

Abstract
Approximately 170 million people worldwide are infected with hepatitis C virus (HCV). Current standard therapy leads to sustained viral elimination in only about 50% of patients treated. Telaprevir, a novel HCV protease inhibitor, has demonstrated substantial antiviral activity in patients with chronic HCV infection. However, drug-resistant variants emerge at frequencies of 5 to 20% of the total virus population as early as the second day after treatment initiation. Here, using probabilistic and viral dynamic models, we show that such rapid emergence of drug resistance is expected. We calculate that all possible single and double mutant viruses preexist before treatment, and that one additional mutation is expected to arise during therapy. Examining data from a clinical trial of telaprevir therapy for HCV infection in detail, we show that our model fits the observed dynamics of both drug-sensitive and -resistant viruses, and argue that combination therapy of direct antivirals will require drug combinations that have a genetic barrier of four or more mutations.
Introduction
Current therapy for HCV infection consists of pegylated interferon (IFN) and ribavirin (RBV) (1, 2). However, less than 50% of treated patients infected with HCV genotype 1 achieve sustained virologic response (SVR) or a cure of the infection (1, 2). Treatment options currently in development include drugs that target the HCV-encoded NS3-4A serine protease and the NS5B RNA-dependent RNA polymerase (RdRp) (3, 4). These drugs have been evaluated in early-phase clinical trials alone and in combination with pegylated IFN and/or RBV (5, 6).
Several protease inhibitors appear to be effective in suppressing viral loads in the early stage of treatment (7-9). One agent in this group, telaprevir, has been well studied (10-12). In a phase I trial, 750 mg telaprevir given orally every 8 hours induced a large decline (median of 4.4 log10 IU/mL) in plasma HCV RNA concentrations in patients infected with genotype 1 HCV after 2 weeks of treatment (10). Some patients, however, experienced viral breakthrough during treatment that was associated with selection of HCV variants with decreased susceptibility to telaprevir (10). Amino acid substitutions in the HCV NS3-4A protease catalytic domain conferred different levels of drug resistance to telaprevir (13). Selection of these resistance substitutions was further confirmed in a subsequent kinetic analysis of HCV variants in patients treated with telaprevir alone or telaprevir plus PEG-IFN-α-2a for 14 days (11). The four genotype 1a patients treated with telaprevir alone had viral breakthrough during therapy (14) (Fig. S1). Virus isolated from these patients 2 days after the initiation of treatment contained drug-resistant variants with single-nucleotide mutations at a frequency of 5 to 20% of the total virus population, which increased in frequency at days 6 and 10, and most were replaced by high-level resistant double-nucleotide variants by day 13 (11). The appearance of these HCV variants at high frequencies such a short time after the start of therapy was not expected, especially since such rapid phenotypic drug resistance has not been seen with monotherapy for human immunodeficiency virus (HIV), hepatitis B virus (HBV), or any other tested pathogen (15).
Here we analyze the emergence of drug-resistant HCV variants in patients treated with telaprevir, using published data (11) to develop a model to inform future treatment paradigms. By calculating the generation rates of HCV variants, we show that the preexistence and selection of drug-resistant variants is expected and estimate the number of substitutions a combination of direct antivirals would need to overcome to be successful. We also develop a model to examine the dynamics of telaprevir-resistant virus after drug administration and show that the model fits patient data well.
Results
Preexistence of drug-resistant variants in HCV patients: an inevitable consequence of HCV biology
A large number of HCV virions (on the order of 1012) are produced each day in an infected, untreated patient (16). Each HCV RNA molecule is made by the NS5B RdRp, which has an error rate (μ) estimated to be 10-5 to 10-4 per copied nucleotide (17, 18). The entire HCV genome has approximately 9600 nucleotides. If we assume μ =10−5 per copied nucleotide, the average number of changes per genome is 0.096 per replication cycle. In generating a new virion, at least two rounds of replication are needed (positive strand to negative strand and negative strand to positive strand). We use the single round mutation rate, which is conservative, to estimate the probabilities of generation of HCV variants. According to the binomial distribution or its Poisson approximation, if a person is infected with wild-type virus that is fully sensitive to a given drug, when a new virion is generated it has a probability of 91% to carry an unmutated genome, 8.7% to carry one substitution, 0.42% to carry two substitutions, 0.013% to carry three substitutions, and so on (see Materials and Methods and Table 1).
Table 1.
Probabilities and rates of generation of various HCV mutants.
| Time | Number of nucleotide changes | Probability | Number of virions generated per day | Number of all possible mutants | Fraction of all possible mutants created per day |
|---|---|---|---|---|---|
| Before therapy | 0 | 0.91 | 9.1×1011 | ||
| 1 | 0.087 | 8.7×1010 | 2.9×104 | 1 | |
| 2 | 0.0042 | 4.2×109 | 4.1×108 | 1 | |
| 3 | 0.00013 | 1.3×108 | 4.0×1012 | 3.4×10-5 | |
| End of first day of therapy* | 0 | 0.91 | 9.1×106 | ||
| 1 | 0.087 | 8.7×105 | 2.9×104 | 1 | |
| 2 | 0.0042 | 4.2×104 | 4.1×108 | 1.0×10-4 | |
| 3 | 0.00013 | 1.3×103 | 4.0×1012 | 3.4×10-10 |
Additional drug-resistant or compensatory mutation after a 5-log10 decrease in the HCV RNA production during treatment.
Thus, of the 1012 virions made per day on average 8.7 × 1010 and 4.2×109 mutants would be generated with single- and double-nucleotide changes, respectively. Since the total number of possible single and double mutants is 2.9×104 and 4.1×108, respectively, all possible single and double mutants are predicted to be generated multiple times each day (Table 1). Because virus is cleared with a half-life of about 3 hours (16), variants generated more than 8 times a day are likely to be constantly present. Many of these might not be observed because they are lethal or confer reduced fitness and are eliminated (19). Because a single-nucleotide change or a number of substitution combinations may be associated with resistance (13), these calculations would predict that all viable single and double mutants that confer drug resistance preexist and may compete with the wild-type virus during therapy.
Only a small fraction (3.4×10-5) of all possible triple mutants are generated each day. Thus, it is unlikely that any particular three-nucleotide mutant arises spontaneously. However, such mutants can be selected by sequential mutations when single or double mutants replicate. In fact, even if therapy is extremely potent and can induce a 5-log10 decrease in HCV RNA after the first day of treatment (that is, decrease the number of total virions produced per day from 1012 to 107), all additional one-nucleotide mutations would be generated during therapy (Table 1), potentially generating drug-resistant variants or compensatory substitutions that improve existing resistant virus fitness.
Whether these generated variants grow during therapy depends on their fitness relative to other preexisting virions. We have studied their dynamics with a model Eq. (1) in Materials and Methods and its schematic representation in Fig. 1] and examined the prevalence of drug-resistant variants before therapy and their development after treatment.
Figure 1.
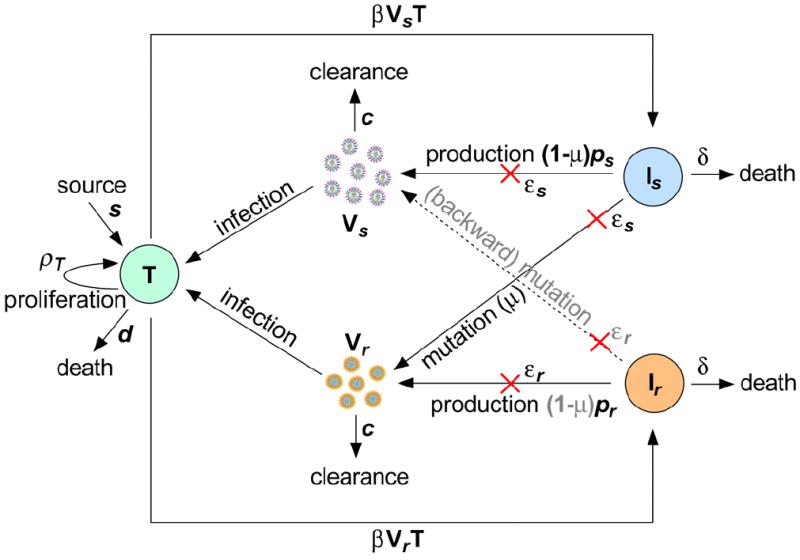
Schematic representation of the viral dynamic model. There are five variables: target cells (T), drug-sensitive virus (Vs), drug-resistant virus (Vr), cells infected with drug-sensitive virus (Is), and cells infected with resistant virus (Ir). s, ρT and d are the recruitment rate, maximum proliferation rate and death rate of target cells, respectively; β is the infection rate of target cells by virus; δ is the death rate of infected cells; and ps and pr are the viral production rates of the two strains; εs and εr are the drug efficacies of telaprevir in reducing viral production; μ is the mutation rate from the drug-sensitive to drug-resistant strain; and c is the viral clearance rate. The red crosses represent the effect of treatment in blocking viral production.
Coexistence of quasispecies variants before therapy
Before therapy, both drug-sensitive (wild-type) virus and mutant variants resistant to a given class of drugs are expected to coexist at steady state concentrations (14). The pretreatment frequency of single-mutants is determined by mutation-selection balance and is given by Γ=μ/(1−r), where μ is the mutation rate and r = Rr / Rs, with Rr and Rs being the basic reproductive ratios of drug-resistant and drug-sensitive strains, respectively (14). Further, the frequency of preexisting i-mutants (mutants with i substitutions) is proportional to μi (20). When Rr is greater than 1, we expect drug-resistant virus will grow and compete with wild-type virus for target cells (hepatocytes at risk for HCV infection). The ratio r represents the relative fitness of drug-resistant to drug-sensitive virions in the absence of treatment, and we expect r < 1 because of resistance-associated loss of fitness (13). Thus, although both drug-sensitive and -resistant virions coexist before therapy, the frequency of drug-resistant virus is low because μ ≪ 1 (17), in agreement with the results in (21).
Rapid emergence of drug resistance after treatment initiation
We first assumed that the number of target cells remained at its baseline value for a few days after drug administration (16, 22). Furthermore, we ignored generation of new drug-resistant mutants during therapy [the term μ(1−εs)psIs in Eq. (1)] because the mutation rate μ is small and telaprevir is very effective in shutting off production of drug-sensitive virus (εs is close to 1) (11, 23). Using constant drug efficacy for each strain (Materials and Methods), we solved the simplified system for the drug-sensitive and -resistant viral loads, and calculated the proportion of mutant virus in the total virus population over time. We considered that different single mutations confer different levels of resistance to telaprevir (13). For example, the variant V36A/M (Fig. 2A,B) confers ~3.5-fold resistance, whereas A156V/T (Fig. 2C,D) confers ~466-fold resistance to telaprevir (13). According to our model, in both cases, the frequency of the mutant virus underwent a substantial increase from the preexisting undetectable low value (<1%) to > 5% within days of therapy initiation (Fig. 2A,C). This is in agreement with the experimental observation that single mutants detected at day 2, such as V36A/M and A156V/T, account for 5 to 20% of the virus population (11) (Fig. S1). Such a rapid and dramatic increase of the mutant frequency, however, does not necessarily imply that drug-resistant HCV variants grow rapidly during treatment. In fact, further investigation of the model showed that during the period in which the number of target cells remained relatively constant, both drug-sensitive and -resistant virions undergo a two-phase decline (Materials and Methods). The duration of the first-phase decline of drug-sensitive virus (ts) is always longer than that of drug-resistant virus (tr) (Fig. 2B,D) (14). It is this increased loss of drug-sensitive virus that causes the rapid increase in the mutant frequency after treatment initiation. Moreover, the higher the fold-increase in resistance of the variants, the shorter the first-phase decline of drug-resistant virus (Fig. 2B,D).
Figure 2.
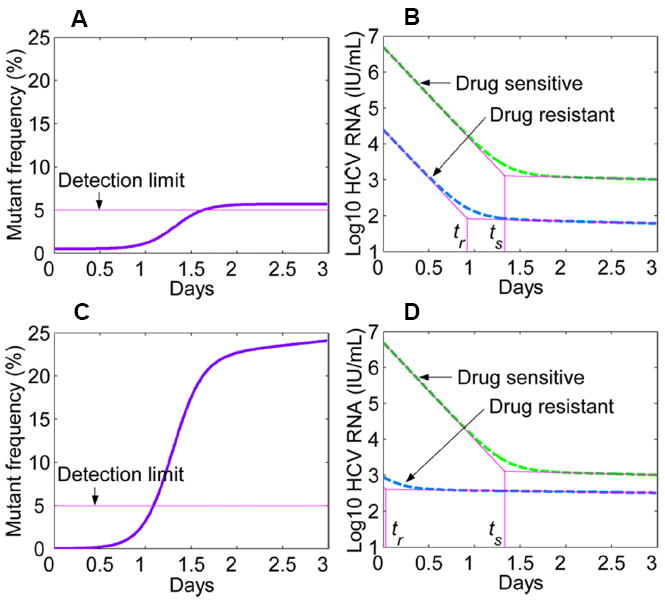
Model predictions of the mutant frequency and viral load decay profiles after drug administration. (A) Predicted frequency of the mutant virus (V36A/M) that induces ~3.5-fold increase in IC50 from wild-type and has a relative fitness of r ≈ 0.98 (13). (B) The two-phase decrease of both drug-sensitive virus (green dashed) and resistant V36A/M (blue dashed) after drug dosing. ts and tr represent the time at which drug-sensitive and drug-resistant virions start the second-phase decline, respectively, and tr is always smaller than ts (14). (C) Predicted frequency of the mutant virus (A156V/T) that induces ~466-fold increase in IC50 and has a relative fitness of r ≈ 0.45 (13). (D) As for B, except with the variant A156V/T. Parameters used are c = 6.2 day-1 (16), δ = 0.14 day-1 (16), μ = 10−4 per copied nucleotide (17), εs = 0.9997 (23), and the Hill coefficient h = 2 (64).
Comparison of model predictions with viral kinetic data
Our model predicts that there will be some suppression of preexisting mutant virus even if it is very drug-resistant (Fig. 2D). This behavior is due to the assumption of a constant target cell number, which may not remain valid at longer times after drug administration. If we remove this assumption and describe the dynamics of target cells as in Eq. (1) in Material and Methods, then drug-resistant virus may grow and ultimately dominate the virus population under certain conditions. The reproductive ratios of the two strains under treatment are and . Because telaprevir is very effective against wild-type virus, usually becomes less than 1 during treatment and wild-type virus is successfully suppressed. Variants with low-level drug resistance to telaprevir will also be suppressed. However, for variants with high-level drug resistance (for which εr is small), may be greater than 1 and the preexisting drug-resistant virus will outcompete wild-type virus.
We have applied Eq. (1) to an analysis of the experimental data from the four patients who had viral breakthrough during the 14-day telaprevir monotherapy (see data fitting in Materials and Methods). The model provides excellent fits (Fig. 3) for both the drug-sensitive and - resistant viral kinetics (Fig. S1). Parameter estimates based on the fits are listed in Table 2. The estimated efficacy of telaprevir against drug-sensitive virus is 0.9986 ± 0.0021, which confirms the previous estimate of telaprevir effectiveness, 0.9997 (23); whereas the estimated efficacy against telaprevir-resistant virus is 0.011±0.017, supporting the notion that these HCV variants have significantly reduced susceptibility to telaprevir.
Figure 3.
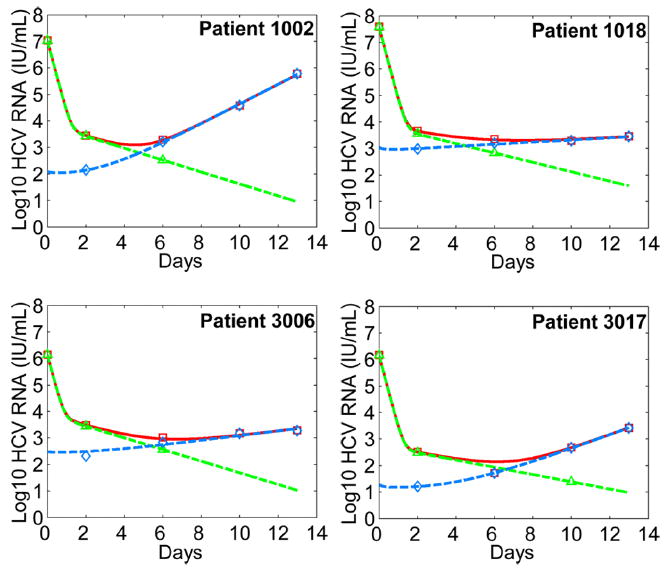
Comparison between model predictions and patient data during telaprevir monotherapy. We employed the pretreatment steady-state values (14) as the initial conditions of the model. We fitted Vs (green dashed) and Vr (blue dashed) in Eq. (1) to the drug-sensitive (green triangle) and drug-resistant (blue diamond) viral load data simultaneously, where Vr is the sum of viral loads of all drug-resistant strains. Since we ignored the drug-sensitive viral load data when they were below the detection limit of the sequencing assay (< 5%) (11), we included fitting Vs + Vr (red solid) to the total viral load data (red square). The best-fit parameter values for each patient are listed in Table 2. Note here day 0 is the time of initiation of telaprevir therapy, whereas in the original study telaprevir therapy was started at day 2 (11, 12).
Table 2.
Best-fit parameter values obtained from comparisons of model predictions [Eq. (1)] with data from the HCV genotype 1a infected patients who were given telaprevir alone.
| Patient | ρT (day-1) | δ (day-1) | μ (10-6) | εs | εr | β (10-8 mL day-1 virion-1) | ps (virions cell-1 day-1) | r | Infected cells at baseline* (%) | Increase in total hepatocytes (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1002 | 1.91 | 0.52 | 2.68 | 0.99943 | 0.001 | 4.30 | 44.36 | 0.80 | 16 | 32 |
| 1018 | 2.38 | 0.41 | 13.91 | 0.99982 | 0.036 | 0.58 | 131.08 | 0.70 | 16 | 11 |
| 3006 | 2.50 | 0.50 | 5.98 | 0.99548 | 0.003 | 11.21 | 6.60 | 0.97 | 11 | 7 |
| 3017 | 1.21 | 0.32 | 1.99 | 0.99965 | 0.002 | 19.41 | 6.17 | 0.84 | 16 | 32 |
| Average±SD | 2.00±0.59 | 0.44±0.09 | 6.14±5.46 | 0.99860±0.00210 | 0.011±0.017 | 8.88±8.29 | 47.05±58.81 | 0.83±0.11 | 15±2.5 | 21±13 |
During data fitting, we assumed that half the maximum number of hepatocytes are not targets of HCV infection (N=Tmax/2). With this assumption, about 15% of hepatocytes were infected before treatment, consistent with the experimental results in (25, 26). Using a smaller N, e.g., N=Tmax/4, we will have a higher percentage of infected cells at baseline and an unrealistic increase in the total number of hepatocytes after 14-day treatment. Using a larger N, e.g., N=3Tmax/4, we could not obtain good fits because of limited replication space for drug-resistant virus.
Although our model fits the patient data very well, we looked at additional predictions that provide further opportunities to disprove the model. From Table 2, we observed that the estimates of the productively infected cell death rate, δ, are higher than previous estimates under daily IFN therapy (16), as has been reported by another group for telaprevir therapy (24). We calculated the percentage of infected hepatocytes at baseline. About 15% of the hepatocytes are predicted to be infected before therapy (Table 2), in agreement with experimental measurements in previous studies (25, 26). After the two-week treatment, the total number of hepatocytes [T + Is + Ir + N in Eq. (1)] is predicted to increase by 21±13% (Table 2), and considering that about 30 to 35% of cells in an uninfected liver are non-parenchymal (27), the total number of liver cells is predicted to increase by 14 to 15%. This is mainly due to the increase in the number of uninfected target cells (Table S1), which suggests that during effective therapy uninfected cells replace infected cells. These predictions remain to be experimentally confirmed.
We have also examined the effect of combination PEG-IFN-α-2a and telaprevir. The model [see (14)] provided excellent fits (Fig. 4) to the viral load data (Fig. S2) from eight patients who received combination therapy (11). Parameter estimates are listed in Table 3. For patient 3011 there appears to be a time period in which HCV RNA declined slowly or remained constant. Such a triphasic decline can be explained by incorporating proliferation of infected cells into our model [see (14) and Fig. S3]. With combination therapy, the estimated total drug efficacy against telaprevir-resistant variants was 0.82 ± 0.11(Table 3), significantly greater than (P=0.006) that of telaprevir alone (0.011±0.017) (Table 2). Thus, HCV variants with reduced susceptibility to telaprevir still remain sensitive to pegylated IFN.
Figure 4.
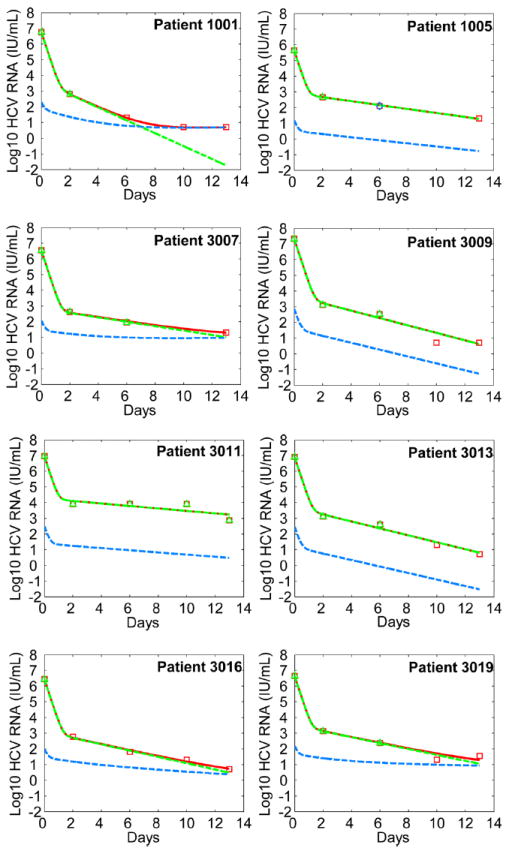
Comparison between model predictions and patient data during combination therapy. We fitted the model Eq. (S1) in (14)] to the viral load data from patients receiving both PEG-IFN-α-2a and telaprevir for 14 days. This model generalizes Eq. (1) by incorporating an effect of IFN in partially blocking viral production. The fitting procedure and the symbols used are the same as those in Fig. 3. The best-fit parameter values for each patient are listed in Table 3. Note that the two fits of the drug-sensitive (green dashed) and total viral load (red solid) overlap in a few patients.
Table 3.
Best-fit parameter values obtained from comparisons of model predictions Eq. (S1) in (14)] with data from patients treated with telaprevir plus PEG-IFN-α-2a for two weeks.
| Patient | ρT (day-1) | δ (day-1) | β (10-8 mL day-1 virion-1) | ps (virions cell-1 day-1) | Infected cells at baseline* (%) | Increase in total hepatocytes (%) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1001 | 0.83 | 0.93 | 0.99951 | 0.68 | 4.77 | 80.07 | 5.4 | 49 | ||
| 1005 | 0.31 | 0.29 | 0.99832 | 0.82 | 7.27 | 5.90 | 4.2 | 8 | ||
| 3007 | 1.12 | 0.33 | 0.99984 | 0.77 | 8.34 | 18.47 | 14 | 40 | ||
| 3009 | 1.76 | 0.55 | 0.99980 | 0.95 | 0.57 | 132.52 | 8.3 | 8 | ||
| 3011 | 0.92 | 0.18 | 0.99810 | 0.91 | 0.76 | 38.36 | 12 | 7 | ||
| 3013 | 2.20 | 0.51 | 0.99950 | 0.95 | 1.27 | 52.17 | 8.0 | 5 | ||
| 3016 | 1.24 | 0.46 | 0.99961 | 0.70 | 9.63 | 13.31 | 13 | 28 | ||
| 3019 | 1.65 | 0.44 | 0.99937 | 0.78 | 11.29 | 20.57 | 15 | 43 | ||
| Average±SD | 1.25±0.60 | 0. 46±0.23 | 0.99930±0.00067 | 0.82±0.11 | 5.49±4.26 | 45.17±42.79 | 10±4.1 | 24±19 |
We assumed that half the maximum number of hepatocytes are not targets of HCV infection during data fitting. Similar sensitivity analysis of data fitting to the choice of N was addressed in the remarks of Table 2.
Discussion
The NS3-4A protease of HCV is essential for generating components of the viral RNA replication complex (28) and is a popular drug target (3). Telaprevir and boceprevir are protease inhibitors that have shown promise in clinical trials (6). For both agents, however, HCV variants emerge rapidly when monotherapy is utilized (11, 29). A number of variants with mutations confer resistance to protease inhibitors (30) particularly for genotype 1a HCV (31).
The selection speed of drug resistance in HCV is strikingly faster than that seen in HIV or HBV. We calculated in untreated patients the numbers of various HCV variants that would be produced per day and their fraction of all possible mutants. The genome sizes are similar for HCV and HIV. The fidelity of reverse transcriptase for HIV is higher than that of RdRp for HCV (19), and thus the intrinsic HCV mutation rate may be higher than that of HIV. To be conservative we used an HCV mutation rate of 10-5 per copied base, although the rate may be as much as 10-fold higher (19). Still, we calculated that all possible single and double HCV mutants can be generated each day, while only ~0.74% of all possible double HIV mutants are generated daily (32). We can also calculate the average waiting time before a specific mutant is generated (14). Because mutations in HCV can occur each time a viral RNA is copied by the RdRp, while in HIV most mutations occur during reverse transcription, drug-resistant variants with changes at particular nucleotides are generated more rapidly in HCV than in HIV and hence have a higher probability of preexisting. This can explain the faster appearance of drug resistance in HCV patients than in HIV patients during treatment.
The speed of drug resistance selection can also be affected by the turnover of the viral nucleic acid that serves as a source of new viral genomes (15). The viral genetic material of HBV (covalently closed circular DNA) can persist as long as the lifespan of infected hepatocytes [several weeks to months (33, 34)]. This also applies to the proviral DNA of HIV integrated into the chromosomes of infected CD4+ T cells [with a half life of ~1 day in most productively infected cells (35), weeks to months in long-lived infected cells (36), and 6 months or more in latently infected cells (37, 38)]. In contrast, there is no such genetic reservoir for HCV. The HCV RNA strands, with a half life on the order of 11 to 19 hours (39, 40), act as templates for generating new HCV virions. In addition, unlike HBV and HIV, the HCV genome does not use overlapping reading frames. Thus, changes at one position do not affect the structure/function of other viral proteins, resulting in a lower frequency of deleterious mutations in HCV.
We developed a viral dynamic model to examine the change in the frequency of resistant mutants after treatment initiation. The model is different from those used in HIV in that hepatocyte regeneration is included, which provides the replication space needed for mutant virus expansion. Assuming that the number of target cells remains at the baseline value over a short time period after treatment, we found that the mutant frequency undergoes a substantial increase within a few days of drug administration, consistent with the results in clinical studies (10, 11). This observation may simply be a consequence of a rapid and profound decline of wild-type virus (Fig. 2B,D), unveiling preexisting HCV variants (11). Over a longer time interval, the tradeoff between the reduced susceptibility to protease inhibitors and resistance-associated fitness loss determines whether HCV mutants can dominate the virus population.
The model can also be employed to investigate the development of drug resistance during treatment with polymerase inhibitors. The HCV NS5B RdRp is crucial for viral RNA synthesis (28) and is another attractive drug target (3). Many polymerase inhibitors have been developed, including the non-nucleoside inhibitors HCV-796, VCH-759, and nucleoside inhibitors NM283 (prodrug of NM107), R1626 (prodrug of R1479) and R7128 (prodrug of PSI-6130) (6). Drug-resistant variants emerged as rapidly in HCV-796 (41) or VCH-759-treated patients (42) as in those treated with telaprevir (11) or boceprevir (29). However, there was no evidence of drug resistance with the nucleoside polymerase inhibitors NM283 (43), R1626 (44), and R7128 (45). Lack of resistance to these nucleoside inhibitors can be explained by the fitness disadvantage they induce. Dynamics of the mutant virus are determined by its fitness during therapy [ in (14)], which depends, among other parameters, on the drug sensitivity (εr) and viral production capacity (pr). The active site of the NS5B polymerase is highly conserved (4) and thus any mutations in this region may inhibit the ability of the virus to replicate. In fact, although many NS5B mutations, such as S282T for PSI-6130 and S96T or S96T/N142T for R1479, confer a 3- to 5-fold loss in sensitivity to the nucleoside polymerase inhibitors in the replicon system, they also result in a > 85% reduction in replication capacity (corresponding to a significantly reduced pr) (46). If this is also true in vivo, then these viral strains would not have enough fitness advantage over wild-type, even in the presence of therapeutic agents, to be selected from the preexisting quasispecies. In contrast, telaprevir and HCV-796 resistance generally incurs a lower replication capacity cost (46), which gives the variants a relatively higher fitness and thus makes them easier to be selected during treatment. Indeed, nucleoside polymerase inhibitors seem to have a higher genetic barrier (the number of mutations required to overcome drug-selective pressure) to resistance than either protease or non-nucleoside polymerase inhibitors in the replicon system (46), which highlights the clinical importance of nucleoside polymerase inhibitors in future HCV therapies. The fitness disadvantage induced by resistance mutations may also explain the hierarchy in the emergence of detectable drug resistant variants during HIV treatment, with less costly mutants appearing first. For example, resistant variants to non-nucleoside reverse transcriptase (RT) inhibitors such as nevirapine are very likely to exist prior to therapy (47), and can be detected as early as 1 week after treatment initiation (48). However, when the nucleoside RT inhibitor lamivudine is used as monotherapy for HIV or HBV, drug resistance develops after 2 to 3 weeks (49) or after several months to years of therapy (50), respectively.
Four genotype 1b patients receiving telaprevir monotherapy had a continuous decline in HCV RNA throughout the 14-day therapy (11). One explanation could be that for genotype 1b two nucleotide changes are required to create the V36M (GTC to ATG) (51) and the R155K (CGG to AAG) variants (52). In contrast, for genotype 1a, only one nucleotide change is required to created the V36M (GTG to ATG) or R155K (AGG to AAG) mutation (51, 52). Thus, the probability of generating the same amino acid change for genotype 1b (10-8 to 10-10) is much lower than that for 1a (10-4 to 10-5). Also the frequency of variants with two-nucleotide changes at baseline is very low (on the order of μ2), and the one-nucleotide mutant intermediates should have no fitness advantage in the presence of drug, which together may explain why no or few resistant variants were observed in genotype 1b patients during 14-day monotherapy with telaprevir (11).
The rapid appearance of HCV drug resistance suggests that treatment failure with monotherapy using a protease inhibitor or a non-nucleoside polymerase inhibitor is likely inevitable, especially in genotype 1a infection. The lessons from HIV infection treatment indicate that combinations of drugs with different mechanisms of action can be an attractive strategy (4). Data from in vitro studies (46, 53), animal models (54), early-stage clinical trials (55), and the present modeling efforts suggest that combinations of direct antivirals should also be beneficial to HCV patients. Extension of Eq. (1) to analyze data from patients on therapy with telaprevir and pegylated IFN also supports the utility of combination therapy of direct antivirals with IFN for HCV, in agreement with recent clinical trials (29, 56, 57). Based on our estimation that all viable single and double mutants preexist before treatment and that one additional mutation is expected to arise during therapy, we predict that combination therapy of direct antivirals will require drug combinations with a genetic barrier of four or more mutations. The number of drugs needed in a combination need not be four and would depend on the number of mutations required to generate high-level resistance to each component of the drug combination. Some drug-resistant variants may be non-viable, replication-deficient or immunogenic variants rapidly eliminated by the immune system. Thus in some circumstances the genetic barrier could be lower. Further, other factors, such as the baseline viral load before treatment (58), infection with genotype 1b HCV (11), genetic variation in IL28B (59, 60) and differences in the potency of drugs chosen for a treatment regime (11, 61), can also affect viral suppression and the development of drug resistance.
Pegylated IFN and RBV can be used in a lead-in period before treatment with direct antivirals with the goal of reducing the frequency of preexisting direct antiviral-resistant variants (62, 63). Our calculations can be used to make predictions about the possible success of lead-in strategies. For example, a patient who responds well to the lead-in treatment and in whom viral load drops 3 logs from 107 at baseline to 104 RNA IU/mL is expected to produce approximately 109 virions per day at the start of therapy with direct antivirals (16). Extrapolating from Table 1, on average 8.7×107 and 4.2×106 mutants would be generated with single- and double-nucleotide changes, respectively, suggesting the preexistence of all possible viable single mutants and only 1% of the double mutants after the lead-in phase. Thus, for this patient, a combination of two potent direct antivirals with different resistance patterns, such as a protease and polymerase inhibitor, might be able to suppress drug resistance. (However, if we assume that the mutation rate of the HCV RdRp is 10-4 per base copied rather than 10-5, then over 40% of all possible double mutants would still be generated daily at the end of the lead-in phase, and therapy with a higher genetic barrier for resistance development would be required.) For patients who do not respond as well, e.g. only have a 1-log drop during lead-in, all possible single and double mutants would still be generated daily and thus lead-in would provide little or no benefit. Thus, lead-in may need to be followed by individualized treatment with direct antivirals, an idea that remains to be tested in clinical trials.
Overall, this study predicts that rapid emergence of HCV protease inhibitor resistance in patients, particularly those with genotype 1a infection and with high viral loads, is expected. Combination therapies of direct antivirals with/without IFN+/-RBV would be promising to combat drug resistance. However, as with HIV, they need to be chosen carefully and with regard to both preexisting and on-treatment generated drug-resistant variants.
Unlike HIV and HBV, HCV is a curable disease and clinical studies will eventually show how many patients can achieve SVR and how many direct antivirals are needed. In addition, the antiviral activity of the direct acting agents used, the genetic barrier to resistance of the single compounds, HCV genotype, baseline viral load, host genetic factors, and the duration of therapy will all play important roles in determining whether cure will be achieved.
Materials and Methods
Probability of generation of HCV mutants
Assuming that the HCV genome is L nucleotide long and that the RNA polymerase introduces errors into the genome at rate μ, the probability of having i substitutions (i-mutant) in one replication event is . Because each of the L nucleotides in HCV can mutate to any of 3 other nucleotides, there are a total of possible sequences with i substitutions.
Modeling the development of HCV protease inhibitor resistance
We investigate the development of drug resistance during telaprevir therapy using a model (Fig. 1), which is described by the following equations:
| (1) |
Drug-sensitive and drug-resistant HCV virions, and Vs and Vr, infect target cells, T, to create productively infected cells, and Is and Ir, at rates βVsT and βVrT, respectively. Target cells are generated at rate s, die at rate d, and can proliferate with maximum proliferation rate ρT. Tmax is the hepatocyte carrying capacity of the liver. Due to the effect of IFN, lack of receptors needed for viral entry, or other biological heterogeneity, some hepatocytes (N) may not be targets (28). For simplicity we assume N is constant. Effectively, ρT(1 − N/Tmax) is the maximum proliferation rate of target cells and Tmax − N is the target cell carrying capacity. Is and Ir are lost at per capita rate, δ, and are assumed to produce virions at rates ps and pr, respectively, which are then cleared with rate constant c. The efficacy of telaprevir in reducing viral production from cells infected with drug-sensitive and -resistant virus is εs and εr, respectively, where 0 ≤ εs, εr ≤ 1 with 0 corresponding to no treatment effect and 1 to 100% effective drug. We assume that Is with probability μ generates drug-resistant virus. Backward mutation is neglected because wild-type virus dominates the population before therapy. We also neglect proliferation of infected cells since we estimate their proliferation rate is small based on data fitting to a model with proliferation of both uninfected and infected hepatocytes (14).
Drug efficacy against each strain
The effectiveness of a drug can be related to its concentration, C(t), by εs (t) = C(t)h /[IC50h + C(t)h], where IC50 is the drug concentration needed to inhibit viral production by 50%, and h is the Hill coefficient representing the steepness of the concentration-effect curve (64). Based on the pharmacodynamics of telaprevir, the median εs exceeds 0.999 (23), which is consistent with the 3 to 4 log10 first-phase drop of plasma HCV RNA levels in patients undergoing telaprevir monotherapy (10). If mutation is associated with an n-fold increase of IC50, then the effectiveness of drug against the resistant strain is εr (t) = C(t)h /[(n · IC50)h + C(t)h] = εs (t)/[εs(t) + (1 − εs (t))nh]. Because telaprevir is dosed frequently, as has been done previously (24), we assume that εs and εr are constants.
Two-phase viral decay and the mutant frequency during monotherapy
Assuming the target cell concentration remains at the baseline value, cδ/[(1 − μ)βps], for a few days after treatment initiation, and ignoring μ(1 − εs)psIs in the Vr equation of Eq. (1), we solved the simplified system and obtained Vs (t) = C1eλ1t + C2eλ2t, Vr(t) = C3eλ3t + C4eλ4t, where Ci, i = 1,2,3,4, are positive constants and λi, i = 1,2,3,4, are the eigenvalues and are all negative. Thus, both drug-sensitive and drug-resistant viral loads undergo a two-phase decline after treatment initiation. The mutant frequency after drug administration is Γ(t) = Vr (t)/[Vs (t) + Vr (t)], which depends on the parameters c, δ, μ, r, εs, εr, and the time t.
Data fitting
We fitted Eq. (1) to experimental data (Fig. S1) from patients who received telaprevir alone and had viral breakthrough during the 14-day treatment (Fig. 3). Using Berkeley-Madonna, Version 8.3.9 (http://www.berkeleymadonna.com), Vs and Vr were fitted to the drug-sensitive and drug-resistant viral load data simultaneously. Due to lack of data on target cells, we fixed d, Tmax and s for all patients. The half-life of uninfected target cells was estimated to be a few hundred days in animal studies (65). Because of the uncertainty and possible changes in humans, we chose d to 0.01 day-1 (assume the half-life of uninfected hepatocytes is ~70 days). Supposing that there are maximally 2×1011 hepatocytes in a normal human liver (66) and that HCV can distribute throughout the 15 liters of extracellular fluid in a 70 kg person, we set Tmax = 2×1011 /15000=1.3×107 cells/mL. Some hepatocytes may not be targets of HCV infection. The proportion of these non-target cells is unknown (28), although recent measurements suggest that only 5 to 20% of hepatocytes are infected in the absence of therapy (25, 26). We fixed N = 6.5×106 cells/mL, that is, half the maximum number of hepatocytes are not targets of HCV infection. With this assumption, we show that the percentage of infected hepatocytes before treatment (Tables 2 and 3) is consistent with the experimental results in (25, 26). If N is chosen to be 25% of the maximum number of hepatocytes, we can also fit the data well (not shown), with a higher percentage of infected cells at baseline (~25%) and a larger increase in the total number of hepatocytes (~50% increase after 14-day therapy, which is unlikely to be realistic). If we choose a larger N (e.g., 75% of the maximum number of cells), we could not obtain good fits because of limited replication space for drug-resistant virus. Since changing s does not have a noticeable effect on our fits, we set s = 0. Because the viral clearance rate c is determined by the first-phase viral decline, we estimated c as done in (16) using the median change of frequent plasma HCV RNA measurements during the first several days after telaprevir treatment (11), and used the estimate c = 6.28 day-1 for all patients. Note that the estimate of μ (Table 2) is smaller than that in (17) (10-5 to 10-4) because here it represents the average mutation rate of both single and double mutants.
We also fitted the model with combination therapy of PEG-IFN-α-2a and telaprevir Eq. (S1) in (14)] to the viral load data (Fig. S2) from patients who were treated with combination therapy (Fig. 4). We fixed, based on the previous fits (Table 2), the following parameters for all patients: d = 0.01 day-1, Tmax = 1.3×107 cells/mL, s = 0, N = 6.5×106 cells/mL, μ = 6.14×10−6, r = 0.83, c = 6.28 day-1.
Statistical analyses
We calculated the significance of differences between the drug efficacy of telaprevir alone and combination therapy with a Mann-Whitney test. We considered a difference as significant when the P value was < 0.05. We used an F-test to compare results obtained from fitting Eqs. (1) and (S2) to the same patient data.
Supplementary Material
Acknowledgments
We thank T.L. Kieffer for providing the patient data, and J.G. McHutchison, J.-M. Pawlotsky, R.T. Schooley, and K.E. Sherman for helpful comments.
Funding: Portions of this work were performed under the auspices of the U.S. Department of Energy under contract DE-AC52-06NA25396, and supported by NIH grants P30-EB011339 (L.R.), P20-RR18754 (R.M.R. and H.D.), RR06555-18, AI28433-19, and AI065256 (A.S.P.) and the University of Illinois Walter Payton Liver Center GUILD (H.D.).
Footnotes
This manuscript has been accepted for publication in Science Translational Medicine. This version has not undergone final editing. Please refer to the complete version of record at http://www.sciencetranslationalmedicine.org/. The manuscript may not be reproduced or used in any manner that does not fall within the fair use provisions of the Copyright Act without the prior, written permission of AAAS.
Author contributions: L.R. and A.S.P. conceived and designed research. L.R., H.D., R.M.R., and A.S.P. performed research. L.R. and H.D. analyzed data. L.R., H.D., R.M.R., and A.S.P. wrote the paper.
Competing interests: The authors have no competing interests to declare.
List of Supplementary Material
Materials and Methods
Figs S1 to S3
Tables S1 and S2
References
References and Notes
- 1.Fried MW, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med. 2002;347:975–982. doi: 10.1056/NEJMoa020047. [DOI] [PubMed] [Google Scholar]
- 2.Manns MP, et al. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet. 2001;358:958–965. doi: 10.1016/s0140-6736(01)06102-5. [DOI] [PubMed] [Google Scholar]
- 3.De Francesco R, Migliaccio G. Challenges and successes in developing new therapies for hepatitis C. Nature. 2005;436:953–960. doi: 10.1038/nature04080. [DOI] [PubMed] [Google Scholar]
- 4.Manns MP, et al. The way forward in HCV treatment--finding the right path. Nat Rev Drug Discov. 2007;6:991–1000. doi: 10.1038/nrd2411. [DOI] [PubMed] [Google Scholar]
- 5.Pawlotsky JM, Chevaliez S, McHutchison JG. The hepatitis C virus life cycle as a target for new antiviral therapies. Gastroenterology. 2007;132:1979–1998. doi: 10.1053/j.gastro.2007.03.116. [DOI] [PubMed] [Google Scholar]
- 6.Thompson AJ, McHutchison JG. Review article: investigational agents for chronic hepatitis C. Aliment Pharmacol Ther. 2009;29:689–705. doi: 10.1111/j.1365-2036.2009.03927.x. [DOI] [PubMed] [Google Scholar]
- 7.Lamarre D, et al. An NS3 protease inhibitor with antiviral effects in humans infected with hepatitis C virus. Nature. 2003;426:186–189. doi: 10.1038/nature02099. [DOI] [PubMed] [Google Scholar]
- 8.Lin K, Perni RB, Kwong AD, Lin C. VX-950, a novel hepatitis C virus (HCV) NS3-4A protease inhibitor, exhibits potent antiviral activities in HCV replicon cells. Antimicrob Agents Chemother. 2006;50:1813–1822. doi: 10.1128/AAC.50.5.1813-1822.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Malcolm BA, et al. SCH 503034, a mechanism-based inhibitor of hepatitis C virus NS3 protease, suppresses polyprotein maturation and enhances the antiviral activity of alpha interferon in replicon cells. Antimicrob Agents Chemother. 2006;50:1013–1020. doi: 10.1128/AAC.50.3.1013-1020.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reesink HW, et al. Rapid decline of viral RNA in hepatitis C patients treated with VX-950: a phase Ib, placebo-controlled, randomized study. Gastroenterology. 2006;131:997–1002. doi: 10.1053/j.gastro.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 11.Kieffer TL, et al. Telaprevir and pegylated interferon-alpha-2a inhibit wild-type and resistant genotype 1 hepatitis C virus replication in patients. Hepatology. 2007;46:631–639. doi: 10.1002/hep.21781. [DOI] [PubMed] [Google Scholar]
- 12.Forestier N, et al. Antiviral activity of telaprevir (VX-950) and peginterferon alfa-2a in patients with hepatitis C. Hepatology. 2007;46:640–648. doi: 10.1002/hep.21774. [DOI] [PubMed] [Google Scholar]
- 13.Sarrazin C, et al. Dynamic hepatitis C virus genotypic and phenotypic changes in patients treated with the protease inhibitor telaprevir. Gastroenterology. 2007;132:1767–1777. doi: 10.1053/j.gastro.2007.02.037. [DOI] [PubMed] [Google Scholar]
- 14.Supplementary materials and methods are available as supporting online material.
- 15.Soriano V, Perelson AS, Zoulim F. Why are there different dynamics in the selection of drug resistance in HIV and hepatitis B and C viruses? J Antimicrob Chemother. 2008;62:1–4. doi: 10.1093/jac/dkn175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Neumann AU, et al. Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-alpha therapy. Science. 1998;282:103–107. doi: 10.1126/science.282.5386.103. [DOI] [PubMed] [Google Scholar]
- 17.Domingo E. Biological significance of viral quasispecies. Viral Hepatitis Rev. 1996;2:247–261. [Google Scholar]
- 18.Cuevas JM, Gonzalez-Candelas F, Moya A, Sanjuan R. Effect of ribavirin on the mutation rate and spectrum of hepatitis C virus in vivo. J Virol. 2009;83:5760–5764. doi: 10.1128/JVI.00201-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duffy S, Shackelton LA, Holmes EC. Rates of evolutionary change in viruses: patterns and determinants. Nat Rev Genet. 2008;9:267–276. doi: 10.1038/nrg2323. [DOI] [PubMed] [Google Scholar]
- 20.Ribeiro RM, Bonhoeffer S, Nowak MA. The frequency of resistant mutant virus before antiviral therapy. AIDS. 1998;12:461–465. doi: 10.1097/00002030-199805000-00006. [DOI] [PubMed] [Google Scholar]
- 21.Kuntzen T, et al. Naturally occurring dominant resistance mutations to hepatitis C virus protease and polymerase inhibitors in treatment-naive patients. Hepatology. 2008;48:1769–1778. doi: 10.1002/hep.22549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dixit NM, Layden-Almer JE, Layden TJ, Perelson AS. Modelling how ribavirin improves interferon response rates in hepatitis C virus infection. Nature. 2004;432:922–924. doi: 10.1038/nature03153. [DOI] [PubMed] [Google Scholar]
- 23.Chu HM, et al. Pharmacokinetics of VX-950, and its effect on hepatitis C viral dynamics. Hepatology. 2005;42(Suppl 1):694A. [Google Scholar]
- 24.Adiwijaya BS, et al. Rapid decrease of wild-type hepatitis C virus on telaprevir treatment. Antivir Ther. 2009;14:591–595. [PubMed] [Google Scholar]
- 25.Rodriguez-Inigo E, et al. Percentage of hepatitis C virus-infected hepatocytes is a better predictor of response than serum viremia levels. J Mol Diagn. 2005;7:535–543. doi: 10.1016/S1525-1578(10)60585-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liang Y, et al. Visualizing hepatitis C virus infections in human liver by two-photon microscopy. Gastroenterology. 2009;137:1448–1458. doi: 10.1053/j.gastro.2009.07.050. [DOI] [PubMed] [Google Scholar]
- 27.Boyer TD, Wright TL, Manns MP. Zakim and Boyer’s Hepatology: A Textbook of Liver Disease. 5. Saunders; 2006. [Google Scholar]
- 28.Lindenbach BD, Rice CM. Unravelling hepatitis C virus replication from genome to function. Nature. 2005;436:933–938. doi: 10.1038/nature04077. [DOI] [PubMed] [Google Scholar]
- 29.Sarrazin C, et al. SCH 503034, a novel hepatitis C virus protease inhibitor, plus pegylated interferon alpha-2b for genotype 1 nonresponders. Gastroenterology. 2007;132:1270–1278. doi: 10.1053/j.gastro.2007.01.041. [DOI] [PubMed] [Google Scholar]
- 30.Thompson AJ, McHutchison JG. Antiviral resistance and specifically targeted therapy for HCV (STAT-C) J Viral Hepat. 2009;16:377–387. doi: 10.1111/j.1365-2893.2009.01124.x. [DOI] [PubMed] [Google Scholar]
- 31.McCown MF, Rajyaguru S, Kular S, Cammack N, Najera I. GT-1a or GT-1b subtype-specific resistance profiles for hepatitis C virus inhibitors telaprevir and HCV-796. Antimicrob Agents Chemother. 2009;53:2129–2132. doi: 10.1128/AAC.01598-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perelson AS, Essunger P, Ho DD. Dynamics of HIV-1 and CD4+ lymphocytes in vivo. AIDS. 1997;11(Suppl A):S17–24. [PubMed] [Google Scholar]
- 33.Tsiang M, Rooney JF, Toole JJ, Gibbs CS. Biphasic clearance kinetics of hepatitis B virus from patients during adefovir dipivoxil therapy. Hepatology. 1999;29:1863–1869. doi: 10.1002/hep.510290626. [DOI] [PubMed] [Google Scholar]
- 34.Nowak MA, et al. Viral dynamics in hepatitis B virus infection. Proc Natl Acad Sci USA. 1996;93:4398–4402. doi: 10.1073/pnas.93.9.4398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ramratnam B, et al. Rapid production and clearance of HIV-1 and hepatitis C virus assessed by large volume plasma apheresis. Lancet. 1999;354:1782–1785. doi: 10.1016/S0140-6736(99)02035-8. [DOI] [PubMed] [Google Scholar]
- 36.Perelson AS, et al. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature. 1997;387:188–191. doi: 10.1038/387188a0. [DOI] [PubMed] [Google Scholar]
- 37.Zhang L, et al. Quantifying residual HIV-1 replication in patients receiving combination antiretroviral therapy. N Engl J Med. 1999;340:1605–1613. doi: 10.1056/NEJM199905273402101. [DOI] [PubMed] [Google Scholar]
- 38.Finzi D, et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat Med. 1999;5:512–517. doi: 10.1038/8394. [DOI] [PubMed] [Google Scholar]
- 39.Pause A, et al. An NS3 serine protease inhibitor abrogates replication of subgenomic hepatitis C virus RNA. J Biol Chem. 2003;278:20374–20380. doi: 10.1074/jbc.M210785200. [DOI] [PubMed] [Google Scholar]
- 40.Dahari H, Sainz B, Jr, Perelson AS, Uprichard SL. Modeling subgenomic hepatitis C virus RNA kinetics during treatment with alpha interferon. J Virol. 2009;83:6383–6390. doi: 10.1128/JVI.02612-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Villano S, et al. Analysis of HCV NS5B genetic variants following monotherapy with HCV-796, a non-nucleoside polymerase inhibitor, in treatment-naive HCV-infected patients. Hepatology. 2006;44(Suppl 1):607A. [Google Scholar]
- 42.Cooper C, et al. Antiviral activity of the non-nucleoside polymerase inhibitor, VCH-759, in chronic hepatitis C patients: results from a randomized, doubleblind, placebo-controlled, ascending multiple dose study. Hepatology. 2007;46(Suppl 1):864A. [Google Scholar]
- 43.Godofsky E, et al. A phase I/II dose escalation trial assessing tolerance, pharmacokinetics, and antiviral activity of NM283, a novel antiviral treatment for hepatitis C. Gastroenterology. 2004;126(Suppl 2):A681. [Google Scholar]
- 44.Roberts SK, et al. Robust antiviral activity of R1626, a novel nucleoside analog: a randomized, placebo-controlled study in patients with chronic hepatitis C. Hepatology. 2008;48:398–406. doi: 10.1002/hep.22321. [DOI] [PubMed] [Google Scholar]
- 45.Reddy R, et al. Antiviral activity, pharmacokinetics, safety, and tolerability of R7128, a novel nucleoside HCV RNA polymerase inhibitor, following multiple, ascending, oral doses in patients with HCV genotype 1 infection who have failed prior interferon therapy. Hepatology. 2007;46(Suppl 1):862A. [Google Scholar]
- 46.McCown MF, et al. The hepatitis C virus replicon presents a higher barrier to resistance to nucleoside analogs than to nonnucleoside polymerase or protease inhibitors. Antimicrob Agents Chemother. 2008;52:1604–1612. doi: 10.1128/AAC.01317-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Havlir DV, Eastman S, Gamst A, Richman DD. Nevirapine-resistant human immunodeficiency virus: kinetics of replication and estimated prevalence in untreated patients. J Virol. 1996;70:7894–7899. doi: 10.1128/jvi.70.11.7894-7899.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Richman DD, et al. Nevirapine resistance mutations of human immunodeficiency virus type 1 selected during therapy. J Virol. 1994;68:1660–1666. doi: 10.1128/jvi.68.3.1660-1666.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schuurman R, et al. Rapid changes in human immunodeficiency virus type 1 RNA load and appearance of drug-resistant virus populations in persons treated with lamivudine (3TC) J Infect Dis. 1995;171:1411–1419. doi: 10.1093/infdis/171.6.1411. [DOI] [PubMed] [Google Scholar]
- 50.Lai CL, et al. A one-year trial of lamivudine for chronic hepatitis B. Asia Hepatitis Lamivudine Study Group. N Engl J Med. 1998;339:61–68. doi: 10.1056/NEJM199807093390201. [DOI] [PubMed] [Google Scholar]
- 51.Zhou Y, et al. Phenotypic characterization of resistant Val36 variants of hepatitis C virus NS3-4A serine protease. Antimicrob Agents Chemother. 2008;52:110–120. doi: 10.1128/AAC.00863-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou Y, et al. Phenotypic and structural analyses of hepatitis C virus NS3 protease Arg155 variants: sensitivity to telaprevir (VX-950) and interferon alpha. J Biol Chem. 2007;282:22619–22628. doi: 10.1074/jbc.M610207200. [DOI] [PubMed] [Google Scholar]
- 53.Koev G, et al. Antiviral interactions of an HCV polymerase inhibitor with an HCV protease inhibitor or interferon in vitro. Antiviral Res. 2007;73:78–83. doi: 10.1016/j.antiviral.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 54.Olsen DB, et al. HCV antiviral activity and resistance analysis in chronically infected chimpanzees treated with NS3/4A protease and NS5B polymerase inhibitors. 42nd Annual Meeting of the European Association for the Study of the Liver (EASL); Barcelona, Spain. April 11-15, 2007. [Google Scholar]
- 55.Gane EJ, et al. First-in-man demonstration of potent antiviral activity with a nucleoside polymerase (R7128) and protease (R7227/ITMN-191) inhibitor combination in HCV: safety, pharmacokinetics, and virologic results from INFORM-1. J Hepatol. 2009;50(Suppl 1):S380. [Google Scholar]
- 56.Hezode C, et al. Telaprevir and peginterferon with or without ribavirin for chronic HCV infection. N Engl J Med. 2009;360:1839–1850. doi: 10.1056/NEJMoa0807650. [DOI] [PubMed] [Google Scholar]
- 57.McHutchison JG, et al. Telaprevir with peginterferon and ribavirin for chronic HCV genotype 1 infection. N Engl J Med. 2009;360:1827–1838. doi: 10.1056/NEJMoa0806104. [DOI] [PubMed] [Google Scholar]
- 58.Suzuki F, et al. Sustained virological response in a patient with chronic hepatitis C treated by monotherapy with the NS3-4A protease inhibitor telaprevir. J Clin Virol. 2010;47:76–78. doi: 10.1016/j.jcv.2009.09.029. [DOI] [PubMed] [Google Scholar]
- 59.Thomas DL, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature. 2009;461:798–801. doi: 10.1038/nature08463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ge D, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature. 2009;461:399–401. doi: 10.1038/nature08309. [DOI] [PubMed] [Google Scholar]
- 61.Susser S, et al. Characterization of resistance to the protease inhibitor boceprevir in hepatitis C virus-infected patients. Hepatology. 2009;50:1709–1718. doi: 10.1002/hep.23192. [DOI] [PubMed] [Google Scholar]
- 62.Kwo P, et al. HCV Sprint-1 final results: SVR 24 from a phase 2 study of boceprevir plus PegIntron™ (peginterferon alfa-2b)/ribavirin in treatment-naive subjects with genotype-1 chronic hepatitis C. J Hepatol. 2009;50(Suppl 1):S4. [Google Scholar]
- 63.Sulkowski M, et al. SILEN-C2: early antiviral activity and safety of BI 201335 combined with pegintergeron alfa-2a and ribavirin (PEGIFN/RBV) in chronic HCV genotype-1 patients with non-response to PEGIFN/RBV. J Hepatol. 2010;52(Suppl 1):S462–S463. [Google Scholar]
- 64.Powers KA, et al. Modeling viral and drug kinetics: hepatitis C virus treatment with pegylated interferon alfa-2b. Semin Liver Dis. 2003;23(Suppl 1):13–18. doi: 10.1055/s-2003-41630. [DOI] [PubMed] [Google Scholar]
- 65.Macdonald RA. “Lifespan” of liver cells. Autoradio-graphic study using tritiated thymidine in normal, cirrhotic, and partially hepatectomized rats. Arch Intern Med. 1961;107:335–343. doi: 10.1001/archinte.1961.03620030023003. [DOI] [PubMed] [Google Scholar]
- 66.Mackay IR. Hepatoimmunology: a perspective. Immunol Cell Biol. 2002;80:36–44. doi: 10.1046/j.1440-1711.2002.01063.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.