

Abstract
There is uncertainty regarding the association of cognitive decline in Alzheimer disease (AD) with classic histopathologic features—neurofibrillary tangles (NFTs) and “neuritic” amyloid plaques (NPs). This uncertainty fuels doubts about the diagnostic importance of NFTs and NPs and leads to confusion regarding hypotheses of AD pathogenesis. Three hundred ninety subjects who underwent longitudinal premortem clinical workup and postmortem quantitative neuropathologic assessment served as the group to address this issue. Subjects with concomitant brain disease(s) were analyzed independently to more accurately assess the contribution of distinct pathologies to cognitive decline. More than 60% of patients of all age groups had important non-AD brain pathologies. However, subjects without superimposed brain diseases showed strong correlations between AD-type pathology counts (NFTs > NPs) and premortem Mini-Mental State Examination scores. The observed correlation was stronger in isocortex than in allocortex and was maintained across age groups including patients older than 90 years. A theoretical model is proposed in which our results are interpreted to support the “amyloid cascade hypothesis” of AD pathogenesis. Our data show that there are many important contributory causes to cognitive decline in older persons. However, NFTs and NPs should not be dismissed as irrelevant in AD based on clinicopathologic correlation.
Keywords: Alzheimer disease, Amyloid, Cerebrovascular, Neurofibrillary tangles (NFTs), Plaques
Introduction
Neurofibrillary tangles (NFTs) and neuritic amyloid plaques (NPs) were described by Alois Alzheimer (1) over a century ago and remain the pathologic hallmarks of Alzheimer disease (AD) (Fig. 1). NFTs are argyrophilic inclusions composed of filamentous tau protein aggregates. NPs are extracellular amyloid deposits invested by swollen, degenerating neurites. Fibrillary polymers of the Aβ peptide comprise the structural core of NPs.
FIGURE 1.
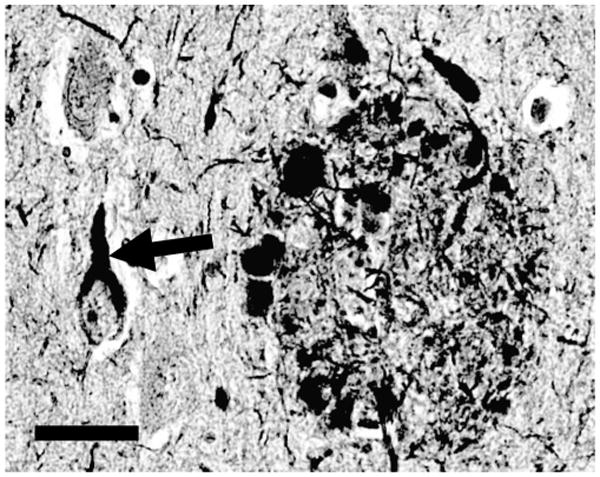
Histopathologic hallmarks of Alzheimer disease (AD). A neurofibrillary tangle (diagonal arrows) and neuritic plaque (oval-shaped structure on right) are shown in a photomicrograph from human AD brain section stained with the silver-impregnation Bielschowsky technique. Neurofibrillary tangles are composed of insoluble and protease-resistant fibrils, and develop intracellularly. Neuritic plaques are extracellular fibrillary amyloid deposits, surrounded by swollen, degenerating, silver-impregnated neurites. Scale bar = 50 μm.
AD is for all practical purposes unique to humans. Hence, careful clinicopathologic research must guide experimental models and potential therapies. Many hypotheses have been put forward to explain the biologic basis for AD. These hypotheses tend to incorporate the development of NPs and NFTs; however, in AD brain there often are many abnormalities present in addition to NFTs and NPs. Furthermore, prior studies on the correlation of NFTs and NPs to cognitive deterioration in AD have yielded somewhat differing results (see Discussion). For these reasons, some researchers have suggested recently that NFTs and NPs may contribute only marginally to dementia (2–8), or that NFTs and NPs are less relevant to cognitive decline in older patients with AD than other pathologic processes (9, 10). These seemingly contradictory results and hypotheses have produced uncertainty in the research and clinical communities about how the severity of cognitive impairment is associated with the pathognomonic features of the disease.
Linking premortem cognitive measures with postmortem neuropathology is challenging. Prospective clinicopathologic correlations require recruiting and evaluating patients over many years, during which time the parameters of evaluation must remain detailed and consistent. Ideally, each patient should be evaluated proximal to death to relate the degree of cognitive impairment to the neuropathologic features found at autopsy. In addition, technical variables often confound the assessment of NFTs and NPs in situ. Standard neuropathologic metrics for NFTs and NPs (Braak staging, Consortium to Establish a Registry for Alzheimer's Disease [CERAD], and National Institute on Aging-Reagan criteria) (11–16) are only semiquantitative, thereby further limiting our ability to make accurate correlations of antemortem cognitive status and the severity of neuropathologic findings. Confounding disease processes that coexist with AD include cerebrovascular disease (CVD), dementia with Lewy bodies, argyrophilic grain disease, and hippocampal sclerosis (HS). Only recently have the contributions of these discrete pathologic processes to the cognitive decline seen in patients with AD been recognized. Despite these challenges, clinicopathologic correlations in AD are important and should be viewed in the light of current pathogenetic hypotheses.
The University of Kentucky (UK) Alzheimer's Disease Center (ADC) follows a large clinical cohort (target n = 700 subjects) longitudinally with yearly in-depth mental status assessments. All subjects enrolled have agreed to brain donation after death. Detailed neuropathologic assessment is performed on >90% of this cohort and includes quantitative assessment of NFTs and NPs, CVD, cortical Lewy bodies (LBs), argyrophilic grains (AGs), and HS for all cases. The present study includes detailed analyses of correlations between premortem cognitive status and quantitative measures of NFTs and NPs and coexisting pathologic features in AD. A theoretical model supported by the present data is described that relates the presence of NPs and NFTs to cognitive status.
Materials and Methods
Subjects
Research protocols were approved by the UK Internal Review Board. The details of UK ADC inclusion criteria, recruitment, and mental status testing have been described previously (17). Briefly, this group of older adults was recruited originally by letter and telephone contact from a volunteer pool of approximately 4,000 individuals maintained by the UK Sanders-Brown Center on Aging. This volunteer pool initially was developed from a list of registered voters aged 60 years and older in the Lexington, KY area. Other volunteers were then recruited through local media coverage of the research activities of the UK ADC and from personal referrals by individuals already enrolled in the project. The inclusion criteria for this project (on enrollment) were 1) absence of National Institute for Neurological and Communicative Disorders and Stroke-Alzheimer's Disease and Related Disorders Association criteria for AD (18), 2) absence of medical and psychiatric conditions that affect cognition and age older than 60 years, 3) initial mental status examination scores above standard clinical cutpoints for dementia (e.g. Mini-Mental State Examination [MMSE]) (18), 4) willingness to complete annual mental status examinations, and 5) brain donation at death. The assessment of cognition in these volunteers was completed on an annual basis. Tests of cognition were selected to provide data that would be comparable to those of other ADC projects. All of the subjects without dementia in this study were contacted at 6-month intervals, had detailed annual mental status testing, and had neurologic and physical examinations at least biannually. Since 2005, all subjects have had annual neuropsychologic testing and physical and neurologic examinations. The subjects in this report had been followed for at least 2 years and most for more than 10 years. Any patient who developed dementia continued to receive annual mental status testing and neurologic examinations.
Mental Status Testing
The extensive mental status testing of our subjects has been described previously (17). The present study focuses on the MMSE conducted closest to the date of death as the “severity metric” for analyses related to the extensive pathologic data presented. The MMSE was chosen as it was the most consistently available measure obtained from both normal subjects and subjects with dementia over the course of their evaluations.
Pathologic Assessments
The gross neuropathologic evaluation was carried out at the time of autopsy. Left hemisphere specimens were taken from the following neocortical regions: frontal pole (Brodmann area 10), middle frontal gyrus (MFG) (area 9), superior and middle temporal gyri (SMT) (areas 21 and 22), temporal pole (area 38), inferior parietal lobule (IPL) (areas 39 and 40), occipital lobe including the primary visual area (Occ) (areas 17 and 18), anterior cingulate gyrus (area 24), and posterior cingulate gyrus (area 23). Specimens were also taken from the olfactory bulbs, hippocampus at the level of the lateral geniculate nucleus, entorhinal cortex, ambient gyrus, gyrus rectus, amygdala, basal ganglia, nucleus basalis of Meynert, thalamus, midbrain, pons, medulla, cerebellum, and choroid plexus. The specimens were immediately fixed in 10% formaldehyde, and selected specimens were fixed in 4% paraformaldehyde. Most of the specimens described were also sampled from the right hemisphere. Care was taken to evaluate any area suspicious for infarct. A total of at least 25 sections were taken from each brain. All specimens were processed in the standard manner (13), and sections were cut at 8-μm thickness. Sections were stained with hematoxylin and eosin and the modified Bielschowsky method. The Gallyas stain was used on sections of the entorhinal cortex, hippocampus, and amygdala. If AGs or AG-like pathology were found with the Gallyas stain in ventromedial temporal lobe structures, this stain was then used on the ambient gyrus, insula, gyrus rectus, and anterior cingulate gyrus to search for AGs. All sections of cortical and ventromedial temporal lobe structures were immunostained with monoclonal antibody NCL-β-amyloid (Novocastra, Newcastle, UK) for β-amyloid peptide, and the amygdala, medulla, and olfactory bulb were immunostained for α-synuclein (Novocastra). β-Amyloid immunohistochemistry was performed after formic acid and pepsin (Biomeda, Foster City, CA) pretreatment. α-Synuclein immunohistochemistry was performed after antigen retrieval at pH 8.0 (Cell Marque, Rocklin, CA) in a Decloaking Chamber (Biocare Medical, Concord CA). Secondary antibodies were from Vector Laboratories (Burlingame, CA), including the ABC Standard Elite kit. If LBs or Lewy neurites were found in any of these sections or in the midbrain or pons, α-synuclein immunostaining was performed on neocortical, entorhinal, and hippocampal sections.
As described previously (17), comparison of the modified Bielschowsky-impregnated sections and the Gallyas-stained sections in ventromedial temporal lobe structures revealed that the Gallyas stain detected more NFTs; however, this result reached statistical significance only in the entorhinal cortex for those cases. Thus, we did not use the Gallyas stain to quantify NFTs in the isocortical structures. Senile plaques were counted using a 10× objective (field size, 2.35 mm2) in the 5 most involved fields in each section of the regions described earlier. Plaque counts were “capped” at 250 for each area. The most severely affected fields were determined by studying the whole section and marking it. Senile plaques were separated into DPs (plaques without neurites) and NPs (plaques with degenerating neurites) in each region. NFTs, DPs, and NPs were counted using Bielschowsky-impregnated sections of the MFG, SMT, IPL, and Occ, and the Gallyas-stained sections were used to count amygdala, hippocampus, and entorhinal cortex. NFTs were counted with a 20× objective (field size, 0.586 mm2) in the 5 most severely affected fields of each section of the regions described earlier. An arithmetic mean was calculated from the count of the 5 most involved fields for DPs (number of DPs per 2.35 mm2), NPs (number of NPs per 2.35 mm2), and NFTs (number of NFTs per 0.586 mm2) for each region.
Groups With and Without Non-AD Pathology
All patients with subcortical or cortical infarct(s), cortical LBs, HS, AGs, and/or frontotemporal dementia (FTD) were assigned to Group II (with non-AD pathology); patients lacking these pathologic features were assigned to Group I. The following criteria were used in this division. Because cerebral amyloid angiopathy (CAA) does not apparently contribute to dementia independently of AD or without frank infarcts (19), CAA (however advanced) was not used as a separate criterion for non-AD pathology in the absence of discrete (>1 mm) infarcts or white matter rarefaction. However, when microinfarcts were identified the patient was assigned to Group II. The diagnoses of infarcts and HS were dependent on the neuropathologic final diagnosis reports as in prior studies (20, 21). Otherwise the criteria for non-AD pathology were as follows: the presence of any α-synuclein-immunoreactive LBs outside of amygdala constituted “cortical LBs,” and a case was designated “FTD” if it was a non-AD tauopathy (e.g. Pick disease, progressive supranuclear palsy, or corticobasal degeneration) or dementia lacking distinct histopathology, compatible with the consensus neuropathologic guidelines for FTDs (22). AGs were defined as described above. Excluded entirely from analyses were patients with Down syndrome, normal pressure hydrocephalus, brain infection, primary brain tumors or metastatic cancer in brain parenchyma.
For clinicopathologic correlations, the following criteria were applied for each patient in Group I. “Dementia” is defined as MMSE score of 20 or lower. The criterion for “NFTs present” is as follows: sum of the mean NFTs per 20× field for each of the 4 neocortical fields (MFG, MTG, IPL, and Occ); if the sum of the 4 means is >25 NFTs, then NFTs are termed “present” in neocortex. “NPs present” is defined as >2 areas in cerebral isocortex (MFG, SMT, IPL, and Occ) in which there are >10 NPs counted per 20× field.
Analysis
Consecutive subjects were evaluated from a database of patients (n = 438) followed at the UK ADC. After subjects were excluded on the basis of the above criteria, 390 subjects remained for analysis. Patients were separated into Group I or Group II according to whether or not the brain showed evidence of important non-AD neuropathology as defined above (Fig. 2). In descending order of frequency, the non-AD neuropathology consisted of infarcts (n = 167), cortical LBs (n = 64), HS (n = 28), AG (n = 23), and FTD (n = 11). Slightly less than two-thirds of patients were assigned to Group II. Seventy-six subjects (19% of total cohort) had more than 2 non-AD pathologies.
FIGURE 2.

(A) Almost two-thirds of aged human brains contain non-Alzheimer disease (AD)-type neuropathology. (B) The subtypes of non-AD pathology are shown in order of prevalence. In the cohort seen at the University of Kentucky Alzheimer's Disease Center, cerebral infarcts are the most prevalent non-AD pathology, followed by cortical Lewy bodies.
Ordinary least-squares regression was conducted to assess the relationship between MMSE and NFTs, NPs, and DPs and with NFTs, NPs, and DPs predicting MMSE scores. Age-stratified analyses were also conducted. Group comparisons involved paired t-tests or analysis of variance for mean differences, and χ2 analyses for frequencies. Statistical analyses were conducted using SAS 9.1 and MS Excel.
Results
Groups I and II did not differ significantly with regard to demographics (Table 1) except for the MMSE scores closest to death for which Group II was lower (p < 0.01). Apolipoprotein E (ApoE) allele frequencies for Group I (n = 250 alleles tested) were 5% for ApoE2 allele, 75% for ApoE3 allele, and 20% for ApoE4 allele. For group II (n = 450 alleles tested), the ApoE allele frequencies were 7% for ApoE2 allele, 68% for ApoE3 allele, and 25% for ApoE4 allele. These frequencies were not significantly different by group (χ2; p = 0.16).
TABLE 1. Cohort Demographics.
| Group | N | Gender F/M (Ratio) | Mean Age (years) | Mean Education (years) | Mean MMSE (Last Evaluation) | Median Months Between Last MMSE, Death |
|---|---|---|---|---|---|---|
| Group I (No cortical LBs, AGs, HS, infarcts, or FTD) | 134 | 83:51 (1.63) | 82.8 | 14.7 | 20.6 | 10.4 |
| Group II (includes cortical LBs, AGs, HS, infarcts, and/or FTD) | 256 | 153:103 (1.49) | 83.1 | 15.0 | 17.7 | 12.0 |
| p value for difference | N/A | NS | NS | NS | 0.003 | NS |
MMSE, Mini-Mental State Examination; LB, Lewy body; AGs, argyrophilic grains; HS, hippocampal sclerosis; FTD, frontotemporal dementia. N/A, not applicable; NS, not significant.
The impact of the presence of non-AD pathology is shown in Figure 3. NFT counts were stratified by MMSE scores. NFT counts in brains with either infarct(s) or cortical LBs showed significantly lower mean NFT counts relative to patients with comparable dementia with AD only. We then assessed the relationship between NFTs, NPs, and DPs, and MMSE in Group I and Group II patients using regression analyses (Fig. 4). The best pathologic predictor of MMSE was NFTs in Group I (R2 correlation coefficient ∼0.73). In contrast, R2 for the regression of MMSE versus NFTs in Group II was ∼0.31. R2 was lower for NPs and DPs, but as with NFTs the fit was better in Group I than for Group II. Separating Group I from Group II improved the R2 coefficient for predicting MMSE with NFTs, NPs, and DPs, regardless of the patient's age group, and the general pattern of R2 coefficients (NFTs > NPs > DPs) remained fairly constant across all the age groups (Table 2). Results were not significantly altered when the MMSE was adjusted to account for the time from the last test administration to death (data not shown). To address whether the isocortical areas were the best neuroanatomical regions in which to correlate the presence of NFTs with MMSE, we compared the R2 coefficients for NFT counts versus antemortem MMSE scores for different areas of cerebral cortex (Table 3). The results show that for mesial temporal lobe structures, either in isolation or summed together, NFT counts are consistently poorer for predicting MMSE scores than isocortical structures.
FIGURE 3.

Patients with impaired cognition and findings of Alzheimer disease (AD) only have more neurofibrillary tangles (combined counts from superior and middle temporal gyri, middle frontal gyrus, inferior parietal lobule, and occipital lobe including the primary visual area) than cohorts with similar levels of cognitive function and concurrent non-AD pathologic findings (Group II). Patients are grouped according to MMSE scores. Blue bars represent Group I no Lewy bodies (LBs), argyrophilic grains (AGs), infarct(s), hippocampal sclerosis (HS), or frontotemporal dementia (FTD). The red bars and yellow bars refer to all Group II cases with cortical LBs or infarct(s), respectively. Results of 2-tailed, 2-sample Student t-test: *, p < 0.05; **, p < 0.001.
FIGURE 4.

Correlating histopathologic lesions of Alzheimer disease (AD) with premortem cognitive decline as quantified with the Mini-Mental State Examination (MMSE). (A, B) Results for neurofibrillary tangles (NFTs), (C, D) results for neuritic amyloid plaques (NPs), and (E, F) results for diffuse plaques (DPs) in Group I (A, C, E) and Group II (with Lewy bodies, argyrophilic grains, infarct[s], hippocampal sclerosis, and/or frontotemporal dementia) (B, D, F). Note that the correlation coefficients are consistently higher for Group I versus Group II, and for both groups the correlation coefficients are NFTs > NPs > DPs. There are a few Group I patients with dementia who lack NFTs and NPs. SMT, superior and middle temporal gyri; MFG, middle frontal gyrus; IPL, inferior parietal lobule; PrV, primary visual cortex.
TABLE 2. R2 Correlation Coefficients.
| Complete Cohort | Group I only (no cortical LBs, AGs, HS, infarcts, or EID) | |||||||
|---|---|---|---|---|---|---|---|---|
| Patient Age (years) | n | NFTs | NPs | DPs | n | NFTs | NPs | DPs |
| <70 | 36 | 0.12 | 0.06 | 0.09 | 13 | 0.58 | 0.33 | 0.44 |
| 70s | 73 | 0.60 | 0.40 | 0.24 | 29 | 0.82 | 0.62 | 0.34 |
| 80s | 196 | 0.41 | 0.37 | 0.22 | 66 | 0.70 | 0.54 | 0.31 |
| >90 | 85 | 0.52 | 0.34 | 0.13 | 26 | 0.80 | 0.49 | 0.17 |
Mini-Mental State Examination versus neurofibrillary tangles (NFTs), neuritic amyloid plaques (NPs), and diffuse plaques (DPs) stratified by age. AGs, argyrophilic grains.
TABLE 3. R2 Correlation Coefficients for Group I.
| Neuroanatomical Area or Grouping | MMSE vs NFT R2 counts |
|---|---|
| Entorhinal cortex | 0.43 |
| Hippocampus (CA1) | 0.51 |
| Hippocampus (subiculum) | 0.47 |
| Amygdala | 0.57 |
| Summed counts, mesial temporal cortical structures | 0.60 |
| Superior and middle temporal gyrus | 0.64 |
| Occipital cortex | 0.55 |
| Parietal cortex | 0.67 |
| Middle frontal gyrus | 0.68 |
| Summed counts, isocortical structures | 0.73 |
Mini-Mental State Examination (MMSE) versus neurofibrillary tangle (NFT) counts in different areas of human brain (individual areas or summed counts).
Group I and Group II differed in the proportion of patients with a MMSE score of 30 (no evidence of dementia by MMSE): 16% of Group I (n = 22) versus 5% for Group II (n = 13). To determine whether these cases constituted a confound that biased Group I to have a better correlation between MMSE and NFTs, we also performed the regressions of MMSE versus NFTs on Group I cases omitting all patients with MMSE = 30. After dropping these cases from analysis, the R2 value for the association between Group I NFTs and MMSE was still 0.71. Of patients with MMSE ≥28, 47 were in Group II. Most of these (33) had cerebral infarct(s); however, 11 had AG.
We also evaluated Group I patients with CAA or amygdala LBs to determine whether eliminating these groups from Group I increased the correlation between MMSE and AD pathology. This would be expected if CAA or amygdala LBs were associated with lower MMSE for a given amount of AD pathology. When these cases (n = 34) were removed from Group I, the Group I R2 values increased for NFTs (0.80) but decreased for NPs (0.57) and for DPs (0.29).
Because prior research has indicated that distinct patterns of AD-related pathology are observed in patients of different age groups (4, 9), the data were stratified to determine whether there were important differences between age groups in the neuropathology of patients in Group II (Table 4). The data show that the types of non-AD pathology changed according to age. Patients who died younger had a higher likelihood of FTD and a lower likelihood of infarcts or AG. The overall proportion of patients in Group II trended higher in older patients with patients older than 90 years being almost 70% in Group II. Within Group II, more than three-quarters (76%) of the oldest patients (age 90+ years) had CVD and almost one-quarter (24%) of these patients had more than 1 type of non-AD neuropathology.
TABLE 4. Group II (Non-AD) Neuropathology by Age Groups.
| Patient Age (years) | n | Group II % of Total Cohort | % with Infarcts | % with Cortical LBs | % with FTD | % with AGs | % with HS |
|---|---|---|---|---|---|---|---|
| <70 | 23 | 64 | 48 | 35 | 30 | 9 | 13 |
| 70s | 44 | 60 | 57 | 34 | 5 | 7 | 9 |
| 80s | 130 | 66 | 75 | 28 | 2 | 17 | 12 |
| >90 | 59 | 69 | 76 | 25 | 0 | 19 | 20 |
AD, Alzheimer disease; LBs, Lewy bodies; FTD, frontotemporal dementia; AGs, argyrophilic grains; HS, hippocampal sclerosis.
Clinicopathologic Correlation in a Theoretical Context
By identifying a group of patients who lacked superimposed neurodegenerative and CVDs (which includes some patients with NPs and NFTs but without dementia), we have the opportunity to examine the hypothesis that NPs and NFTs contribute to cognitive decline in AD. As demonstrated in Figure 5, our data show that most patients either do not have dementia and NPs and NFTs (41.8%) or have dementia with abundant NPs and NFTs (32.8%). However, most cases outside of those 2 groups (>20%) are likely to represent patients with “subclinical” disease that are theoretically highly likely to be represented in any sample of aged brains.
FIGURE 5.

Clinical and pathologic data are assessed in the context of prevalent hypotheses about the pathogenesis of Alzheimer disease (AD). (A) The amyloid cascade hypothesis is that genetic and environmental influences contribute to the formation of neuritic amyloid plaques (NPs), which in turn potentiate neurofibrillary tangle (NFT) formation, and together, NFTs and NPs contribute to the synapse elimination and cell death seen in AD. (B) The chart relates the clinical features of AD to the pathology. As NPs and NFTs accumulate and cognition deteriorates there is a time during which no overt dementia is detectable. (C) A simple chart to evaluate whether the hypotheses about AD pathogenesis are borne out by clinicopathologic correlation. (D) It would be counter to the hypotheses in A and B, if the table positions in C designated w, x, y, and z were highly represented in the population. (E) Empirical data from Group I patients are highly compatible with the hypothesis shown in A and B, because the vast majority of patients either have no dementia with no pathology (∼42%), have dementia with NPs and NFTs (∼33%), or are in some transitional condition (∼20%).
Discussion
The goal of this study was to use a well-characterized clinical cohort to assess how aged persons' premortem cognitive status (severity) was associated with postmortem counts of NPs and NFTs. The results show strong relationships between AD pathology (NFTs > NPs > DPs) and the extent of premortem cognitive decline. This association held for each age group as long as patients with appreciable non-AD pathology were excluded. The pattern of pathology observed in this sample of patients with and without dementia is highly compatible with the prevalent hypothesis of AD pathogenesis (23, 24).
Clinicopathologic correlation in the context of the aged brain is difficult. There are some limitations to our study to consider. First, we used the MMSE as the sole indicator of cognitive ability. Other measures may be more sensitive and specific for early cognitive decline, because they exhibit fewer ceiling or floor effects (17, 18, 25). However, the use of the MMSE allows our results to be interpreted in the context of other existing clinicopathologic cohort databases that have relied on the MMSE for cross-study comparisons (26–30). Also, using the MMSE as a metric for disease severity allows continued assessment of disease progression over time. Our analyses are not able to account fully for subtle cognitive changes of mild cognitive impairment that may be clinically relevant yet poorly understood. Second, unavoidable ascertainment bias exists in the UK ADC cohort. This cohort represents a convenience sample rather than an epidemiologic cohort. Sample demographic biases include a relatively advanced age at death (mean >80 years), gender bias (ratio of female to male of 3:2), and advanced educational level (∼15 years). Patients with neurodegenerative diseases tend not to die in nondementia or “transitional” phases, so there is under-representation of more cognitively intact patients (25, 31). Of the 390 patients reported in this study, however, 123 had MMSE scores of 28 or higher. Hence, patients without dementia were relatively well represented in this study. Furthermore, as shown in Table 1, Group I and Group II subjects showed nearly identical age, sex, time since last MMSE, and educational parameters. A third potential limitation of this study involves the difficulties inherent to quantifying neuropathologic lesions in neurodegenerative diseases (12, 32). These methods have been published previously and used for productive studies in AD (17). Consensus diagnostic guidelines (13, 33) recommend semiquantitative methods rather than lesion counts because these are less laborious with satisfactory inter-rater reliability when counts between different laboratories are compared (12, 14, 34). However, if performed consistently, counts of NFTs, NPs, and DPs produce a more finely graded readout that can be linked to other clinical and pathologic variables.
The Group I/Group II assignment criteria do not fully address the many changes present in aged human brains. Disease processes other than cerebral infarcts, dementia with LBs, AGs, FTDs, and HS occur in many aged persons. For example, brain pathology and clinical dementia are associated with psychiatric illness, heart surgery, alcoholism/substance abuse, diabetes, and other metabolic disorders (30, 35). Patients from each of these groups were present in Group I. In the future, more refined clinicopathologic correlation may show an even greater correlation between NFTs and NPs and the development of AD.
Although cases with non-AD pathology (Group II) were separated for clinicopathologic correlation, this separation does not imply that AD-related pathology is absent from Group II cases. In fact, the results shown in Figure 4 suggest that NFTs and NPs do contribute to cognitive decline in many Group II cases.
Because clinicopathologic correlation is important for diseases without natural animal models, prior researchers have also attempted to assess critically the correlation of NFTs/NPs and cognitive decline in AD. The ground-breaking studies correlating AD neuropathology with antemortem clinical decline area were performed by Tomlinson, Blessed, and Roth in the 1960s (36, 37). However, these studies were performed before the importance of other neurodegenerative diseases (e.g. synucleinopathies, many FTDs including tauopathies, and argyrophilic grain disease) was appreciated.
Our results indicate that the number of NFTs in cerebral isocortex correlates best with AD cognitive decline. The contradistinction between isocortical and allocortical (mesial temporal lobe) pathology is important. In the first place, NFTs are seen relatively frequently in the entorhinal cortex, hippocampus, and amygdala of aged persons without dementia (18, 38–40). But just as important, data from a number of prior studies show evidence of a nonlinear “ceiling effect” relationship between dementia and the number of NFTs in medial temporal structures in the later stages of AD (39, 41–45). The late AD plateau of NFT densities may indicate that there is turnover of NFTs in, for example, the entorhinal cortex in late-stage AD. Unfortunately, it is impossible to either prove or disprove this hypothesis in humans at this time. Yet there is a discrepancy worth noting: the hippocampus, amygdala, and entorhinal cortex are anatomical areas associated with the pathology of AD, although these medial temporal structures are not where neuropathology correlates best with dementia at any stage of the disease.
Numerous studies have shown that isocortical NFT numbers correlate relatively well with clinical dementia severity (11, 16, 17, 26, 27, 46–54). Some of these studies are hampered by semiquantitative metrics of dementia, pathology, or both. For example, when neurofibrillary pathology is stratified by Braak and Braak staging, there are only 3 stages (Braak stages IV, V, and VI) that encompass the progression of neurofibrillary pathology in the cerebral isocortex. Braak and Braak stages correlate satisfactorily with clinical dementia levels (15, 54); however, quantitative counts provide a more textured clinicopathologic correlation. Most prior studies have shown a relatively poor correlation of amyloid plaques to dementia (18, 26, 27, 46–49, 52, 55–57; however, see also 58, 59). The general pattern of “noncorrelation” is that aged brains can sustain numerous isocortical NPs and (fewer) NFTs with negligible apparent deficits or only a mild loss of episodic memory (24, 38). A substantial subgroup of “plaque only” AD (60) has not been borne out by large clinicopathologic studies, and there is not such a group in our series.
Although NP counts correlate less well than NFTs with dementia severity, these data do not necessarily argue against the central importance of NPs in the disease process. The data can best be interpreted to imply that AD pathology does not begin to diminish cognitive reserve until both NPs and NFTs are prevalent in isocortex. Prior studies have also shown evidence that NPs may mediate their effect via NFT formation (52). The manner in which pathology and dementia correlate in AD brains in no way proves, but is absolutely compatible with, the most prevalent scientific hypothesis about AD pathogenesis, the “amyloid cascade hypothesis” (23). According to this hypothesis, primary genetic and/or environmental factors contribute to overproduction of “toxic” plaque amyloid, which then leads to neurofibrillary pathology. Together, NPs and NFTs may contribute to the cell loss and synapse elimination that culminate in dementia. The amyloid cascade hypothesis is supported by data derived from genetics, animal models, and cell culture studies (24); however, the compatibility of neuropathologic data has been debated (61). Our data are incorporated into a theoretical framework and presented in Figure 5. In the progression of the amyloid cascade hypothesis, a significant minority of aged persons without dementia are predicted to harbor isocortical NPs and some NFTs. Hence, the theory and the data are compatible with each other.
Complementing the clinicopathologic correlation, our data underscore the point that it is impossible to study AD pathology without rigorous assessment of other prevalent types of neurodegeneration in the aged brain. The relationships of AD pathology and vascular pathology to dementia in older persons have been investigated (20, 40, 62–64), but currently no validated, overarching qualitative or quantitative classification system for vascular brain pathology provides precise clinicopathologic correlations. However, when older patients with brain vascular pathology are grouped together, they usually exhibit a relatively rapid progression of dementia (21, 28, 65, 66), and AD lesions are less severe for a given extent of dementia (67–69). Notwithstanding the common risks associated with the ApoE4 polymorphism, vascular risk and AD risk are apparently independent (20, 70–72). From a practical standpoint, the presence of any brain infarct—even subcortical infarcts or microvascular disease— increases the amount of expected premortem dementia (21, 28, 66, 73, 74), so correlations of AD pathology with dementia must include careful neuropathologic analyses of the entire brain.
CVD may be an important variable in the controversy about whether or not AD pathology in the “oldest old” persons has a different relationship to dementia in comparison to persons who died at a younger age (9, 10). Of persons who die aged >90 years old, 50% to 85% can be expected to show appreciable CVD (68, 75, 76). However, in the current and prior studies (15), the correlation of AD histopathology with dementia was the same in Group I patients from all age categories. There is a need for better clinicopathologic assessment scales to grade CVD contributions to dementia (77), and further work remains to be performed in this area.
In addition to cases with CVD, we removed from Group I cases with cortical LBs, AGs, HS, or FTD. Prior studies have shown that these subtypes of pathology are quite common in aged brains (78–80). LBs are considered “incidental” if present in AD amygdala only (81–84); therefore, cases with LBs in the amygdala only were assigned to Group I for this study. However, for the current study, the R2 correlation coefficient for NFTs versus MMSE improved from 0.73 to 0.80 when cases with amygdala LBs and CAA (total n = 34) were removed from Group I. This indicates that amygdala LBs and/or CAA may, even without the presence of other CVD, alter the correlation of AD pathology with dementia severity.
In summary, this study helps to solidify 3 important points about the connection between AD pathology and premortem cognitive decline: first, to evaluate AD pathology one must carefully exclude or control inclusion of brains with prevalent concomitant pathology; second, the numbers of isocortical NFTs and NPs correlate well with premortem cognitive decline; and last, careful clinicopathologic correlation supports the prevalent model of AD pathogenesis. Although none of these “prove” directly a causal connection, it is important to conceptualize that clinicopathologic correlation in AD supports the importance of NFTs and NPs in the clinicobiologic sense. Until better surrogate biologic markers of AD are discovered, refined, and proved, plaques and tangles will remain the sine qua non of AD.
Acknowledgments
We are profoundly grateful to all of the participants in our longitudinal aging study and to the patients with Alzheimer disease in the research clinic of our Alzheimer's Disease Center. We thank Ela Patel, Ann Tudor, Paula Thomason, and Sonya Anderson for technical support and Charles Smith, MD, Gregory Cooper, MD, PhD, Nancy Stiles, MD, and Allison Caban-Holt, PhD, for clinical evaluations.
This study was supported by Grant 5-P30-AG028383 and K08 NS050110 from the National Institutes of Health, Bethesda, MD, and a grant from the Abercrombie Foundation.
References
- 1.Alzheimer A. Uber egenartige Krankheitsfalle des spateren Alters. Z Ges Neurol. 1911;4:356–85. [Google Scholar]
- 2.Lee HG, Zhu X, Castellani RJ, et al. Amyloid-β in Alzheimer disease: The null versus the alternate hypotheses. J Pharmacol Exp Ther. 2007;321:823–29. doi: 10.1124/jpet.106.114009. [DOI] [PubMed] [Google Scholar]
- 3.Wirths O, Multhaup G, Bayer TA. A modified β-amyloid hypothesis: Intraneuronal accumulation of the β-amyloid peptide—the first step of a fatal cascade. J Neurochem. 2004;91:513–20. doi: 10.1111/j.1471-4159.2004.02737.x. [DOI] [PubMed] [Google Scholar]
- 4.Marchesi VT. An alternative interpretation of the amyloid Aβ hypothesis with regard to the pathogenesis of Alzheimer's disease. Proc Natl Acad Sci USA. 2005;102:9093–98. doi: 10.1073/pnas.0503181102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lesne S, Koh MT, Kotilinek L, et al. A specific amyloid-β protein assembly in the brain impairs memory. Nature. 2006;440:352–57. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 6.Robinson SR, Bishop GM. Aβ as a bioflocculant: Implications for the amyloid hypothesis of Alzheimer's disease. Neurobiol Aging. 2002;23:1051–72. doi: 10.1016/s0197-4580(01)00342-6. [DOI] [PubMed] [Google Scholar]
- 7.Savory J, Ghribi O, Herman MM. Is amyloid β-peptide neurotoxic or neuroprotective and what is its role in the binding of metal ions? Neurobiol Aging. 2002;23:1089–92. doi: 10.1016/s0197-4580(02)00037-4. [DOI] [PubMed] [Google Scholar]
- 8.Castellani RJ, Lee HG, Zhu X, et al. Neuropathology of Alzheimer disease: Pathognomonic but not pathogenic. Acta Neuropathol (Berl) 2006;111:503–9. doi: 10.1007/s00401-006-0071-y. [DOI] [PubMed] [Google Scholar]
- 9.Imhof A, Kovari E, von Gunten A, et al. Morphological substrates of cognitive decline in nonagenarians and centenarians: A new paradigm? J Neurol Sci. 2007;257:72–79. doi: 10.1016/j.jns.2007.01.025. [DOI] [PubMed] [Google Scholar]
- 10.Prohovnik I, Perl DP, Davis KL, et al. Dissociation of neuropathology from severity of dementia in late-onset Alzheimer disease. Neurology. 2006;66:49–55. doi: 10.1212/01.wnl.0000191298.68045.50. [DOI] [PubMed] [Google Scholar]
- 11.Duyckaerts C, Hauw JJ. Diagnosis and staging of Alzheimer disease. Neurobiol Aging. 1997;18:S33–42. doi: 10.1016/s0197-4580(97)00067-5. [DOI] [PubMed] [Google Scholar]
- 12.Mirra SS, Gearing M, McKeel DW, Jr, et al. Interlaboratory comparison of neuropathology assessments in Alzheimer's disease: A study of the Consortium to Establish a Registry for Alzheimer's Disease (CERAD) J Neuropathol Exp Neurol. 1994;53:303–15. doi: 10.1097/00005072-199405000-00012. [DOI] [PubMed] [Google Scholar]
- 13.Mirra SS. The CERAD neuropathology protocol and consensus recommendations for the postmortem diagnosis of Alzheimer's disease: A commentary. Neurobiol Aging. 1997;18:S91–94. doi: 10.1016/s0197-4580(97)00058-4. [DOI] [PubMed] [Google Scholar]
- 14.Hogervorst E, Bandelow S, Combrinck M, et al. The validity and reliability of 6 sets of clinical criteria to classify Alzheimer's disease and vascular dementia in cases confirmed post-mortem: Added value of a decision tree approach. Dement Geriatr Cogn Disord. 2003;16:170–80. doi: 10.1159/000071006. [DOI] [PubMed] [Google Scholar]
- 15.Jellinger KA. Clinical validity of Braak staging in the oldest-old. Acta Neuropathol (Berl) 2000;99:583–84. [Google Scholar]
- 16.Braak H, Braak E, Bohl J. Staging of Alzheimer-related cortical destruction. Eur Neurol. 1993;33:403–8. doi: 10.1159/000116984. [DOI] [PubMed] [Google Scholar]
- 17.Markesbery WR, Schmitt FA, Kryscio RJ, et al. Neuropathologic substrate of mild cognitive impairment. Arch Neurol. 2006;63:38–46. doi: 10.1001/archneur.63.1.38. [DOI] [PubMed] [Google Scholar]
- 18.Schmitt FA, Davis DG, Wekstein DR, et al. “Preclinical” AD revisited: Neuropathology of cognitively normal older adults. Neurology. 2000;55:370–76. doi: 10.1212/wnl.55.3.370. [DOI] [PubMed] [Google Scholar]
- 19.Attems J, Quass M, Jellinger KA, et al. Topographical distribution of cerebral amyloid angiopathy and its effect on cognitive decline are influenced by Alzheimer disease pathology. J Neurol Sci. 2007;257:49–55. doi: 10.1016/j.jns.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 20.White L, Petrovitch H, Hardman J, et al. Cerebrovascular pathology and dementia in autopsied Honolulu-Asia Aging Study participants. Ann NY Acad Sci. 2002;977:9–23. doi: 10.1111/j.1749-6632.2002.tb04794.x. [DOI] [PubMed] [Google Scholar]
- 21.Snowdon DA, Greiner LH, Mortimer JA, et al. Brain infarction and the clinical expression of Alzheimer disease: The Nun Study. JAMA. 1997;277:813–17. [PubMed] [Google Scholar]
- 22.Cairns NJ, Bigio EH, Mackenzie IR, et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: Consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol (Berl) 2007;114:5–22. doi: 10.1007/s00401-007-0237-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: Progress and problems on the road to therapeutics. Science. 2002;297:353–56. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 24.Hardy J. Alzheimer's disease: The amyloid cascade hypothesis: An update and reappraisal. J Alzheimers Dis. 2006;9:151–53. doi: 10.3233/jad-2006-9s317. [DOI] [PubMed] [Google Scholar]
- 25.Kryscio RJ, Schmitt FA, Salazar JC, et al. Risk factors for transitions from normal to mild cognitive impairment and dementia. Neurology. 2006;66:828–32. doi: 10.1212/01.wnl.0000203264.71880.45. [DOI] [PubMed] [Google Scholar]
- 26.Giannakopoulos P, Herrmann FR, Bussiere T, et al. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer's disease. Neurology. 2003;60:1495–500. doi: 10.1212/01.wnl.0000063311.58879.01. [DOI] [PubMed] [Google Scholar]
- 27.Tiraboschi P, Hansen LA, Thal LJ, et al. The importance of neuritic plaques and tangles to the development and evolution of AD. Neurology. 2004;62:1984–89. doi: 10.1212/01.wnl.0000129697.01779.0a. [DOI] [PubMed] [Google Scholar]
- 28.Mungas D, Reed BR, Ellis WG, et al. The effects of age on rate of progression of Alzheimer disease and dementia with associated cerebrovascular disease. Arch Neurol. 2001;58:1243–47. doi: 10.1001/archneur.58.8.1243. [DOI] [PubMed] [Google Scholar]
- 29.Mendiondo MS, Ashford JW, Kryscio RJ, et al. Modelling Mini-Mental State Examination changes in Alzheimer's disease. Stat Med. 2000;19:1607–16. doi: 10.1002/(sici)1097-0258(20000615/30)19:11/12<1607::aid-sim449>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 30.Notzold A, Michel K, Khattab AA, et al. Diabetes mellitus increases adverse neurocognitive outcome after coronary artery bypass grafting surgery. Thorac Cardiovasc Surg. 2006;54:307–12. doi: 10.1055/s-2006-924089. [DOI] [PubMed] [Google Scholar]
- 31.Salazar JC, Schmitt FA, Yu L, et al. Shared random effects analysis of multi-state Markov models: Application to a longitudinal study of transitions to dementia. Stat Med. 2007;26:568–80. doi: 10.1002/sim.2437. [DOI] [PubMed] [Google Scholar]
- 32.Geddes JW, Tekirian TL, Soultanian NS, et al. Comparison of neuropathologic criteria for the diagnosis of Alzheimer's disease. Neurobiol Aging. 1997;18:S99–105. doi: 10.1016/s0197-4580(97)00063-8. [DOI] [PubMed] [Google Scholar]
- 33.Bancher C, Paulus W, Paukner K, et al. Neuropathologic diagnosis of Alzheimer disease: Consensus between practicing neuropathologists? Alzheimer Dis Assoc Disord. 1997;11:207–19. [PubMed] [Google Scholar]
- 34.Halliday G, Ng T, Rodriguez M, et al. Consensus neuropathological diagnosis of common dementia syndromes: Testing and standardising the use of multiple diagnostic criteria. Acta Neuropathol (Berl) 2002;104:72–78. doi: 10.1007/s00401-002-0529-5. [DOI] [PubMed] [Google Scholar]
- 35.Harper C. The neuropathology of alcohol-specific brain damage, or does alcohol damage the brain? J Neuropathol Exp Neurol. 1998;57:101–10. doi: 10.1097/00005072-199802000-00001. [DOI] [PubMed] [Google Scholar]
- 36.Blessed G, Tomlinson BE, Roth M. The association between quantitative measures of dementia and of senile change in the cerebral grey matter of elderly subjects. Br J Psychiatry. 1968;114:797–811. doi: 10.1192/bjp.114.512.797. [DOI] [PubMed] [Google Scholar]
- 37.Roth M, Tomlinson BE, Blessed G. Correlation between scores for dementia and counts of ‘senile plaques’ in cerebral grey matter of elderly subjects. Nature. 1966;209:109–10. doi: 10.1038/209109a0. [DOI] [PubMed] [Google Scholar]
- 38.Bennett DA, Schneider JA, Arvanitakis Z, et al. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology. 2006;66:1837–44. doi: 10.1212/01.wnl.0000219668.47116.e6. [DOI] [PubMed] [Google Scholar]
- 39.Berg L, McKeel DW, Jr, Miller JP, et al. Clinicopathologic studies in cognitively healthy aging and Alzheimer's disease: Relation of histologic markers to dementia severity, age, sex, and apolipoprotein E genotype. Arch Neurol. 1998;55:326–35. doi: 10.1001/archneur.55.3.326. [DOI] [PubMed] [Google Scholar]
- 40.Xuereb JH, Brayne C, Dufouil C, et al. Neuropathological findings in the very old: Results from the first 101 brains of a population-based longitudinal study of dementing disorders. Ann NY Acad Sci. 2000;903:490–96. doi: 10.1111/j.1749-6632.2000.tb06404.x. [DOI] [PubMed] [Google Scholar]
- 41.Mitchell TW, Mufson EJ, Schneider JA, et al. Parahippocampal tau pathology in healthy aging, mild cognitive impairment, and early Alzheimer's disease. Ann Neurol. 2002;51:182–89. doi: 10.1002/ana.10086. [DOI] [PubMed] [Google Scholar]
- 42.von Gunten A, Kovari E, Bussiere T, et al. Cognitive impact of neuronal pathology in the entorhinal cortex and CA1 field in Alzheimer's disease. Neurobiol Aging. 2006;27:270–77. doi: 10.1016/j.neurobiolaging.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 43.Giannakopoulos P, Hof PR, Surini M, et al. Quantitative immunohistochemical analysis of the distribution of neurofibrillary tangles and senile plaques in the cerebral cortex of nonagenarians and centenarians. Acta Neuropathol (Berl) 1993;85:602–10. doi: 10.1007/BF00334669. [DOI] [PubMed] [Google Scholar]
- 44.Giannakopoulos P, Hof PR, Giannakopoulos AS, et al. Regional distribution of neurofibrillary tangles and senile plaques in the cerebral cortex of very old patients. Arch Neurol. 1995;52:1150–59. doi: 10.1001/archneur.1995.00540360028012. [DOI] [PubMed] [Google Scholar]
- 45.Haroutunian V, Purohit DP, Perl DP, et al. Neurofibrillary tangles in nondemented elderly subjects and mild Alzheimer disease. Arch Neurol. 1999;56:713–18. doi: 10.1001/archneur.56.6.713. [DOI] [PubMed] [Google Scholar]
- 46.Grober E, Dickson D, Sliwinski MJ, et al. Memory and mental status correlates of modified Braak staging. Neurobiol Aging. 1999;20:573–79. doi: 10.1016/s0197-4580(99)00063-9. [DOI] [PubMed] [Google Scholar]
- 47.Bierer LM, Hof PR, Purohit DP, et al. Neocortical neurofibrillary tangles correlate with dementia severity in Alzheimer's disease. Arch Neurol. 1995;52:81–88. doi: 10.1001/archneur.1995.00540250089017. [DOI] [PubMed] [Google Scholar]
- 48.Nagy Z, Esiri MM, Jobst KA, et al. Relative roles of plaques and tangles in the dementia of Alzheimer's disease: Correlations using three sets of neuropathological criteria. Dementia. 1995;6:21–31. doi: 10.1159/000106918. [DOI] [PubMed] [Google Scholar]
- 49.Arriagada PV, Growdon JH, Hedley-Whyte ET, et al. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology. 1992;42:631–39. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- 50.Bancher C, Braak H, Fischer P, et al. Neuropathological staging of Alzheimer lesions and intellectual status in Alzheimer's and Parkinson's disease patients. Neurosci Lett. 1993;162:179–82. doi: 10.1016/0304-3940(93)90590-h. [DOI] [PubMed] [Google Scholar]
- 51.Bancher C, Jellinger K, Lassmann H, et al. Correlations between mental state and quantitative neuropathology in the Vienna Longitudinal Study on Dementia. Eur Arch Psychiatry Clin Neurosci. 1996;246:137–46. doi: 10.1007/BF02189115. [DOI] [PubMed] [Google Scholar]
- 52.Bennett DA, Schneider JA, Wilson RS, et al. Neurofibrillary tangles mediate the association of amyloid load with clinical Alzheimer disease and level of cognitive function. Arch Neurol. 2004;61:378–84. doi: 10.1001/archneur.61.3.378. [DOI] [PubMed] [Google Scholar]
- 53.Guillozet AL, Weintraub S, Mash DC, et al. Neurofibrillary tangles, amyloid, and memory in aging and mild cognitive impairment. Arch Neurol. 2003;60:729–36. doi: 10.1001/archneur.60.5.729. [DOI] [PubMed] [Google Scholar]
- 54.Braak H, Braak E. Staging of Alzheimer's disease-related neurofibrillary changes. Neurobiol Aging. 1995;16:271–78. doi: 10.1016/0197-4580(95)00021-6. discussion 8-84. [DOI] [PubMed] [Google Scholar]
- 55.Davies L, Wolska B, Hilbich C, et al. A4 amyloid protein deposition and the diagnosis of Alzheimer's disease: Prevalence in aged brains determined by immunocytochemistry compared with conventional neuropathologic techniques. Neurology. 1988;38:1688–93. doi: 10.1212/wnl.38.11.1688. [DOI] [PubMed] [Google Scholar]
- 56.Wolf DS, Gearing M, Snowdon DA, et al. Progression of regional neuropathology in Alzheimer disease and normal elderly: Findings from the Nun study. Alzheimer Dis Assoc Disord. 1999;13:226–31. doi: 10.1097/00002093-199910000-00009. [DOI] [PubMed] [Google Scholar]
- 57.Mann DM, Jones D, South PW, et al. Deposition of amyloid β protein in non-Alzheimer dementias: Evidence for a neuronal origin of parenchymal deposits of A protein in neurodegenerative disease. Acta Neuropathol (Berl) 1992;83:415–19. doi: 10.1007/BF00713534. [DOI] [PubMed] [Google Scholar]
- 58.McKeel DW, Jr, Price JL, Miller JP, et al. Neuropathologic criteria for diagnosing Alzheimer disease in persons with pure dementia of Alzheimer type. J Neuropathol Exp Neurol. 2004;63:1028–37. doi: 10.1093/jnen/63.10.1028. [DOI] [PubMed] [Google Scholar]
- 59.Price JL, Davis PB, Morris JC, et al. The distribution of tangles, plaques and related immunohistochemical markers in healthy aging and Alzheimer's disease. Neurobiol Aging. 1991;12:295–312. doi: 10.1016/0197-4580(91)90006-6. [DOI] [PubMed] [Google Scholar]
- 60.Tiraboschi P, Sabbagh MN, Hansen LA, et al. Alzheimer disease without neocortical neurofibrillary tangles: “A second look”. Neurology. 2004;62:1141–47. doi: 10.1212/01.wnl.0000118212.41542.e7. [DOI] [PubMed] [Google Scholar]
- 61.Lee HG, Castellani RJ, Zhu X, et al. Amyloid-β in Alzheimer's disease: The horse or the cart? Pathogenic or protective? Int J Exp Pathol. 2005;86:133–38. doi: 10.1111/j.0959-9673.2005.00429.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chui H. Neuropathology lessons in vascular dementia. Alzheimer Dis Assoc Disord. 2005;19:45–52. doi: 10.1097/01.wad.0000155065.31561.0a. [DOI] [PubMed] [Google Scholar]
- 63.Chui H, Skoog I. Advances in vascular cognitive impairment 2005. Stroke. 2006;37:323–25. doi: 10.1161/01.STR.0000200556.18993.9a. [DOI] [PubMed] [Google Scholar]
- 64.Dickson DW. Neuropathology of Alzheimer's disease and other dementias. Clin Geriatr Med. 2001;17:209–28. doi: 10.1016/s0749-0690(05)70066-5. [DOI] [PubMed] [Google Scholar]
- 65.Sheng B, Cheng LF, Law CB, et al. Coexisting cerebral infarction in Alzheimer's disease is associated with fast dementia progression: Applying the National Institute for Neurological Disorders and Stroke/Association Internationale pour la Recherche et l'Enseignement en Neurosciences Neuroimaging Criteria in Alzheimer's disease with concomitant cerebral infarction. J Am Geriatr Soc. 2007;55:918–22. doi: 10.1111/j.1532-5415.2007.01171.x. [DOI] [PubMed] [Google Scholar]
- 66.Chui HC, Zarow C, Mack WJ, et al. Cognitive impact of subcortical vascular and Alzheimer's disease pathology. Ann Neurol. 2006;60:677–87. doi: 10.1002/ana.21009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nagy Z, Esiri MM, Jobst KA, et al. The effects of additional pathology on the cognitive deficit in Alzheimer disease. J Neuropathol Exp Neurol. 1997;56:165–70. doi: 10.1097/00005072-199702000-00007. [DOI] [PubMed] [Google Scholar]
- 68.Petrovitch H, Ross GW, Steinhorn SC, et al. AD lesions and infarcts in demented and non-demented Japanese-American men. Ann Neurol. 2005;57:98–103. doi: 10.1002/ana.20318. [DOI] [PubMed] [Google Scholar]
- 69.Corder EH, Woodbury MA, Volkmann I, et al. Density profiles of Alzheimer disease regional brain pathology for the Huddinge brain bank: Pattern recognition emulates and expands upon Braak staging. Exp Gerontol. 2000;35:851–64. doi: 10.1016/s0531-5565(00)00147-9. [DOI] [PubMed] [Google Scholar]
- 70.Launer LJ, Petrovitch H, Ross GW, et al. AD brain pathology: Vascular origins? Results from the HAAS autopsy study. Neurobiol Aging. 2007 Apr 25; doi: 10.1016/j.neurobiolaging.2007.03.008. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schneider JA, Wilson RS, Bienias JL, et al. Cerebral infarctions and the likelihood of dementia from Alzheimer disease pathology. Neurology. 2004;62:1148–55. doi: 10.1212/01.wnl.0000118211.78503.f5. [DOI] [PubMed] [Google Scholar]
- 72.Bennett DA, Schneider JA, Bienias JL, et al. Mild cognitive impairment is related to Alzheimer disease pathology and cerebral infarctions. Neurology. 2005;64:834–41. doi: 10.1212/01.WNL.0000152982.47274.9E. [DOI] [PubMed] [Google Scholar]
- 73.Schneider JA, Boyle PA, Arvanitakis Z, et al. Subcortical infarcts, Alzheimer's disease pathology, and memory function in older persons. Ann Neurol. 2007;62:59–66. doi: 10.1002/ana.21142. [DOI] [PubMed] [Google Scholar]
- 74.Petrovitch H, White LR, Ross GW, et al. Accuracy of clinical criteria for AD in the Honolulu-Asia Aging Study, a population-based study. Neurology. 2001;57:226–34. doi: 10.1212/wnl.57.2.226. [DOI] [PubMed] [Google Scholar]
- 75.White L, Small BJ, Petrovitch H, et al. Recent clinical-pathologic research on the causes of dementia in late life: Update from the Honolulu-Asia Aging Study. J Geriatr Psychiatry Neurol. 2005;18:224–27. doi: 10.1177/0891988705281872. [DOI] [PubMed] [Google Scholar]
- 76.Mizutani T, Shimada H. Neuropathological background of twenty-seven centenarian brains. J Neurol Sci. 1992;108:168–77. doi: 10.1016/0022-510x(92)90047-o. [DOI] [PubMed] [Google Scholar]
- 77.Giannakopoulos P, Gold G, Kovari E, et al. Assessing the cognitive impact of Alzheimer disease pathology and vascular burden in the aging brain: The Geneva experience. Acta Neuropathol (Berl) 2007;113:1–12. doi: 10.1007/s00401-006-0144-y. [DOI] [PubMed] [Google Scholar]
- 78.Jicha GA, Parisi JE, Dickson DW, et al. Neuropathologic outcome of mild cognitive impairment following progression to clinical dementia. Arch Neurol. 2006;63:674–81. doi: 10.1001/archneur.63.5.674. [DOI] [PubMed] [Google Scholar]
- 79.Jicha GA, Petersen RC, Knopman DS, et al. Argyrophilic grain disease in demented subjects presenting initially with amnestic mild cognitive impairment. J Neuropathol Exp Neurol. 2006;65:602–9. doi: 10.1097/01.jnen.0000225312.11858.57. [DOI] [PubMed] [Google Scholar]
- 80.Petersen RC, Parisi JE, Dickson DW, et al. Neuropathologic features of amnestic mild cognitive impairment. Arch Neurol. 2006;63:665–72. doi: 10.1001/archneur.63.5.665. [DOI] [PubMed] [Google Scholar]
- 81.Arai Y, Yamazaki M, Mori O, et al. α-Synuclein-positive structures in cases with sporadic Alzheimer's disease: Morphology and its relationship to tau aggregation. Brain Res. 2001;888:287–96. doi: 10.1016/s0006-8993(00)03082-1. [DOI] [PubMed] [Google Scholar]
- 82.Hansen LA, Masliah E, Terry RD, et al. A neuropathological subset of Alzheimer's disease with concomitant Lewy body disease and spongiform change. Acta Neuropathol (Berl) 1989;78:194–201. doi: 10.1007/BF00688209. [DOI] [PubMed] [Google Scholar]
- 83.Lippa SM, Lippa CF, Mori H. α-Synuclein aggregation in pathological aging and Alzheimer's disease: The impact of β-amyloid plaque level. Am J Alzheimers Dis Other Demen. 2005;20:315–18. doi: 10.1177/153331750502000506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tsuang DW, Riekse RG, Purganan KM, et al. Lewy body pathology in late-onset familial Alzheimer's disease: A clinicopathological case series. J Alzheimers Dis. 2006;9:235–42. doi: 10.3233/jad-2006-9302. [DOI] [PubMed] [Google Scholar]