

Abstract
Rashid ST, Corbineau S, Hannan N, Marciniak SJ, Miranda E, Alexander G, Huang-Doran I, Griffin J, Ahrlund-Richter L, Skepper J, Semple R, Weber A, Lomas DA, Vallier L. Modeling inherited metabolic disorders of the liver using human induced pluripotent stem cells. J Clin Invest. 2010 Sep 1;120(9):3127–36.
Human induced pluripotent stem (iPS) cells hold great promise for advancements in developmental biology, cell-based therapy, and modeling of human disease. Here, we examined the use of human iPS cells for modeling inherited metabolic disorders of the liver. Dermal fibroblasts from patients with various inherited metabolic diseases of the liver were used to generate a library of patient-specific human iPS cell lines. Each line was differentiated into hepatocytes using what we believe to be a novel 3-step differentiation protocol in chemically defined conditions. The resulting cells exhibited properties of mature hepatocytes, such as albumin secretion and cytochrome P450 metabolism. Moreover, cells generated from patients with 3 of the inherited metabolic conditions studied in further detail (alpha1-antitrypsin deficiency, familial hypercholesterolemia, and glycogen storage disease type 1a) were found to recapitulate key pathological features of the diseases affecting the patients from which they were derived, such as aggregation of misfolded alpha1-antitrypsin in the endoplasmic reticulum, deficient LDL receptor-mediated cholesterol uptake, and elevated lipid and glycogen accumulation. Therefore, we report a simple and effective platform for hepatocyte generation from patient-specific human iPS cells. These patient-derived hepatocytes demonstrate that it is possible to model diseases whose phenotypes are caused by pathological dysregulation of key processes within adult cells.
Espejel S, Roll GR, McLaughlin KJ, Lee AY, Zhang JY, Laird DJ, Okita K, Yamanaka S, Willenbring H. Induced pluripotent stem cell-derived hepatocytes have the functional and proliferative capabilities needed for liver regeneration in mice. J Clin Invest. 2010 Sep 1;120(9):3120–6.
The ability to generate induced pluripotent stem (iPS) cells from a patient’s somatic cells has provided a foundation for organ regeneration without the need for immune suppression. However, it has not been established that the differentiated progeny of iPS cells can effectively reverse failure of a vital organ. Here, we examined whether iPS cell-derived hepatocytes have both the functional and proliferative capabilities needed for liver regeneration in mice with fumarylacetoacetate hydrolase deficiency. To avoid biases resulting from random genomic integration, we used iPS cells generated without viruses. To exclude compensation by hepatocytes not derived from iPS cells, we generated chimeric mice in which all hepatocytes were iPS cell derived. In vivo analyses showed that iPS cells were intrinsically able to differentiate into fully mature hepatocytes that provided full liver function. The iPS cell-derived hepatocytes also replicated the unique proliferative capabilities of normal hepatocytes and were able to regenerate the liver after transplantation and two-thirds partial hepatectomy. Thus, our results establish the feasibility of using iPS cells generated in a clinically acceptable fashion for rapid and stable liver regeneration.
Comment
One of the most revolutionary recent discoveries in the field of biological science, first reported by Takahashi and Yamanaka (1), was that somatic cells could be engineered to pluripotency via epigenetic reprogramming by expressing four well-defined transcription factors. The significance of this process is enhanced by the fact that expression of these factors is required only transiently; thus, cellular reprogramming can be accomplished without leaving a lasting genetic footprint (2). Induced pluripotent (iPS) cells generated from readily obtainable somatic cells from individual patients can be a powerful new tool that should help in better understanding the mechanisms of inherited human disease and variability of clinical disease phenotype, facilitate drug discovery, and provide new screening tools for drug toxicity. While many animal models generated by genetic modification have been critical for understanding disease pathophysiology, significant biological and physiological differences between mice and humans have limited their use in clinical translation, and may have, at least partially, accounted for the failure of some clinical trials (3). Recapitulation of human disease by differentiating patient-specific iPS cells into diseased somatic cells could provide much new information concerning a broad spectrum of diseases. iPS cells have been generated from patients with various neurological disorders and juvenile diabetes mellitus (4). Because the liver is a primary site of numerous metabolic processes, generating iPS cells from patients with inherited metabolic disorders and differentiating them into hepatocytes are of particular interest. Animal models available for transplanting iPS-generated human hepatocytes to create human-rodent liver chimeras could be used to better recapitulate a range of inherited diseases where primary metabolic defects in the liver may cause hepatic and/or extrahepatic disease. In the long run, hepatocytes generated from somatic cells of individual patients may be a platform for ex vivo gene therapy, without the need for immune suppression.
Two recent reports published in the Journal of Clinical Investigation show the feasibility of using human iPS-derived hepatocytes for modeling inherited metabolic human diseases in cell culture systems (5) and demonstrate the ability of the iPS cells to differentiate into functional hepatocytes in vivo (6). In the first report, Rashid et al derived iPS cell lines from skin fibroblasts of patients with alpha-1 anti-trypsin deficiency (A1ATD), glycogen storage disease type 1a (GSD1a), familial hypercholesterolemia (FH), Crigler-Najjar syndrome type 1 and hereditary tyrosinemia. The iPS cell lines were then differentiated into a hepatic phenotype by a three-step process, and characterized with special attention to the phenotypic properties specific to the corresponding diseases. The relevance of such iPS-derived hepatocytes lies in the fact that the characteristic phenotypic expression of a genetic disorder may become manifest only in the context of other cell-type specific proteins. Therefore, it is important to determine how the gene expression profile of these cells compares quantitatively with that of primary human hepatocytes. For A1ATD, the authors demonstrate accumulation of the polymeric AAT protein in the endoplasmic reticulum; for familial hypercholesterolemia, the iPS-derived hepatocytes were shown to have reduced low-density lipoprotein (LDL) uptake by immunofluorescence and flow analysis; and for glycogen storage disease type 1a, the iPS-derived hepatocytes were shown to have high levels of intracellular glycogen and lipid content, and lactate production. The authors further demonstrated that after glucagon stimulation, canonical glucagon-target genes were upregulated. This study is an important first step in modeling liver diseases directly from patient’s cells. While the study is one of the first to create iPS cell lines from such a broad array of liver-based metabolic disorders, future studies will need to address critical additional considerations for in vitro liver disease modeling. Only a single patient sample was used to make iPS cell lines for all but A1ATD. Studies have shown that there is substantial variability between iPS cell lines both from the same source as well as different sources, even with the same mutation(7, 8). Over the course of differentiation, the cell lines were shown to express many appropriate stage-specific markers, but it is not clear how quantitatively similar the final “mature” hepatocytes were to primary human hepatocytes in terms of gene expression. The residual high alpha-fetoprotein expression and the presence of two distinct cell populations, as evidenced in the flow analysis for albumin expression, make it uncertain how well the differentiation process recapitulated normal hepatocyte development or function. Although all three disease-specific iPS-derived hepatocyte samples were analyzed for a mature hepatocyte phenotype, there were substantial differences among the final differentiated cells, particularly for albumin secretion. Further, although the authors examined several characteristics of the disease phenotypes, the immunocytochemical staining did not show the characteristic globular inclusions of polymerized alpha-1 antitrypsin Z, which are the hallmark of A1ATD pathophysiology. It is also not known whether the degree of accumulation of mutant A1AT proteins in hepatocytes correlates with the severity of liver disease in humans. Thus, the amount of polymers does not necessarily translate to modeling severity of disease. For the FH-iPS-derived hepatocytes, the cells showed albumin expression and glycogen storage, but their albumin secretion and CYP3A4 activity were significantly lower than that of the A1ATD iPS-derived hepatocytes. Additionally, in the intracellular LDL comparisons, there were significant differences between what was seen grossly by immune fluorescence and what was measured by computerized fluorescence analysis, where there was not as striking a difference between the control and diseased cells. Finally, the glucagon-induced gene expression data derived from the GSD1a-iPS derived hepatocytes were similar to those derived from HepG2 cells, but it is unclear how this result would compare to that obtained from primary mature hepatocytes. Despite these limitations, the large array of iPS cell lines created in this study should permit more in-depth studies of disease phenotypes.
In the second report, Espejel et al transferred wildtype mouse iPS cells into the embryos of fumarylacetoacetate hydrolase (FAH)-deficient mice (a model for human tyrosinemia type 1) to demonstrate the ability of iPS cells to develop into hepatocytes in vivo, and to protect the FAH-deficient mice from developing hepatic failure. In doing so, the authors were able to circumvent the current limitations of in vitro hepatocyte differentiation programs by allowing the iPS cells to undergo normal ontogenic development into mature hepatocytes in vivo. To accomplish this goal, the authors created iPS cells by repeatedly transfecting mouse embryonic fibroblasts with plasmids expressing Oct4, Sox2, Klf4 and Myc. The resultant iPS cells contained no foreign DNA integration, eliminating the risk of future reactivation of genes whose expression could potentially lead to tumor formation or abnormal somatic cell function. The iPS cells were then injected into FAH-deficient blastocysts to generate a number of mosaic offspring. Because liver cells from FAH-deficient mice are dependent on the presence of the drug 2-(2-nitro-4-fluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC) for survival, a small number of hepatocytes originating from the wildtype iPS cells can replace the entire liver of these animals following NTBC withdrawal. Thus, upon withdrawal of NTBC, the chimeric offspring underwent near complete liver cell repopulation with iPS-derived cells that the authors showed were not the consequence of a fusion event. The animals with iPS-derived chimeric livers were protected from liver failure that would be normally associated with NTBC withdrawal. Importantly, the iPS-derived hepatocytes were able to respond to post-natal liver injury with the same efficiency as primary hepatocytes. This finding of equivalence is critically important if pluripotent stem cells are to be useful in regenerative medicine. Interestingly, the renal proximal tubulopathy associated with FAH-deficiency was also corrected because iPS-derived proximal tubular epithelial cells substantially replaced the FAH-deficient cells by a similar process of positive selection and expansion. Although the correction of a metabolic disease by blastocyst injection of iPS cells cannot be translated to the treatment of human metabolic diseases, this study clearly demonstrates that iPS cells can give rise to fully functional somatic cells in vivo. Application of the iPS cell technology to study human disease in experimental animals will require transplantation of in vitro-differentiated human iPS-derived hepatocytes.
Together these two novel proof-of-principle studies demonstrate the great potential that exists for using iPS-derived hepatocytes in investigating the pathophysiology of metabolic liver diseases, discovering new drugs and devising strategies for tissue regeneration. Because most inherited liver disease phenotypes are observed only in lineage-committed or fully differentiated cells, the degree to which disease-specific iPS cells can be differentiated into hepatocytes will affect the extent to which the disease can be modeled in vitro. Moreover, variability in response to differentiation among iPS cell lines, derived either from a single individual or from different individuals, will need to be carefully addressed. The study reported by Espejel et al. proves that mouse iPS cells are able to follow a normal developmental pathway and are able to fully differentiate into mature hepatocytes in chimeric mice. Whether human hepatocytes generated by differentiating iPS cells in culture will exhibit similar levels of function after transplantation into experimental animals remains to be evaluated. Similarly, to model human disease, functional and mitotic equivalence of the human cells present in the “humanized” liver chimeras to primary human hepatocytes will need to be documented. Undoubtedly, significant challenges remain; however, the remarkable progress in iPS cell technology through the effort of a large number of innovative investigators will impact our ability to understand liver diseases and to develop novel therapeutic interventions for years to come.
Figure.
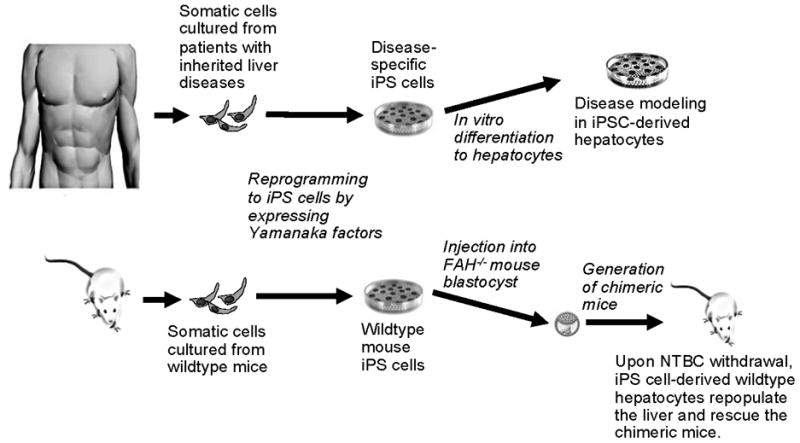
The potential of iPS cell technology to improve disease modeling and therapy. Rashid et al created pluripotent stem (iPS) cells from patients with a number of inherited metabolic disorders by reprogramming with Yamanaka factors. The cells were then differentiated in vitro into hepatocytes for disease modeling (top). Espejel et al demonstrated that mouse iPS cells from wildtype mice could give rise to fully functional hepatocytes. They injected the wildtype iPS cells directly into the blastocyst of FAH null mice. After withdrawal of NTBC, a drug that allows FAH null hepatocytes to survive, the liver in these chimeric mice is repopulated with wildtype hepatocytes derived from the original mouse wildtype iPS cells, thus demonstrating that mouse iPS cells have the potential to differentiate into fully-intact hepatocytes in vivo (bottom).
Acknowledgments
Supported by NY Stem Cell Foundation CO24346 and Oxalosis and Hyperoxaluria Foundation 9526-6675 (JRC) and NIH/NIDDK DK48794 (IJF).
References
- 1.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 2.Yamanaka S, Blau HM. Nuclear reprogramming to a pluripotent state by three approaches. Nature. 465:704–712. doi: 10.1038/nature09229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rubin LL. Stem cells and drug discovery: the beginning of a new era? Cell. 2008;132:549–552. doi: 10.1016/j.cell.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 4.Kiskinis E, Eggan K. Progress toward the clinical application of patient-specific pluripotent stem cells. J Clin Invest. 120:51–59. doi: 10.1172/JCI40553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rashid ST, Corbineau S, Hannan N, Marciniak SJ, Miranda E, Alexander G, Huang-Doran I, et al. Modeling inherited metabolic disorders of the liver using human induced pluripotent stem cells. J Clin Invest. 120:3127–3136. doi: 10.1172/JCI43122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Espejel S, Roll GR, McLaughlin KJ, Lee AY, Zhang JY, Laird DJ, Okita K, et al. Induced pluripotent stem cell-derived hepatocytes have the functional and proliferative capabilities needed for liver regeneration in mice. J Clin Invest. 120:3120–3126. doi: 10.1172/JCI43267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hu BY, Weick JP, Yu J, Ma LX, Zhang XQ, Thomson JA, Zhang SC. Neural differentiation of human induced pluripotent stem cells follows developmental principles but with variable potency. Proc Natl Acad Sci U S A. 107:4335–4340. doi: 10.1073/pnas.0910012107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kulkeaw K, Horio Y, Mizuochi C, Ogawa M, Sugiyama D. Variation in hematopoietic potential of induced pluripotent stem cell lines. Stem Cell Rev. 6:381–389. doi: 10.1007/s12015-010-9150-5. [DOI] [PubMed] [Google Scholar]