

Abstract
Background
Systemic lupus erythematosus (SLE) is a chronic, multiorgan, autoimmune disease that affects people of all ages and ethnicities.
Objectives
To explore the relationship between age at disease onset and many of the diverse manifestations of SLE. Additionally, to determine the relationship between age of disease onset and genetic risk in patients with SLE.
Methods
The relationship between the age at disease onset and SLE manifestations were explored in a multiracial cohort of 1317 patients. Patients with SLE were genotyped across 19 confirmed genetic susceptibility loci for SLE. Logistic regression was used to determine the relationships between the number of risk alleles present and age of disease onset.
Results
Childhood-onset SLE had higher odds of proteinuria, malar rash, anti-dsDNA antibody, haemolytic anaemia, arthritis and leucopenia (OR=3.03, 2.13, 2.08, 2.50, 1.89, 1.53, respectively; p values <0.0001, 0.0004, 0.0005, 0.0024, 0.0114, 0.045, respectively). In female subjects, the odds of having cellular casts were 2.18 times higher in childhood-onset than in adult-onset SLE (p=0.0027). With age of onset ≥50, the odds of having proteinuria, cellular casts, anti-nRNP antibody, anti-Sm antibody, anti-dsDNA antibody and seizures were reduced. However, late adult-onset patients with SLE have higher odds of developing photosensitivity than early adult-onset patients. Each SLE-susceptibility risk allele carried within the genome of patients with SLE increased the odds of having a childhood-onset disease in a race-specific manner: by an average of 48% in Gullah and 25% in African-Americans, but this was not significant in Hispanic and European-American lupus patients.
Conclusions
The genetic contribution towards predicting early-onset disease in patients with SLE is quantified for the first time. A more severe SLE phenotype is found in patients with early-onset disease in a large multi-racial cohort, independent of gender, race and disease duration.
Introduction
Systemic lupus erythematosus (SLE) is a chronic, persistent, autoimmune disease with diverse manifestations. It is most common in women of reproductive age, but occurs in people of all ages. Typically, there is a 9:1 female to male sex bias in SLE, and it is still unclear why this phenomenon occurs. Evidence suggests that a dose effect of the X-chromosome may have a role in this female preponderance as can be seen by the increased prevalence of SLE in male subjects with Klinefelter's disease (47, XXY). 1 There is also strong evidence that genetics have a critical role in the pathogenesis of SLE, as can be seen in the strong familial aggregation of this disease. 2–4 SLE disease severity varies profoundly by race. 5 6 Disease onset occurs in adolescence in approximately 15–20% of patients, and is known to be more aggressive than adult-onset SLE. 7 8 It is understood that SLE disease manifestations accumulate with increasing disease duration. 9
Herein, we explore the independent association of the age at SLE onset with the many clinical and serological manifestations of the disease in a large patient cohort. We further examine the relationship between genetic risk for SLE and patient's age at disease onset. Our data indicate that early disease onset is associated with a more severe phenotype in patients with SLE. We also show that childhood-onset disease is predicted by the presence of more genetic susceptibility loci for SLE compared with adult-onset disease in patients of African descent.9
Patients and Methods
Patients and clinical data
Our study population included 1317 patients with SLE enrolled in the Lupus Family Registry and Repository (LFRR) at the Oklahoma Medical Research Foundation. Patient enrolment in the LFRR has previously been described. 4 All patients met at least four of the 11 American College of Rheumatology criteria for classification of SLE. 10 11 Patients completed a questionnaire that described their disease history. Medical records were also obtained, and relevant information was extracted and reviewed by a nurse, doctor's assistant, or a doctor. Age of disease onset was determined as the age at which the fourth American College of Rheumatology SLE criterion had been met (table 1). Race was self-reported. Our study population included 43% European-American, 27% African-American, 12% Hispanics, 10% Gullah, 5% Native-American and 2% Asian lupus patients. Principal component analysis (PCA) was used to determine population substructure as described below, and outliers were identified before genetic data analysis. Our studies were approved by the institution review boards of our institutes and each patient signed a written informed consent before participation in this study.
Table 1. Age at disease onset and gender distribution of the lupus patients included in this study.
| Age group | Gender | Total | |
|---|---|---|---|
| Male | Female | ||
| N (% of group) | N (% of group) | ||
| <18 | 20 (18.0) | 91 (82.0) | 111 |
| 18–49 | 83 (8.0) | 955 (92.0) | 1038 |
| ≥50 | 23 (13.7) | 145 (86.3) | 168 |
| Total (%) | 126 (9.6) | 1191 (90.4) | 1317 |
Serology
Serum was obtained from all patients upon LFRR enrolment. All serological testing was performed at the Oklahoma Medical Research Foundation clinical immunology laboratory. Oüchterlony immunodiffusion assays were performed to assess the presence of extractable nuclear antigens (anti-Sm, nRNP, Ro, La, ribosomal P). 12 Anti-dsDNA antibodies were assayed using indirect immunofluorescence by Crithidia luciliae (Inova Diagnostics, San Diego, California, USA). The presence of IgG and IgM anticardiolipin antibodies (anti-aCL) was determined by ELISA.
Genetic susceptibility quantification
Female patients with SLE were genotyped for 19 common single nucleotide polymorphisms (SNPs) that represent confirmed SLE susceptibility loci (table 2). These represent common genetic susceptibility loci that have been confirmed in at least two independent cohorts of lupus patients and controls. Rare variants such as susceptibility loci in TNFAIP3 and TREX1 were not examined. For each genetic susceptibility locus examined, a representative associated SNP that has been previously confirmed in lupus patients was selected for genotyping, except for the human leucocyte antigen region and interferon regulatory factor 5 as they are known to harbour multiple independent genetic effects. For the HLA region, the two SNPs that were recently shown to have independent genetic effects using logistic regression analysis of a large number of lupus-associated SNPs in the HLA region 13 were genotyped in our study. In the interferon regulatory factor 5 locus, three independent genetic effects have been previously identified, 14 and three tag SNPs representing these independent effects were genotyped in our study.
Table 2. Single nucleotide polymorphisms (SNPs) representing 19 previously established and confirmed independent susceptibility loci for systemic lupus erythematosus that were genotyped in our study population.
| Gene/region | Chromosome | Genotyped SNP | Risk allele |
|---|---|---|---|
| BANK1 | 4q24 | rs10516487 | G |
| C8orf13-BLK | 8p22–23 | rs13277113 | A |
| CTLA4 | 2q33 | rs231775 | G |
| FCGR2A | 1q23 | rs1801274 | C |
| HLA region 1 | 6p21.33 | rs3131379 | A |
| HLA region 2 | 6p21.32 | rs1270942 | G |
| IL21 | 4q26 | rs907715 | G |
| IRF5 | 7q32 | rs729302 | A |
| IRF5 | 7q32 | rs10954213 | A |
| IRF5 | 7q32 | rs2070197 | C |
| ITGAM | 16p11.2 | rs1143679 | A |
| KIAA1542 | 11p15.5 | rs4963128 | C |
| MBL | 10q11 | rs1800450 | A |
| MECP2 | Xq28 | rs17435 | T |
| PDCD1 | 2q37.3 | rs11568821 | A |
| PTPN22 | 1p13 | rs2476601 | A |
| PXK | 3p14.3 | rs6445975 | C |
| STAT4 | 2q32.2 | rs7574865 | T |
| TNFSF4 | 1q25 | rs2205960 | T |
HLA, human leucocyte antigen; IRF5, interferon regulatory factor 5.
Genotyping was performed using the Illumina iScan System and Illumina Infinium II genotyping assays following the manufacturer's recommendations. Genotyping success rate ranged between 97.6% and 100% for the 19 SNPs genotyped. The total number of SLE-risk alleles within all 19 loci genotyped was determined in each female patient with SLE using an additive model. Patients with any undetermined genotype were excluded from the analysis.
Statistical analyses
Univariable analyses were performed on the age of disease onset and the various clinical and serological disease manifestations. Fisher's exact tests were performed on manifestations with a 2 × 2 table cell value with an expected value <5. These manifestations were excluded from any further analysis owing to sample-size constraints. SLE disease manifestations with p values <0.20 were further characterised.
We fitted two logistic regression models for each SLE disease manifestation: one for childhood-onset (<18 years at onset) versus adult-onset disease (≥18 years at onset), and one for early adult-onset (18–49 years at onset) versus late adult-onset disease (≥50 at onset). Each logistic regression model included the disease manifestation as the dependent variable, and age of onset as the independent variable of interest, while controlling for gender, race and disease duration. We also tested for interaction between age of onset and gender, and fit separate models for male subjects and female subjects as needed. Patients of native Hawaiian, historic Asian, or Pacific Islander descent were not included in the regression analyses because of sample size constraints.
We compared gender distributions between patients with childhood-onset SLE and patients with early adult-onset disease using the χ2 test. We similarly compared the distribution of gender for patients with late adult-onset SLE versus patients with early adult-onset disease.
Logistic regression was also used to compare the number of SLE risk alleles between childhood-onset and adult-onset disease in female patients with SLE. To account for confounding population substructure or admixture in the studied population we employed PCA using genotyping data from 107 ancestry informative markers. We identified one outlier individual who was excluded from the analysis, and a second individual who was self-reported as American Indian or Alaskan Native but clustered as Hispanic, and thus was analysed as Hispanic in assessing gene risk for SLE.
Linear regression assessed the association between the number of SLE risk alleles and the age of disease onset. The number of risk alleles was the dependent variable and the age at onset was the independent variable. Each race was modelled independently as there was evidence of interaction between races. All linear modelling assumptions were assessed and met.
Statistical analysis was performed using SAS v9.1.3.
Results
Malar rash, arthritis, proteinuria, cellular casts, haemolytic anaemia and anti-dsDNA and anti-nRNP antibodies were all independently associated with the age of disease onset when comparing childhood-onset with adult-onset SLE (table 3). Photosensensitivity, oral ulcers, pericarditis, proteinuria, cellular casts, seizures, thrombocytopenia and anti-dsDNA, anti-Sm, and anti-nRNP antibodies were all independently associated with the age of disease onset when comparing early and late-adult onset SLE (table 3).
Table 3. Univariable analyses comparing age at onset and the clinical and serological manifestations of SLE.
| <18 versus ≥18 | χ2 | OR | 95% CI | p Value | 18–49 versus ≥50 | χ2 | OR | 95% CI | p Value | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| LL | UL | LL | UL | ||||||||
| Clinical features | Clinical features | ||||||||||
| Malar rash | 16.77 | 2.25 | 1.51 | 3.34 | <0.0001 | Malar rash | 2.51 | 1.32 | 0.94 | 1.86 | 0.1133 |
| Discoid rash | 0.01 | 0.98 | 0.56 | 1.70 | 0.9404 | Discoid rash | 1.20 | 1.32 | 0.80 | 2.17 | 0.2738 |
| Photosensitivity | 0.75 | 1.19 | 0.80 | 1.77 | 0.3867 | Photosensitivity | 7.50 | 0.63 | 0.46 | 0.88 | 0.0062 |
| Oral ulcer | 0.57 | 1.17 | 0.77 | 1.78 | 0.4500 | Oral ulcer | 4.64 | 1.53 | 1.04 | 2.25 | 0.0312 |
| Arthritis | 4.11 | 1.62 | 1.01 | 2.58 | 0.0426 | Arthritis | 2.17 | 1.29 | 0.92 | 1.82 | 0.1408 |
| Pericarditis | 1.80 | 1.37 | 0.86 | 2.18 | 0.1801 | Pericarditis | 4.32 | 1.66 | 1.02 | 2.69 | 0.0378 |
| Pleuritis | 0.62 | 1.18 | 0.78 | 1.80 | 0.4329 | Pleuritis | 2.24 | 1.34 | 0.91 | 1.96 | 0.1344 |
| Proteinuria | 36.10 | 3.34 | 2.21 | 5.03 | <0.0001 | Proteinuria | 40.33 | 3.80 | 2.46 | 5.88 | <0.0001 |
| Cellular casts | 9.91 | 2.04 | 1.30 | 3.21 | 0.0016 | Cellular casts | 19.42 | 5.36 | 2.34 | 12.31 | <0.0001 |
| Seizures | 1.33 | 1.45 | 0.77 | 2.74 | 0.2487 | Seizures | 6.15 | 3.02 | 1.21 | 7.55 | 0.0131 |
| Psychosis | 0.51 | 0.5770* | Psychosis | 2.15 | 0.2580* | ||||||
| Leucopenia | 3.11 | 1.43 | 0.96 | 2.12 | 0.0780 | Leucopenia | 2.37 | 1.33 | 0.93 | 1.90 | 0.1234 |
| Lymphopenia | 0.26 | 1.11 | 0.75 | 1.64 | 0.6130 | Lymphopenia | 1.42 | 1.23 | 0.88 | 1.72 | 0.2342 |
| Thrombocytopenia | 3.04 | 1.52 | 0.95 | 2.43 | 0.0814 | Thrombocytopenia | 5.15 | 1.83 | 1.08 | 3.09 | 0.0233 |
| Haemolytic anaemia | 12.94 | 2.73 | 1.55 | 4.81 | 0.0003 | Haemolytic anaemia | 0.71 | 1.38 | 0.65 | 2.93 | 0.3993 |
| Autoantibodies | Autoantibodies | ||||||||||
| Anti-dsDNA | 11.22 | 1.95 | 1.31 | 2.90 | 0.0008 | Anti-dsDNA | 5.98 | 1.64 | 1.10 | 2.44 | 0.0144 |
| Anti-Sm | 0.85 | 0.69 | 0.31 | 1.52 | 0.3576 | Anti-Sm | 5.35 | 2.45 | 1.12 | 5.37 | 0.0208 |
| Anti-nRNP | 5.37 | 0.53 | 0.31 | 0.91 | 0.0205 | Anti-nRNP | 5.03 | 1.62 | 1.06 | 2.47 | 0.0249 |
| Anti-Ro | 0.71 | 0.81 | 0.49 | 1.33 | 0.3992 | Anti-Ro | 0.08 | 0.95 | 0.64 | 1.39 | 0.7818 |
| Anti-La | 0.25 | 0.79 | 0.31 | 2.00 | 0.6173 | Anti-La | 1.62 | 0.67 | 0.36 | 1.25 | 0.2035 |
| Anti-ribosomal P | 0.51 | 1.0000* | Anti-ribosomal P | 0.97 | 1.0000* | ||||||
| aCL IgG | 1.74 | 1.31 | 0.88 | 1.95 | 0.1875 | aCL IgG | 1.43 | 1.24 | 0.87 | 1.78 | 0.2313 |
| aCL IgM | 0.06 | 1.09 | 0.57 | 2.08 | 0.8061 | aCL IgM | 0.18 | 1.13 | 0.63 | 2.04 | 0.6739 |
Fisher's exact two-tailed p value.
aCL, anticardiolipin antibodies, LL, lower limit; SLE, systemic lupus erythematosus; UL, upper limit.
After adjusting for gender, race and disease duration, the odds of having proteinuria, malar rash, anti-dsDNA antibodies, haemolytic anaemia, arthritis and leucopenia were all higher in childhood-onset than in adult-onset SLE (table 4). The differences in the frequency of proteinuria, malar rash, anti-dsDNA antibodies and haemolytic anaemia survive a stringent Bonferroni corrected p value of <0.004. There was an interaction between the age of onset and gender when modelling for cellular casts. In male subjects, although not significant, the odds of having cellular casts were higher in adult-onset than in childhood-onset SLE. In female subjects, the odds of having cellular casts were higher in childhood-onset than in adult-onset SLE (p=0.0027).
Table 4. Multivariable analysis comparing age at SLE onset and the clinical and serological manifestations with univariable analysis p values <0.20.
| <18 versus ≥18 | χ2 | OR | 95% CI | p Value | 18–49 versus ≥50 | χ2 | OR | 95% CI | p Value | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| LL | UL | LL | UL | ||||||||
| Proteinuria | 23.90 | 3.03 | 1.94 | 4.72 | <0.0001 | Proteinuria | 22.24 | 2.98 | 1.89 | 4.68 | <0.0001 |
| Malar rash | 12.65 | 2.13 | 1.40 | 3.23 | 0.0004 | Cellular casts | 10.38 | 4.00 | 1.72 | 9.30 | 0.0013 |
| Anti-dsDNA | 12.17 | 2.08 | 1.38 | 3.14 | 0.0005 | Photosensitivity | 6.70 | 0.63 | 0.45 | 0.89 | 0.0097 |
| Haemolytic anaemia | 9.18 | 2.50 | 1.38 | 4.52 | 0.0024 | Anti-nRNP | 6.44 | 1.82 | 1.15 | 2.89 | 0.0112 |
| Arthritis | 6.40 | 1.89 | 1.15 | 3.09 | 0.0114 | Anti-Sm | 6.11 | 2.77 | 1.24 | 6.20 | 0.0134 |
| Leucopenia | 4.04 | 1.53 | 1.01 | 2.31 | 0.0445 | Anti-dsDNA | 5.37 | 1.62 | 1.08 | 2.45 | 0.0205 |
| Anti-nRNP | 2.67 | 0.61 | 0.34 | 1.10 | 0.1020 | Seizures | 4.98 | 2.88 | 1.14 | 7.29 | 0.0256 |
| Pericarditis | 1.44 | 1.34 | 0.83 | 2.17 | 0.2301 | Oral ulcer | 3.47 | 1.46 | 0.98 | 2.18 | 0.0624 |
| Thrombocytopenia | 0.86 | 1.26 | 0.77 | 2.07 | 0.3550 | Thrombocytopenia | 3.42 | 1.67 | 0.97 | 2.87 | 0.0645 |
| aCL IgG | 0.44 | 1.15 | 0.76 | 1.75 | 0.5094 | Malar rash | 2.01 | 1.30 | 0.91 | 1.86 | 0.1564 |
| Pericarditis | 1.97 | 1.43 | 0.87 | 2.34 | 0.1608 | ||||||
| Cellular casts | Arthritis | 1.49 | 1.25 | 0.88 | 1.78 | 0.2224 | |||||
| Men | 1.05 | 0.40 | 0.07 | 2.31 | 0.3057 | Pleuritis | 1.15 | 1.24 | 0.84 | 1.84 | 0.2841 |
| Women | 9.01 | 2.18 | 1.31 | 3.64 | 0.0027 | Leucopenia | 0.51 | 1.15 | 0.79 | 1.67 | 0.4751 |
Adjusted for gender, race and disease duration. p Values are reported without correction for multiple testing.
With a Bonferroni correction a p value of <0.004 is considered significant. p Values <0.05 were considered highly suggestive.
aCL, anticardiolipin antibodies; LL, lower limit; SLE, systemic lupus erythematosus; UL, upper limit.
The odds of having proteinuria, cellular casts, anti-nRNP antibody, anti-Sm antibody, anti-dsDNA antibody and seizures were higher in early adult-onset than in late adult-onset disease after adjusting for gender, race and disease duration, while late adult-onset patients with SLE had higher odds of developing photosensitivity than patients with early adult-onset disease (table 4). The difference in the frequency of renal involvement in early adult-onset compared with late adult-onset disease, as indicated by the presence of proteinuria and cellular casts, survive a Bonferroni corrected p value of <0.004 (table 4).
It should be noted that the serological data we used in this study are based on a single serum sample obtained at the time of patient enrolment in LFRR. Given possible fluctuation in autoantibody levels in lupus patients, data for autoantibody associations should be replicated to be considered confirmed.
Gender proportions are not equal across our population of patients with SLE (table 1). Specifically, the proportion of male subjects is 10.0% higher in the group of patients with age of onset <18 years than in the 18–49-year-old age group (χ2=12.34, p=0.0004). The proportion of male subjects is 5.7% higher in the ≥50 age group than in the 18–49 year-old age group (χ2 =5.85, p=0.0156).
We next hypothesised that childhood-onset SLE is associated with higher genetic risk for the disease, which might explain a more severe disease than with adult-onset disease. We calculated the total number of SLE-risk alleles by genotyping 19 SNPs that represent 19 independent previously confirmed genetic susceptibility loci for SLE. PCA was used to identify outliers and then lupus genetic risk analysis was performed in each race separately. We found a significant difference in the number of carried SLE risk alleles between childhood-onset and adult-onset disease in Gullah and African-American lupus patients with odds ratios (95% CI) of 1.48 (1.07 to 2.04) and 1.25 (1.02 to 1.54), and p values of 0.018 and 0.032, respectively. These data indicate that for each additional risk allele carried, the odds of developing SLE during childhood (<18 years) increases by an average of 48% per risk allele in Gullah patients with SLE, and 25% in African-American patients. There was no association between the number of lupus risk alleles and age of disease onset in Hispanic patients, although a similar trend was observed (OR=1.21 (0.98 to 1.50), p=0.075). There was no association between the number of SLE risk alleles and age of disease onset in European-American lupus patients (figure 1, online supplementary table 1). These findings were further confirmed using linear regression and modelling each race individually to test if the age at disease onset predicts the number of risk alleles present in female patients with SLE (figure 2, online supplementary table 2).
Figure 1.
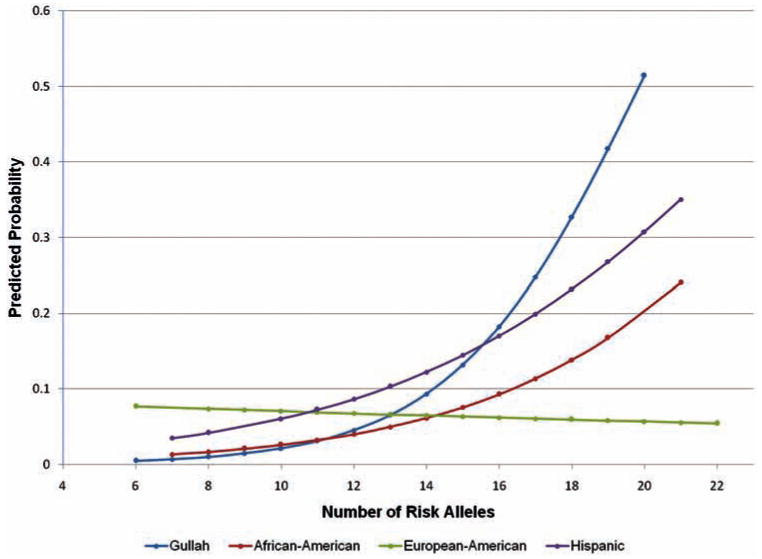
Estimated probability of childhood-onset systemic lupus erythematosus (SLE) predicted by the number of risk alleles present. Logistic regression modelling depicting the probability of developing childhood-onset SLE based on the number of SLE susceptibility alleles carried in patients who are European-American (green), African-American (red), Hispanic (purple), and Gullah (blue). The number of SLE risk alleles is associated with the development of childhood-onset disease in Gullah and African-American, but not in Hispanic and European-American patients with SLE.
Figure 2.
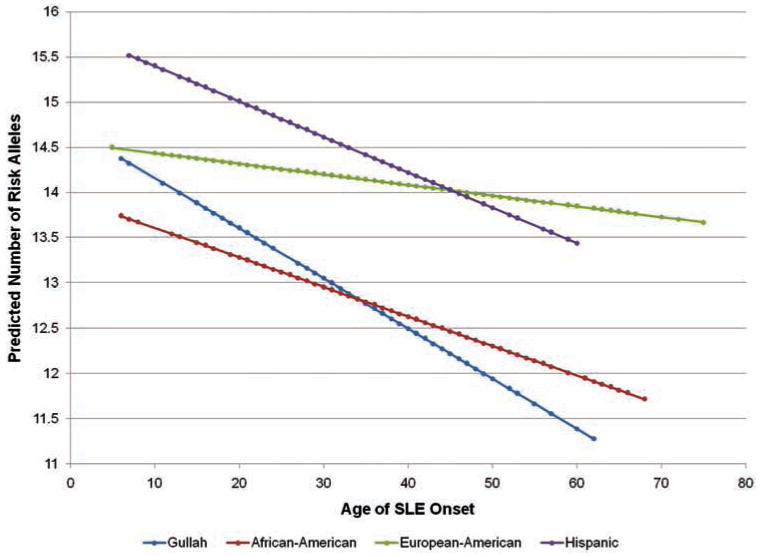
Estimated number of systemic lupus erythematosus (SLE) risk alleles present as predicted by the age of disease onset. Linear regression analysis showing significant reduction in the number of risk alleles needed to develop SLE with increase age at disease onset in Gullah (blue) and African-American (red) patients with SLE (p=0.0044 and 0.011, respectively), but not Hispanic (purple) or European-American (green) patients (p=0.059 and 0.29, respectively).
Discussion
We report the effect of age at disease onset on the various SLE manifestations in a large cohort of racially divergent patients with SLE. Childhood-onset SLE is associated with a more severe disease with higher odds of proteinuria, malar rash, anti-dsDNA antibody, haemolytic anaemia, arthritis and leucopenia than adult-onset disease.
A recent study by Hoffman et αl15 in 2009 had similar findings for proteinuria, cellular casts, haemolytic anaemia and dsDNA antibodies all being more prevalent in childhood-onset SLE than in adult-onset disease. Their study failed to detect differences in the prevalence of malar rash, arthritis or leucopenia, probably owing to the much smaller sample size (56 childhood-onset and 194 adult-onset patients with SLE), and inability to control for other known confounders such as gender, race and disease duration. Our study fails to extend the association they found with anti-ribosomal P autoantibodies in childhood-onset disease.
Kidney involvement and damage has repeatedly been reported to have a higher prevalence in patients with SLE with disease onset during childhood. 8 16 17 These reports are consistent with our findings. Our work further characterises kidney involvement and reports the effects of the age at onset after accounting for confounding gender, race and disease duration. In adult-onset SLE, patients with a disease onset at <50 years of age are about three to four times more likely to have renal involvement than patients with a disease onset ≥50 years of age. Our data are consistent with previous reports that find renal involvement less common in SLE onset after age 50.18–21
Patients in our study population with SLE onset ≥50 years were 1.59 times as likely to develop photosensitivity, compared with SLE onset 18–49 years. This finding opposes that of Wang et al 22 in a study of 695 Chinese patients with SLE where they observed photosensitivity in 16.3% of late-onset and 29.3% of early-onset SLE cases. These differences are probably due to race, as our population has <2% Asian composition.
In 2006, Bertoli et al 19 reported that patients with SLE with late disease onset (≥50 years) were 2.82 times as likely to have neurological involvement after adjusting for gender. We reached different conclusions and found that patients with early adult-onset disease were nearly three times as likely to have a history of seizures. We were unable to find any association with psychosis. These differences might be due to their inclusion of headaches and neurocognitive impairment as neurological involvement. Our data do, however, agree with their findings that the odds of having anti-Sm antibodies are much higher in patients with SLE onset before age 50. 19
One finding that is particularly interesting is that the odds of having anti-nRNP antibodies are higher in adult-onset than in childhood-onset SLE. Also, in adult-onset SLE, the odds of having anti-nRNP antibodies are 1.82 times higher in early adult than in late adult-onset disease. These data suggest that female sex hormones may have a role in the production of these autoantibodies. Indeed, Furukawa et al 23 demonstrated the enhanced binding of keratinocytes to anti-nRNP upon stimulation with β-oestradiol. We also observe a shift in the SLE sex bias of our population. The proportion of female subjects is 10% lower in the childhood-onset group, and 5.7% lower in the late adult-onset group than in early adult-onset SLE in this population.
We report a higher genetic risk in childhood-onset disease than in adult-onset disease in Gullah and African-American patients with SLE. Our findings suggest that genetic risk has a role in determining age of disease onset in patients with SLE of African descent, which is an important predictor of disease severity. However, it should be noted that the absolute difference in the number of risk alleles between childhood-onset and adult-onset disease is rather small, reflecting the fact that the confirmed SLE genetic susceptibility loci known to date, and which were largely discovered by genotyping common genetic variants, explain only a small fraction of the genetic risk for SLE. We suspect that different susceptibility genes for SLE contribute variable proportions towards the overall genetic risk for the disease in different races. This might explain why we did not see an association between the age of disease onset and genetic risk in patients of European descent. It is also plausible that the correlation between increased genetic risk and earlier onset of the disease in Gullah and African-American patients reflects a typically more severe disease in this group.
Although most of the known SLE genetic susceptibility loci were discovered in European-derived patients, more studies to investigate the genetic basis of SLE in minority populations, including African-Americans are underway. We believe that the initial discovery of genetic effects requires less rigorous genotyping in European-derived populations, owing to relatively larger haplotype blocks. More dense genotyping in African-derived populations is more likely to help localise these genetic effects to smaller regions in the genome, owing to relatively smaller haplotype blocks in African populations. Increased severity of SLE in African-Americans compared with European-Americans is well recognised. This suggests that differences in genetic susceptibility might account for differences in the clinical phenotype of the disease, in addition to the difference in disease prevalence, which is more common in African-Americans than in European-Americans. Our data suggest that increased genetic susceptibility predicts an earlier disease onset in African-derived but not European-derived patients. This is consistent with a more severe disease in African-derived patients and in childhood-onset disease. Of interest, it appears that the association between increased genetic risk and childhood-onset disease is proportional to the African component of the population studied, being highest in Gullah patients compared with African-American patients who have more European admixture. This effect then disappears in the European-American patients who have no or minimal African admixture.
We suggest a model that predicts the onset of SLE as a factor of both SLE susceptibility genetic background and environmental factors. We propose that the presence of higher genetic susceptibility for the disease lowers the threshold to develop SLE, allowing for earlier disease onset in the presence of necessary environmental triggers. To our knowledge, our data represent the first quantitative evidence in any complex autoimmune disease to indicate that increased genetic susceptibility is associated with an early disease onset. These data were confirmed in two independent cohorts of Gullah and African-American patients with SLE.
Supplementary Material
Acknowledgments
Funding This work was supported by the NIH grant number R03AI076729 from the National Institute of Allergy and Infectious Diseases, and NIH grants numbers P20-RR015577, P20- RR020143, P30-AR053483, P01-AI083194, P01-AR049084, N01-AR62277, R37-AI024717, R01-AR042460, UL1-RR029882, P60-AR049459, UL1-RR024986, the Lupus Foundation of America, Merit Award from the US Department of Veterans Affairs, the University of Oklahoma Health Sciences Center, the Oklahoma City VA Medical Center and the Oklahoma Medical Research Foundation.
Footnotes
Additional data (supplementary tables) are published online only. To view these files please visit the journal online (http://ard.bmj.com).
Competing interests None.
Ethics approval This study was conducted with the approval of the Oklahoma Medical Research Foundation and the University of Oklahoma Health Sciences Center.
Provenance and peer review Not commissioned; externally peer reviewed.
References
- 1.Scofield RH, Bruner GR, Namjou B, et al. Klinefelter's syndrome (47,XXY) in male systemic lupus erythematosus patients: support for the notion of a gene-dose effect from the X chromosome. Arthritis Rheum. 2008;58:2511–17. doi: 10.1002/art.23701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ramos PS, Kelly JA, Gray-McGuire C, et al. Familial aggregation and linkage analysis of autoantibody traits in pedigrees multiplex for systemic lupus erythematosus. Genes Immun. 2006;7:417–32. doi: 10.1038/sj.gene.6364316. [DOI] [PubMed] [Google Scholar]
- 3.Alarcón-Segovia D, Alarcón-Riquelme ME, Cardiel MH, et al. Familial aggregation of systemic lupus erythematosus, rheumatoid arthritis, and other autoimmune diseases in 1,177 lupus patients from the GLADEL cohort. Arthritis Rheum. 2005;52:1138–47. doi: 10.1002/art.20999. [DOI] [PubMed] [Google Scholar]
- 4.Sestak AL, Shaver TS, Moser KL, et al. Familial aggregation of lupus and autoimmunity in an unusual multiplex pedigree. J Rheumatol. 1999;26:1495–9. [PubMed] [Google Scholar]
- 5.Crosslin KL, Wiginton KL. The impact of race and ethnicity on disease severity in systemic lupus erythematosus. Ethn Dis. 2009;19:301–7. [PubMed] [Google Scholar]
- 6.Hiraki LT, Benseler SM, Tyrrell PN, et al. Ethnic differences in pediatric systemic lupus erythematosus. J Rheumatol. 2009;36:2539–46. doi: 10.3899/jrheum.081141. [DOI] [PubMed] [Google Scholar]
- 7.Arkachaisri T, Lehman TJ. Systemic lupus erythematosus and related disorders of childhood. Curr Opin Rheumatol. 1999;11:384–92. doi: 10.1097/00002281-199909000-00010. [DOI] [PubMed] [Google Scholar]
- 8.Brunner HI, Gladman DD, Ibañez D, et al. Difference in disease features between childhood-onset and adult-onset systemic lupus erythematosus. Arthritis Rheum. 2008;58:556–62. doi: 10.1002/art.23204. [DOI] [PubMed] [Google Scholar]
- 9.Zonana-Nacach A, Camargo-Coronel A, Yáñez P, et al. Measurement of damage in 210 Mexican patients with systemic lupus erythematosus: relationship with disease duration. Lupus. 1998;7:119–23. doi: 10.1191/096120398678919831. [DOI] [PubMed] [Google Scholar]
- 10.Doria A, Vesco P, Zulian F, et al. The 1982 ARA/ACR criteria for the classification of systemic lupus erythematosus in pediatric and adult patients. Clin Exp Rheumatol. 1994;12:689–90. [PubMed] [Google Scholar]
- 11.Tan EM, Cohen AS, Fries JF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25:1271–7. doi: 10.1002/art.1780251101. [DOI] [PubMed] [Google Scholar]
- 12.Clark G, Reichlin M, Tomasi TB., Jr Characterization of a soluble cytoplasmic antigen reactive with sera from patients with systemic lupus erythmatosus. J Immunol. 1969;102:117–22. [PubMed] [Google Scholar]
- 13.Harley JB, Alarcón-Riquelme ME, Criswell LA, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet. 2008;40:204–10. doi: 10.1038/ng.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Graham RR, Kyogoku C, Sigurdsson S, et al. Three functional variants of IFN regulatory factor 5 (IRF5) define risk and protective haplotypes for human lupus. Proc Natl Acad Sci USA. 2007;104:6758–63. doi: 10.1073/pnas.0701266104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoffman IE, Lauwerys BR, De Keyser F, et al. Juvenile-onset systemic lupus erythematosus: different clinical and serological pattern than adult-onset systemic lupus erythematosus. Ann Rheum Dis. 2009;68:412–15. doi: 10.1136/ard.2008.094813. [DOI] [PubMed] [Google Scholar]
- 16.Hersh AO, von Scheven E, Yazdany J, et al. Differences in long-term disease activity and treatment of adult patients with childhood- and adult-onset systemic lupus erythematosus. Arthritis Rheum. 2009;61:13–20. doi: 10.1002/art.24091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tucker LB, Uribe AG, Fernández M, et al. Adolescent onset of lupus results in more aggressive disease and worse outcomes: results of a nested matched case-control study within LUMINA, a multiethnic US cohort (LUMINA LVII) Lupus. 2008;17:314–22. doi: 10.1177/0961203307087875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koh ET, Boey ML. Late onset lupus: a clinical and immunological study in a predominantly Chinese population. J Rheumatol. 1994;21:1463–7. [PubMed] [Google Scholar]
- 19.Bertoli AM, Alarcón GS, Calvo-Alén J, et al. Systemic lupus erythematosus in a multiethnic US cohort. XXXIII. Clinical [corrected] features, course, and outcome in patients with late-onset disease. Arthritis Rheum. 2006;54:1580–7. doi: 10.1002/art.21765. [DOI] [PubMed] [Google Scholar]
- 20.Boddaert J, Huong DL, Amoura Z, et al. Late-onset systemic lupus erythematosus: a personal series of 47 patients and pooled analysis of 714 cases in the literature. Medicine (Baltimore) 2004;83:348–59. doi: 10.1097/01.md.0000147737.57861.7c. [DOI] [PubMed] [Google Scholar]
- 21.Maddison P, Farewell V, Isenberg D, et al. The rate and pattern of organ damage in late onset systemic lupus erythematosus. J Rheumatol. 2002;29:913–17. [PubMed] [Google Scholar]
- 22.Wang J, Yang S, Chen JJ, et al. Systemic lupus erythematosus: a genetic epidemiology study of 695 patients from China. Arch Dermatol Res. 2007;298:485–91. doi: 10.1007/s00403-006-0719-4. [DOI] [PubMed] [Google Scholar]
- 23.Furukawa F, Imamura S, Norris DA. Stimulation of anti-RNP antibody binding to cultured keratinocytes by estradiol. Arch Dermatol Res. 1991;283:258–61. doi: 10.1007/BF01106112. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.