescored. The null distribution was used to compute a p value (probability of random spectral matching) using standard statistical methods. The smaller p value means a better match between the SRM data and spectral library. The peptide that gave a p value less than 0.05 was considered to be confidently confirmed by the library match. We used a cutoff of 0.05 to designate confident identifications; i.e. only composite-MS/MS spectra that had p values lower than 0.05 were listed as identified confidently using the library spectra. This cutoff value was chosen based on empirical evidence where p values between large sets of composite spectra and CID spectra were compared (12). For each targeted peptide that fulfilled the verification criteria, the software computed the integrated peak areas of all primary ions for quantification and calculated coefficients of variation (CVs) (in percent) for the repeated experiments.
RESULTS
Principle of iSRM
The systematic and reliable targeted analysis of proteomics samples has to overcome two main hurdles. First, sufficient selectivity has to be ensured to confidently quantify the analyte of interest in complex biological samples, which are characterized by numerous background interferences, through the concomitant monitoring of multiple transitions. Second, the number of peptides to be analyzed in a given time window has to match the cycle time constraints required for reliable quantification. Two parameters have to be considered: the dwell time of each measurement that may need to be increased for low abundance ions and the number of transitions measured for each analyte. A trade-off between selectivity and sensitivity is generally required if a larger set of peptides, typically exceeding a dozen, is analyzed under such conditions.
The iSRM work flow exploits the specificity of selected reaction monitoring to simultaneously quantify and confirm the identity of the targeted peptides in two ways. The principle of the iSRM acquisition method is illustrated in Fig. 1. A few transitions, typically two or three, corresponding to the most intense fragments in the MS/MS spectra of targeted peptides, referred to here as primary transitions, are monitored continuously during a (predicted or observed) elution time window of usually 2–4 min. For each transition, an intensity threshold is defined by the user that can optionally take into account the expected intensity of the signal. When all the primary transitions satisfy all pretrigger conditions (i.e. following AND logic), a secondary event is initiated (Fig. 1B). It aims to punctually measure additional transitions referred to as secondary transitions that are used to generate a composite MS/MS spectrum (Fig. 1C) to confirm that the signals observed for the primary transitions are indeed corresponding to the authentic targeted peptide as opposed to interfering species. In fact, the acquisition of these transitions is performed when the HPLC profile, usually 25–30 s wide, is reaching its apex. Thus, a delay of 8–10 s, corresponding to a few of cycles, is set before the actual triggering of the data-dependent measurements. This design of the dynamic exclusion allows each secondary SRM acquisition to be triggered only once (or twice if desired) for each eluting LC trace, which yields sufficient structural information to unambiguously confirm the identity of the compound without significantly perturbing the quantification obtained with the primary SRM transition list.
Fig. 1.

Principle of iSRM. A illustrates the iSRM logic in which two transitions of a given peptide are monitored continuously and trigger a data-dependent event if both signals exceed a preset threshold. B and C show the primary and data-dependent (secondary) SRM events. D and E show the channels (and the corresponding ion intensities) of a primary and a secondary iSRM event, respectively.
Generation of iSRM Methods
The design of an SRM experiment encompasses several steps, including the selection of the peptides corresponding to the proteins that are targeted in the study. These tryptic peptides need to be uniquely associated with the proteins of interest (proteotypic). In addition, they need to fulfill some specific criteria, ensuring their detection in the mass spectrometer, such as good ionization efficiency and an appropriate mass-to-charge ratio. For practical reasons, peptides containing amino acids that undergo facile chemical modification (e.g. oxidation or deamidation) are normally excluded. For each selected peptide, specific attributes, including retention time, precursor ion mass-to-charge ratio, fragment ion masses, and collision energy, are required to establish an SRM method. From a practical point of view, existing MS/MS data dramatically facilitate the design of the method and increase the success rate of SRM experiments, particularly if the spectra were acquired on an instrument with similar fragmentation characteristics compared with a triple quadrupole. There is already a wealth of existing information available in publicly accessible proteomics MS/MS repositories, for instance PeptideAtlas (17) or Global Proteome Machine (18). More recently, repositories specifically containing SRM transitions have been established (MRMAtlas) (8) that represent a tremendous primary resource to design such experiments.
Such information is pertinent to establish an iSRM method, more specifically the selection of primary and secondary fragment ions in the context of experiments targeting hundreds of peptides in an automated fashion for keeping the analytical throughput. By using Pinpoint 1.0, we were able to design the iSRM assay based on the data of a spectral library (Fig. 2). In this study, using a yeast cell lysate to demonstrate proof of principle, we automatically extracted the relative fragment ion intensity information of each targeted peptide based on the database of MRMAtlas by using the Pinpoint 1.0 software. First, the software read in the information of fragment ion intensities from the database to build a “local” MS/MS spectral library. Then the information of this spectral library was used to define the primary and secondary ions of each iSRM experiment. The two most intense fragment ions were defined as primary ions, and an additional six fragment ions were defined as secondary ions based on rank of ion intensity per peptide.
Fig. 2.

Work flow used to perform iSRM experiment. A set of predefined proteins defines the experiment; the proteotypic peptides associated with these proteins are the subjects of the actual SRM experiment. The parameters necessary to perform the analysis are extracted from exiting proteomics resources (e.g. MRMAtlas); the method includes primary and secondary transitions (for details, see the text). The results of the LC-MS analysis are subsequently processed in two ways: the quantification is performed using the primary SRM traces, and the confirmation of the identity relies on the fragmentation patterns represented in the composite spectra. Pep, peptide; QQQ, triple quadrupole; I.S., internal standard; Lib, library.
Proof-of-concept Experiment
To evaluate the performance of the iSRM technique in terms of sensitivity, selectivity, and dynamic range in a biological sample, eight isotopically labeled, synthetic yeast peptides were spiked into the yeast digest in a dilution series ranging from 10 amol to 100 fmol injected on the column. The eight endogenous peptides and their corresponding internal standards were targeted using the iSRM method, and each sample was analyzed in triplicate.
The composite MS/MS spectra of the peptides ELASGLSFPVGFK*, LVEDPQVIAPFLGK*, LTILEELR*, GISNEGQNASIK*, ELGQSGVDTYLQDK*, and GAIAAAHYIR* allowed confident confirmation of the identity at the 10-amol level with p values less than 0.05. The identities of the two peptides LGNDDEVILFR* and GILFVGSGVSGGEEGAR* were assertively confirmed by the composite MS/MS spectra at the 100-amol level with p values less than 0.05.
For each positively identified peptide, the integrated peak areas of two primary ions were used for quantification to determine the limit of sensitivity of the method. One of the most significant expected benefits of using iSRM is to increase the instrument throughput and selectivity by triggering the data-dependent SRM scan only for a real eluted peak. As shown in Fig. 3A, although multiple peaks were observed from each primary transition, the data-dependent scan was triggered only for the actual peptide peak when both primary transitions were detected simultaneously. Additionally, the dynamic exclusion design of iSRM assured that the data-dependent scan was triggered only once. With effective instrument time usage, most of the instrument time is devoted to the actual quantification. With a fixed cycle time of 2 s, the acquisition instrument time is essentially used for a longer dwell time of the primary transitions and therefore results in increased sensitivity and signal-to-noise ratio. Response curves for all eight spiked isotopically labeled peptides cover a wide dynamic range using the iSRM method. Fig. 3C shows the calibration curves acquired for the peptides LTILEELR*, GISNEGQNASIK*, and LVEDPQVIAPFLGK* with a dynamic range covering 4 orders of magnitude, ranging from 10 amol to 100 fmol injected on the column. These results demonstrate that the technique preserves all the characteristics of a conventional SRM experiment in terms of selectivity and sensitivity while optimally utilizing the acquisition time to gain additional information, thereby increasing the confidence of the analyses.
Fig. 3.

The use of several traces increases the selectivity of the detection of the analyte of interest (A). The coincidence of two signals is required to trigger the acquisition of secondary transitions that are used to generate a composite MS/MS spectrum (shown in B). The detection of peptides in a complex background occurs over a wide dynamic range (10 amol to 100 fmol injected on the column; see C). Quality composite spectra obtained at low concentration are shown in D and E. Int, intensity, *: primary transitions.
Large scale iSRM Experiment
To test the performance of the approach when applied in the context of a large scale screening experiment, 757 tryptic peptides from the yeast digest mixture (see supplemental Table 2) were targeted using the iSRM method. The peptide sequences and the associated MS/MS information were downloaded from the MRMAtlas database and intended to cover a wide expression range of the yeast proteome. This large scale iSRM experiment was repeated five times for testing the reproducibility and the robustness of the method. The raw data of the five experiments were processed postacquisition (using the Pinpoint software; see “Experimental Procedures”). Among the 757 monitored yeast peptides initially selected across a wide expression range, 673 peptides were actually detected and properly identified from 1 μg of yeast digest injected using the default acquisition parameters typical of an initial experiment (i.e. without adjustment of the dwell times to detect low abundance species). Regardless of the complexity of the experiment, most of the detected peptides (86%) were verified by the composite MS/MS spectra with a p value <0.05. The increased instrument throughput yields enough data points for each primary transition to perform precise quantification for this large scale data set. Very consistent results were obtained with over 80% of the peptides detected exhibiting coefficients of variation below 10%. As shown in Table I, the peptides detected cover a wide range of concentrations with yeast proteins expressed in the range from less than 120 to one million copies per cell as reported previously by Ghaemmaghami et al. (19).
Table I. Peptide analyzed to illustrate protein expression range covered in yeast proteome using iSRM.
| Group no. | Literature valuea | Protein | Peptide | Integrated areab | CV (n = 3) | p value |
|---|---|---|---|---|---|---|
| copies/cell | % | |||||
| 1 | 524,000–1,256,000 | YLR249W | SNFATIADPEAR | 4.01e+06 | 6 | 2.48e−05 |
| LVEDPQVIAPFLGK | 1.78e+06 | 3 | 3.47e−04 | |||
| 2 | 262,000–524,000 | YER091C | NVSGQDVAAALEANAK | 1.42e+06 | 12 | 1.20e−04 |
| AYTYFGEQSNLPK | 1.49e+06 | 3 | 1.48e−03 | |||
| 3 | 131,000–262,000 | YNL178W | ALPDAVTIIEPK | 9.16e+05 | 8 | 7.19e−04 |
| ALPDAVTIIEPKEEEPILAPSVK | 1.18e+05 | 1 | 2.11e−02 | |||
| 4 | 65,000–131,000 | YKL182W | ALDTVTNVLNFIK | 3.91e+05 | 4 | 4.34e−03 |
| ALIENWAADSVSSR | 4.88+e5 | 12 | 1.74e−04 | |||
| 5 | 32,000–65,000 | YLR058C | VLVAGTSAYCR | 1.34+e6 | 7 | 3.67e−03 |
| AVEFAQQVQQSLPK | 2.62e+05 | 9 | 2.48e−05 | |||
| 6 | 16,000–32,000 | YBR249C | ELASGLSFPVGFK | 6.89e+05 | 5 | 8.23e−04 |
| GLINDPDVNNTFNINK | 1.10e+05 | 9 | 2.73e−04 | |||
| 7 | 8,000–16,000 | YJL130C | ELVAPGAIQNLIR | 6.69e+05 | 15 | 1.32e−02 |
| TADLASVLLLTSLQNR | 7.60e+04 | 8 | 1.74e−04 | |||
| 8 | 4,000–8,000 | YKR001C | ELSSQELSGGAR | 2.50e+05 | 5 | 1.14e−03 |
| LAALESPPPVLK | 3.78e+05 | 12 | 1.74e−04 | |||
| 9 | 2,000–4,000 | YFL014W | GVFQGVHDSAEK | 1.28e+06 | 5 | 1.36e−02 |
| LNDAVEYVSGR | 8.26e+05 | 3 | 7.19e−04 | |||
| 10 | 1,000–2,000 | YGL202W | SLANTFLSLDTEGR | 4.05e+05 | 3 | 3.46e−−02 |
| ALQYGFSAGQPELLNFIR | 9.74e+04 | 13 | 3.99e−03 | |||
| 11 | 512–1,024 | YCL030C | LQDAPEESYTR | 4.48e+05 | 2 | 2.21e−03 |
| 12 | 128–256 | YBR117C | LFDFTADGVASR | 2.19e+05 | 7 | 1.24e−04 |
| 13 | <128 | YNL014W | WVPMMSVDNAWLPR | 6.78e+04 | 9 | 2.12e−02 |
| 14 | Not expressed | YJR077C | QLGFFGSFAGLPTR | 9.44e+04 | 3 | 1.74e−04 |
| 15 | Not quantifiable by Western blot | YGR284C | IEGLTDNAVVYK | 7.80e+04 | 10 | 1.14e−02 |
a See Ghaemmaghami et al. (19).
b Sum of two primary ions.
These results were compared with a complementary experiment performed on a subset of 352 peptides that were analyzed using the conventional time-based SRM (t-SRM) method in which all eight transitions were monitored during the specified time window for simultaneous peptide verification and quantification. The number of peptides targeted in this second experiment was determined by the upper practical limit for simultaneously monitoring multiple transitions using t-SRM and maintaining an acceptable dwell time (i.e. over 2000 transitions for a single HPLC run). The results comparing the two methods clearly demonstrate the advantage of iSRM (see Table II) where the acquisition time is more effectively partitioned between true quantification traces and identity confirmatory measurements, thus yielding more data points for more accurate quantification. With the same 40-min-gradient HPLC separation and 4-min time window per peptide, the analytical precision using iSRM to target the same 352 yeast peptides was much improved compared with that of t-SRM as shown in Table II.
Table II. Comparison of iSRM and t-SRM acquisition methods.
Shown are the parameters used for the acquisition and the summary results obtained for 352 yeast peptides, thus illustrating the increased analytical precision. n/a, not applicable.
| Value | t-SRM | iSRM | |
|---|---|---|---|
| Parameters | |||
| Time window (s) | 240 | 240 | 240 |
| No. peptides in window (example) | 20 | ||
| Cycle timea (s) | 2.0 | 2.0 | |
| No. transitions monitored | 8b | n/a | |
| No. primary transitions | n/a | 2 | |
| Dwell time (ms) | 12 | 50 | |
| Actual quantification time (ms) | 480 | 2000 | |
| Additional transitions | n/a | 6 | |
| Additional acquisition time (ms) | n/a | 100 | |
| Acquisition timec (s) | n/a | 1.2 | |
| Time devoted to quantificationd (%) | 25 | 98 | |
| Resultse | |||
| CV > 5% | 143 (41%) | 268 (76%) | |
| CV > 10% | 289 (82%) | 340 (96%) | |
| CV > 15% | 335 (95%) | 352 (100%) | |
| CV > 20% | 352 (100%) |
a Acquisition was performed using a constant cycle time.
b The two most intense transitions were used for the quantification.
c Calculation took into account the acquisition of secondary transitions twice for each peptide in the time window.
d Fraction of time used for quantification in the 240-s time window.
e Quantification was performed on 352 peptides.
Application: TCA Cycle in Yeast
To illustrate the practical use of iSRM method in a biology context, the analysis was focused on the peptides associated with proteins involved in the yeast TCA cycle, also known as the citric acid cycle. The series of enzymes involved in the catalyzed chemical reactions is of central importance in S. cerevisiae and in general all living cells that use oxygen as part of cellular respiration. In yeast and other eukaryotes, the TCA cycle occurs in the matrix of the mitochondrion and is involved in the chemical conversion of carbohydrates, fatty acids, and proteins into carbon dioxide and water to generate energy in the form of ATP. In addition, the TCA cycle provides precursors for the synthesis of amino acids, nucleotides, and lipids and is affected by the growing conditions of the cells. Thus, it is important to establish a reliable and sensitive screening method to rapidly detect and quantify these metabolic enzymes from different cells in the context of systematic, large systems biology experiments (14).
The qualitative and qualitative results for these specific enzymes are summarized in Table III (for details, see supplemental Table 3). To demonstrate the ability of the method to systematically quantify all the peptides present using stable isotope dilution, the experiment was performed by mixing two different yeast digests: the first one was grown under standard conditions, whereas the second was grown in 15N-containing medium to produce uniformly 15N-labeled peptides as a global internal standard. Thus, all peptides will be present in two forms (light and heavy) at approximately equal ratios. The two yeast lysates were mixed in equal amount prior to trypsinization. The final concentration of the digested peptide mixture used for the LC-MS analysis was estimated around 1 μg/μl.
Table III. Quantitative results for the subset of yeast metabolic enzymes analyzed by iSRM.
| Peptide | Ratio 14N/15N pair | Integrated peak area |
CV | p value | |||||
|---|---|---|---|---|---|---|---|---|---|
| n = 1 | n = 2 | n = 3 | n = 4 | n = 5 | Average | ||||
| % | |||||||||
| Pyruvate kinase 1 | |||||||||
| EPVSDWTDDVEAR (14N) | 2.30 | 1.97e+05 | 1.87e+05 | 1.86e+05 | 1.89e+05 | 1.88e+05 | 1.89e+05 | 2.0 | 1.24e−04 |
| EPVSDWTDDVEAR (15N) | 8.80e+04 | 8.25e+04 | 8.77e+04 | 8.42e+04 | 7.79e+04 | 8.41e+04 | 4.0 | 7.19e−04 | |
| IENQQGVNNFDEILK (14N) | 2.20 | 2.48e+06 | 2.52e+06 | 2.37e+06 | 2.39e+06 | 2.33e+06 | 2.42e+06 | 3.0 | 6.03e−03 |
| IENQQGVNNFDEILK (15N) | 1.14e+06 | 1.12e+06 | 1.11e+06 | 1.07e+06 | 1.11e+06 | 1.11e+06 | 2.0 | 1.49e−03 | |
| Phosphoglucose isomerase | |||||||||
| LATELPAWSK (14N) | 2.40 | 6.83e+06 | 6.68e+06 | 6.64e+06 | 6.91e+06 | 6.64e+06 | 6.74e+06 | 2.0 | 7.61e−03 |
| LATELPAWSK (15N) | 2.85e+06 | 2.77e+06 | 2.73e+06 | 2.84e+06 | 2.72e+06 | 2.78e+06 | 2.0 | 1.39e−03 | |
| VVDPETTLFLIASK (14N) | 2.70 | 6.10e+05 | 6.09e+05 | 5.88e+05 | 6.04e+05 | 5.90e+05 | 6.00e+05 | 2.0 | 8.75e−02 |
| VVDPETTLFLIASK (15N) | 2.34e+05 | 2.23e+05 | 2.07e+05 | 2.24e+05 | 2.14e+05 | 2.20e+05 | 4.0 | 7.94e−04 | |
| Glucokinase | |||||||||
| GVLLAADLGGTNFR (14N) | 0.39 | 3.13e+05 | 2.95e+05 | 2.86e+05 | 2.94e+05 | 2.71e+05 | 2.92e+05 | 5.0 | 2.70e−03 |
| GVLLAADLGGTNFR (15N) | 7.69e+05 | 7.76e+05 | 7.47e+05 | 7.39e+05 | 7.29e+05 | 7.52e+05 | 2.0 | 2.70e−03 | |
| HALALSPLGAEGER (14N) | 0.37 | 6.75e+04 | 6.67e+04 | 6.22e+04 | 6.24e+04 | 6.03e+04 | 6.38e+04 | 4.0 | 5.36e−02 |
| HALALSPLGAEGER (15N) | 1.86e+05 | 1.75e+05 | 1.65e+05 | 1.79e+05 | 1.51e+05 | 1.71e+05 | 7.0 | 2.78e−03 | |
| 3-Phosphoglycerate kinase | |||||||||
| ASAPGSVILLENLR (14N) | 3.64 | 2.26e+07 | 2.12e+07 | 2.11e+07 | 2.06e+07 | 2.09e+07 | 2.13e+07 | 3.0 | 1.32e−02 |
| ASAPGSVILLENLR (15N) | 5.85e+06 | 5.99e+06 | 5.79e+06 | 6.08e+06 | 5.54e+06 | 5.85e+06 | 3.0 | 3.97e−03 | |
| TIVWNGPPGVFEFEK (14N) | 3.81 | 1.50e+07 | 1.49e+07 | 1.43e+07 | 1.44e+07 | 1.39e+07 | 1.45e+07 | 3.0 | 3.47e−04 |
| TIVWNGPPGVFEFEK (15N) | 3.94e+06 | 3.88e+06 | 3.76e+06 | 3.79e+06 | 3.65e+06 | 3.80e+06 | 3.0 | 2.78e−03 | |
| Aconitase | |||||||||
| GYDAGENTYQAPPADR 14N) | 0.32 | 1.80e+05 | 1.77e+05 | 1.74e+05 | 1.60e+05 | 1.72e+05 | 1.73e+05 | 4.0 | 1.49e−03 |
| GYDAGENTYQAPPADR (15N) | 5.85e+05 | 5.77e+05 | 5.79e+05 | 5.66e+05 | 5.32e+05 | 5.68e+05 | 3.0 | 7.50e−02 | |
| TIFTVTPGSEQIR (14N) | 0.33 | 1.15e+06 | 1.15e+06 | 1.09e+06 | 1.15e+06 | 1.08e+06 | 1.12e+06 | 3.0 | 5.95e−04 |
| TIFTVTPGSEQIR (15N) | 3.53e+06 | 3.52e+06 | 3.41e+06 | 3.48e+06 | 3.27e+06 | 3.44e+06 | 3.0 | 7.68e−02 | |
| Citrate synthase | |||||||||
| LVSTIYEVAPGVLTK (14N) | 0.12 | 2.95e+05 | 2.83e+05 | 2.64e+05 | 2.66e+05 | 2.37e+05 | 2.69e+05 | 7.0 | 2.48e−05 |
| LVSTIYEVAPGVLTK (15N) | 2.35e+06 | 2.19e+06 | 2.21e+06 | 2.12e+06 | 1.98e+06 | 2.17e+06 | 6.0 | 2.78e−03 | |
| YLWDTLNAGR (14N) | 0.12 | 1.69e+05 | 1.62e+05 | 1.63e+05 | 1.59e+05 | 1.47e+05 | 1.60e+05 | 5.0 | 3.13e−03 |
| YLWDTLNAGR (15N) | 1.34e+06 | 1.21e+06 | 1.29e+06 | 1.23e+06 | 1.27e+06 | 1.27e+06 | 4.0 | 4.17e−03 | |
| Alcohol dehydrogenase isoenzyme type 1 | |||||||||
| ANELLINVK (14N) | 2.40 | 1.68e+07 | 1.70e+07 | 1.67e+07 | 1.71e+07 | 1.76e+07 | 1.70e+07 | 2.0 | 3.83e−02 |
| ANELLINVK (15N) | 7.17e+06 | 7.04e+06 | 7.06e+06 | 7.12e+06 | 7.32e+06 | 7.14e+06 | 1.0 | 4.17e−02 | |
| VVGLSTLPEIYEK (14N) | 2.50 | 2.04e+07 | 2.11e+07 | 2.00e+07 | 2.05e+07 | 2.01e+07 | 2.04e+07 | 2.0 | 1.49e−03 |
| VVGLSTLPEIYEK (15N) | 8.96e+06 | 8.14e+06 | 8.12e+06 | 8.07e+06 | 8.19e+06 | 8.30e+06 | 4.0 | 5.95e−04 | |
| Cytoplasmic malate dehydrogenase | |||||||||
| EINIESGLTPR (14N) | 0.04 | 7.33e+04 | 7.60e+04 | 7.54e+04 | 6.48e+04 | 6.72e+04 | 7.13e+04 | 6.0 | 9.30e−02 |
| EINIESGLTPR (15N) | 1.89e+06 | 1.88e+06 | 1.83e+06 | 1.88e+06 | 1.75e+06 | 1.85e+06 | 3.0 | 2.78e−03 | |
| GVSYVDYDIVNR (14N) | 0.03 | 2.73e+04 | 2.86e+04 | 2.54e+04 | 2.10e+04 | 2.44e+04 | 2.53e+04 | 10.0 | 1.24e−04 |
| GVSYVDYDIVNR (15N) | 9.52e+05 | 9.27e+05 | 8.97e+05 | 9.29e+05 | 8.74e+05 | 9.16e+05 | 3.0 | 2.78e−03 | |
| α subunit succinyl-CoA ligase | |||||||||
| SGTLTYEAVQQTTK (14N) | 0.64 | 8.24e+04 | 8.23e+04 | 8.10e+04 | 7.85e+04 | 7.97e+04 | 8.08e+04 | 2.0 | 1.74e−03 |
| SGTLTYEAVQQTTK (15N) | 1.28e+05 | 1.29e+05 | 1.29e+05 | 1.23e+05 | 1.23e+05 | 1.26e+05 | 2.0 | 3.97e−03 | |
| VIFQGFTGK (14N) | 0.58 | 2.33e+05 | 2.29e+05 | 2.19e+05 | 2.35e+05 | 2.28e+05 | 2.29e+05 | 2.0 | 2.63e−02 |
| VIFQGFTGK (15N) | 3.99e+05 | 3.92e+05 | 3.79e+05 | 4.04e+05 | 3.91e+05 | 3.93e+05 | 2.0 | 1.01e−02 | |
| Aldehyde dehydrogenase | |||||||||
| ANFQGAITNR (14N) | 2.00 | 3.77e+06 | 4.14e+06 | 3.89e+06 | 3.72e+06 | 3.56e+06 | 3.82e+06 | 5.0 | 2.21e−03 |
| ANFQGAITNR (15N) | 1.76e+06 | 1.81e+06 | 1.76e+06 | 1.77e+06 | 1.64e+06 | 1.75e+06 | 3.0 | 9.72e−03 | |
| VGIPAGVVNIVPGPGR (14N) | 2.10 | 1.83e+06 | 1.76e+06 | 1.76e+06 | 1.82e+06 | 1.74e+06 | 1.78e+06 | 2.0 | 2.02e−03 |
| VGIPAGVVNIVPGPGR (15N) | 8.87e+05 | 8.68e+05 | 8.36e+05 | 8.68e+05 | 8.00e+05 | 8.52e+05 | 4.0 | 3.06e−02 | |
The results for a subset of 34 pairs of peptides (both 14N- and 15N-labeled) associated with 18 proteins involved in the TCA cycle (see supplemental Table 3) demonstrate the ability of the iSRM screening method to rapidly detect and quantify a specific subset of proteins. In a previous account, using a conventional SRM method to target these peptides, the three most intense fragment ions from the MRMAtlas database were used for quantification of each peptide (14). In addition, SRM-triggered MS/MS acquisitions were performed separately to verify the identity of targeted peptides and their retention times. By contrast, the iSRM method presented here simultaneously confirmed the peptide identities by composite MS/MS spectra and precisely quantified the verified peptides by integrating the LC-MS peak area of primary fragment ions. The same iSRM analysis was replicated five times to demonstrate reproducibility and assess the analytical precision. As a typical example, the results for protein Aco1p are presented in Fig. 4. The chromatographic traces of the two peptides, TIFTVTPGSEQIR and GYDAGENTYQAPPADR, derived from the protein Aco1p are shown in Fig. 4, B and C. The traces of both the 14N- and 15N-labeled peptides are perfectly co-eluting, and the ion intensity ratios are retained. The identity confirmation of the heavy and light forms relies on their composite MS/MS spectra displayed, and the consistency of the fragmentation patterns is illustrated in Fig. 4, D and E.
Fig. 4.

Application of iSRM to quantify protein of TCA cycle in yeast. A summary of the results obtained for the protein Aco1p is shown in A; the columns indicate five replicated analyses. B and C show the chromatographic traces of two peptides, TIFTVTPGSEQIR and GYDAGENTYQAPPADR. The composite MS/MS spectra of the peptide GYDAGENTYQAPPADR (for both light (top spectra) and heavy (lower spectra) forms are shown in D and E.
The quantitative results presented in Table III illustrate the degree of analytical precision that can be achieved with this method: over 85% of the analytes measured exhibit a CV below 10% in replicated experiments. The relative quantitative results indicate very consistent ratios for the different peptides associated with a given protein; variations between the ratios of different proteins reflect the biological variability in the cell growth conditions. This study demonstrates the capacity of iSRM to comprehensively and reproducibly measure sets of biologically related proteins in one LC-MS analysis with increased precision while improving the confidence in the measurements.
DISCUSSION
A novel data acquisition paradigm was developed and applied to increase the performance of targeted proteomics analyses using triple quadrupole instruments in the selected reaction monitoring mode. The results from the different proteomics experiments performed in this study clearly demonstrate the potential of this approach and its benefits. First, the iSRM technique offers clear advantages over the conventional targeted proteomics analyses by using a dual acquisition mode of SRM. The continuous monitoring of selected transitions within a defined time window yields quality chromatographic traces and hence reliable quantitative results with increased precision. The generation of composite tandem mass spectra acquired in a data-dependent manner allows the definitive confirmation of the analyte identity. The consistent quantitative and qualitative analyses are performed without compromising the performance of SRM in terms of sensitivity and selectivity, thus quantifying peptides in complex biological samples over a wide dynamic range with a limit of detection demonstrated down to the tens of attomole range while maintaining good analytical precision.
Second, the iSRM data acquisition scheme manages the concomitant analysis of a large number of peptides while retaining a degree of selectivity. It dramatically increases the throughput of the SRM assay, allowing a large targeted screen in a single HPLC-MS experiment. The largest assay we used in our experiments consisted of 6050 transitions for targeting 757 peptides. The iSRM instrument control software has the capability to support over 10,000 transitions, which is sufficient to target 1000 peptides in a single experiment.
To exploit fully the capability of the technique and to apply it routinely to the concurrent qualitative and quantitative analysis of large sets of peptides, an integrated work flow was developed. As illustrated in Fig. 2, there are three distinct steps: (i) the design of the analytical method exploiting pre-existing information, (ii) the actual data collection encompassing primary and secondary transitions, and (iii) the subsequent processing of the data and analysis of the results. The combination of the iSRM with the Pinpoint software package allows rapid design of large scale SRM screening experiments.
The design of the experimental method leverages existing peptide MS/MS information that is captured in public databases. The data are automatically downloaded and used to generate the acquisition method. If some peptides are not yet captured in the database, the necessary parameters (retention time and fragment ions) are theoretically predicted. The resulting instrument method is downloaded to perform the actual iSRM experiment that can include the analysis of multiple samples. The raw data collected are subsequently processed automatically in two streams. First, to confirm the identity of the analytes, the composite mass spectrum of each peptide is compared and scored using a local library created during the method design. Second, the quantification is performed using the sets of chromatographic traces of the different primary transitions corresponding to each peptide.
In conclusion, the linear work flow described and used throughout this study is likely to expedite large scale hypothesis-driven proteomics studies, generating reliable quantitative data sets, which are required by systems biology studies, or clinical biomarker validation. This improved technique that exploits specificity through a controlled (narrow) mass selection window, the versatility of the dual acquisition mode, the scheduling capabilities, and the capability of programming as 1000 peptides (i.e. >10,000 transitions) in a single LC-MS experiment open new avenues in targeted proteomics.
Acknowledgments
We thank Dr. Eric Deutsch, Institute for Systems Biology, Seattle, WA for assistance in generating methods using PeptideAtlas and MRMAtlas. We thank Drs. Nathalie Selevsek and Mariette Matondo, ETH, Zurich, Switzerland for helpful discussions.
Footnotes
 This article contains supplemental Tables 1–3.
This article contains supplemental Tables 1–3.
2 Prakash, A., Kiyonami, R., Schoen, A., Nguyen, H. Q., Peterman, S. M., Huhmer, A., Lopez, M. F., Domon, B., and Aebersold, R. (2009) Integrated workflow to design methods and analyze data in large scale SRM experiments, Poster TH695 presented at the 57th American Society for Mass Spectrometry annual conference, Philadelphia (May 31–June 4, 2009).
1 The abbreviations used are:
- SRM
- selected reaction monitoring
- iSRM
- intelligent selected reaction monitoring
- t-SRM
- time-based SRM
- CV
- coefficient of variation.
REFERENCES
- 1. Aebersold R., Mann M. (2003) Mass spectrometry-based proteomics. Nature 422, 198–207 [DOI] [PubMed] [Google Scholar]
- 2. Lin D., Tabb D. L., Yates J. R., 3rd (2003) Large-scale protein identification using mass spectrometry. Biochim. Biophys. Acta 1646, 1–10 [DOI] [PubMed] [Google Scholar]
- 3. Domon B., Aebersold R. (2006) Mass spectrometry and protein analysis. Science. 312, 212–217 [DOI] [PubMed] [Google Scholar]
- 4. Anderson L., Hunter C. L. (2006) Quantitative mass spectrometric multiple reaction monitoring assays for major plasma proteins. Mol. Cell. Proteomics 5, 573–588 [DOI] [PubMed] [Google Scholar]
- 5. Keshishian H., Addona T., Burgess M., Kuhn E., Carr S. A. (2007) Quantitative, multiplexed assays for low abundance proteins in plasma by targeted mass spectrometry and stable isotope dilution. Mol. Cell. Proteomics 6, 2212–2229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kuzyk M. A., Smith D., Yang J., Cross T. J., Jackson A. M., Hardie D. B., Anderson N. L., Borchers C. H. (2009) MRM-based, multiplexed, absolute quantitation of 45 proteins in human plasma. Mol. Cell. Proteomics 8, 1860–1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Picotti P., Rinner O., Stallmach R., Dautel F., Farrah T., Domon B., Wenschuh H., Aebersold R. (2010) High-throughput generation of selected reaction-monitoring assays for proteins and proteomes. Nat. Methods 7, 43–46 [DOI] [PubMed] [Google Scholar]
- 8. Picotti P., Lam H., Campbell D., Deutsch E. W., Mirzaei H., Ranish J., Domon B., Aebersold R. (2008) A database of mass spectrometric assays for the yeast proteome. Nat. Methods 5, 913–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gerber S. A., Rush J., Stemman O., Kirschner M. W., Gygi S. P. (2003) Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc. Natl. Acad. Sci. U.S.A. 100, 6940–6945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Anderson N. L., Anderson N. G., Haines L. R., Hardie D. B., Olafson R. W., Pearson T. W. (2004) Mass spectrometric quantitation of peptides and proteins using stable isotope standards and capture by anti-peptide antibodies (SISCAPA). J. Proteome Res. 3, 235–244 [DOI] [PubMed] [Google Scholar]
- 11. Unwin R. D., Griffiths J. R., Whetton A. D. (2009) A sensitive mass spectrometric method for hypothesis-driven detection of peptide post-translational modifications: multiple reaction monitoring-initiated detection and sequencing (MIDAS). Nat. Protoc. 4, 870–887 [DOI] [PubMed] [Google Scholar]
- 12. Prakash A., Tomazela D. M., Frewen B., Maclean B., Merrihew G., Peterman S., Maccoss M. J. (2009) Expediting the development of targeted SRM assays: using data from shotgun proteomics to automate method development. J. Proteome Res. 8, 2733–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sherwood C. A., Eastham A., Lee L. W., Risler J., Vitek O., Martin D. B. (2009) Correlation between y-type ions observed in ion trap and triple quadrupole mass spectrometers. J. Proteome Res. 8, 4243–4251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Picotti P., Bodenmiller B., Mueller L. N., Domon B., Aebersold R. (2009) Full dynamic range proteome analysis of S. cerevisiae by targeted proteomics. Cell 138, 795–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Krokhin O. V., Craig R., Spicer V., Ens W., Standing K. G., Beavis R. C., Wilkins J. A. (2004) An improved model for prediction of retention times of tryptic peptides in ion-pair reverse-phase HPLC: its application to protein peptide mapping by off-line HPLC-MALDI MS. Mol. Cell. Proteomics 3, 908–919 [DOI] [PubMed] [Google Scholar]
- 16. Pinto Da Costa J. F., Soares C. (2005) A weighted rank measure of correlation. Aust. N. Z. J. Stat. 47, 515–529 [Google Scholar]
- 17. Deutsch E. W., Eng J. K., Zhang H., King N. L., Nesvizhskii A. I., Lin B., Lee H., Yi E. C., Ossola R., Aebersold R. (2005) Human plasma PeptideAtlas. Proteomics 5, 3497–3500 [DOI] [PubMed] [Google Scholar]
- 18. Craig R., Cortens J. P., Beavis R. C. (2004) Open source system for analyzing, validating, and storing protein identification data. J. Proteome Res. 3, 1234–1242 [DOI] [PubMed] [Google Scholar]
- 19. Ghaemmaghami S., Huh W. K., Bower K., Howson R. W., Belle A., Dephoure N., O'Shea E. K., Weissman J. S. (2003) Global analysis of protein expression in yeast. Nature 425, 737–741 [DOI] [PubMed] [Google Scholar]