

Abstract
Rimonabant, the prototypic antagonist of cannabinoid CB1 receptors, has been reported to have inverse agonist properties at higher concentrations, which may complicate its use as a tool for mechanistic evaluation of cannabinoid pharmacology. Consequently, recent synthesis efforts have concentrated on discovery of a neutral antagonist using a variety of structural templates. The purpose of this study was to evaluate the pharmacological properties of the putative neutral cannabinoid CB1 receptor antagonist O-2050, a sulfonamide side chain analog of Δ8-tetrahydrocannabinol. O-2050 and related sulfonamide cannabinoids exhibited good affinity for both cannabinoid CB1 and CB2 receptors. While the other sulfonamide analogs produced cannabinoid agonist effects in vivo (e.g., activity suppression, antinociception, and hypothermia), O-2050 stimulated activity and was inactive in the other two tests. O-2050 also decreased food intake in mice, an effect that was reminiscent of that produced by rimonabant. Unlike rimonabant, however, O-2050 did not block the effects of cannabinoid agonists in vivo, even when administered i.c.v. In contrast, O-2050 antagonized the in vitro effects of cannabinoid agonists in [35S]GTPγS and mouse vas deferens assays without having activity on its own in either assay. Further evaluation revealed that O-2050 fully and dose-dependently substituted for Δ9-tetrahydrocannabinol in a mouse drug discrimination procedure (a cannabinoid agonist effect) and that it inhibited forskolin-stimulated cyclic AMP signaling with a maximum efficacy of approximately half that of the full agonist CP55,940 [(−)-cis-3-[2-hydroxy-4(1,1-dimethyl-heptyl)phenyl]-trans-4-(3-hydroxy-propyl)cyclohexanol]. Together, these results suggest that O-2050 is not a viable candidate for classification as a neutral cannabinoid CB1 receptor antagonist.
Keywords: cannabinoids, CB1 receptor, neutral antagonist, O-2050, structure-activity relationship
1. Introduction
The endocannabinoid system is comprised of endogenous ligands (e.g., anandamide and 2-arachidonoyl glycerol) that interact with at least two major cannabinoid receptors, CB1 and CB2. While both types of cannabinoid receptors are found in the periphery, cannabinoid CB1 receptors are the primary type of cannabinoid receptor in the brain, wherein they play a significant role in an array of physiological processes, including appetite regulation (Cota et al., 2003), brain reward (Solinas et al., 2008), and memory (Lichtman et al., 2002). Original confirmation of cannabinoid CB1 receptor mediation of these processes was made possible by synthesis of rimonabant, the first selective cannabinoid CB1 receptor antagonist (Rinaldi-Carmona et al., 1994). Rimonabant was shown to block in vivo effects of cannabinoid agonists in mice (Compton et al., 1996) and discriminative stimulus effects of Δ9-tetrahydrocannabinol in rodents (McMahon et al., 2007; Wiley et al., 1995). In vitro assessment, however, showed a mixed pattern of effects: its affinity in agonist displacement binding was good (Ki = 2 nM)(Rinaldi-Carmona et al., 1995) and it blocked agonist-stimulated [35S]GTPγgS binding (Selley et al., 1996) and electrically evoked stimulation of the mouse vas deferens (Rinaldi-Carmona et al., 1995), but higher concentrations of rimonabant also possessed inverse agonist properties in some functional assays, including [35S]GTPγgS binding, forskolin-induced cAMP accumulation, and in the guinea pig small intestine (Coutts et al., 2000; Landsman et al., 1997; Mato et al., 2002). Determination of whether rimonabant blockade of behavioral and physiological effects in animals is mediated via inverse agonism or antagonism at cannabinoid CB1 receptors is technically difficult. Rimonabant also produces a few in vivo effects of its own in mice, with the two most prominent being stimulation of motor activity (Compton et al., 1996) and appetite suppression (Wiley et al., 2005). While the exact receptor mechanism(s) for these effects remain unclear, the results of structure-activity relationship analysis suggest that rimonabant-induced hyperactivity is not cannabinoid CB1 receptor-mediated (Bass et al., 2002). Possible cannabinoid CB1 receptor-related mechanisms for the inverse agonist effects of rimonabant include antagonism of endocannabinoid tone or negative modulation of constitutive activity of cannabinoid CB1 receptors (Pertwee, 2005). In order to distinguish these two possibilities, a neutral antagonist is needed.
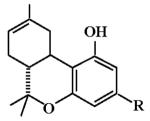
To date, candidates with several structural templates have been proposed as neutral cannabinoid CB1 receptor antagonists, including pyrazole analogs of rimonabant (Chambers et al., 2007), synthetic analogs of tetrahydrocannabinols (Gardner and Mallet, 2006), and analogs of plant-derived cannabinoids such as (−)-cannabidiol (Thomas et al., 2004). Confirmatory evidence for the antagonist properties of these different types of cannabinoid compounds is variable. One candidate that has shown substantial promise as a neutral cannabinoid CB1 receptor antagonist is O-2050, a structural analog of Δ8-tetrahydrocannabinol, in which the pentyl side chain at position C3 is replaced by an acetylene moiety with a terminal sulfonamide group (Table 1). Previous studies have demonstrated that O-2050 blocked preference for a high-fat diet (Higuchi et al., 2010) and reversed Δ9-tetrahydrocannabinol-induced immobility and increase in corticosterone levels in a forced swim model of depression in mice (Sano et al., 2009). The purpose of this study was to investigate further the behavioral effects of O-2050 as well as the nature of its interaction(s) with cannabinoid CB1 receptors.
Table 1.
Cannabinoid CB1 and CB2 Receptor Binding Affinities and Pharmacological Effects of Sulfonamides with Dimethyl and Acetylene Side Chain Substitutions*
 | ||||||
|---|---|---|---|---|---|---|
| Ki (nM) | ED50 | |||||
| ID (MW) |
R | CB1 | CB2 | SA | TF | RT |
| O-2050 (418) |
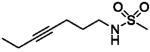
|
2.5 ± 0.4 | 0.2 ± 0.1 | 69% @ 30 |
< 30 | < 30 |
| O-1991 (432) |
|
30 ± 13 | 1.4 ± 0.2 | 2 (1-3) |
0.9 (0.6-1.4) |
0.8 (0.4-1.6) |
| O-1993 (460) |
|
70 ± 10 | 86 ± 7 | 8 (3-16) |
14 (7-27) |
12 (9-15) |
| O-2113 (459) |
|
1.7 ± 0.3 | 0.08 ± 0.02 | 0.4 (0.1-1.8) |
0.3 (0.1-1) |
1.4 (0.3-4.6) |
| JWH-104** (354) |
|
909 ± 121 | 137 ± 6 | 5 (3-10) |
2 (1-3) |
1 (0.7-2) |
Kis are presented as means ± S.E.M. All ED50s are expressed as mg/kg (with 95% confidence limits in parentheses). For compounds that failed to produce either maximal or dose-related effects, the percent effect at the highest dose (mg/kg) or > highest dose tested (in cases where largest response was less than 50% of the typical maximum) is indicated. MW = molecular weight; SA = suppression of spontaneous activity; MPE = % maximum possible antinociceptive effect in tail flick assay; RT = rectal temperature.
From Wiley et al., 2002.
2.0 Materials and methods
2.1 Subjects
Male Institute of Cancer (ICR) outbred albino mice (25-32g), obtained from Harlan (Dublin, VA) and housed in groups of five, were used for assessment of locomotor suppression, antinociception and hypothermia. Separate mice were used for testing each dose of each compound in this battery of procedures. These mice had free access to food when in their home cages. Singly housed ICR mice were used in the feeding experiment. Food access was removed for these mice twenty-four h before each test session and replaced immediately after the session. Singly housed male C57/Bl6J inbred mice (20-25 g) [Jackson Laboratories, Bar Harbor, ME] were used in the drug discrimination experiments. These mice were maintained at 85 - 90% of free-feeding body weights by restricting daily ration of standard rodent chow. All animals were kept in a temperature-controlled (20-22°C) environment with a 12-h light-dark cycle (lights on at 7 a.m.) and had free access to water in their home cages. The in vivo studies reported in this manuscript were carried out in accordance with guidelines published in the Guide for the Care and Use of Laboratory Animals (National Research Council, 1996) and were approved by the Institutional Animal Care and Use Committee of Virginia Commonwealth University.
2.2 Apparatus
Measurement of spontaneous activity in mice occurred in standard activity chambers interfaced with a Digiscan Animal Activity Monitor (Omnitech Electronics, Inc., Columbus, OH). A standard tail-flick apparatus and a digital thermometer (Fisher Scientific, Pittsburgh, PA) were used to measure antinociception and rectal temperature, respectively. For the feeding experiment, weight of food pellets was measured with a Mettler AT261 Delta Range scale (Toledo, OH) at 0.01 mg accuracy.
Eight standard mice operant conditioning chambers that were sound- and light-attenuated (MED Associates, St. Albans, VT) were used for the drug discrimination experiments. Each operant conditioning chamber (18 × 18 × 18 cm) was equipped with a house light, two nose pokes (left and right), and a recessed pellet receptacle centered between the nose pokes. A pellet dispenser delivered a 14 mg sweetened food pellet (Bio-Serv, Frenchtown, NJ) when reinforcement criteria was met. Fan motors provided ventilation and masking noise for each chamber. House lights were illuminated during training and testing sessions. A computer with Logic ‘1’ interface (MED Associates) and MED-PC software (MED Associates) was used to control schedule contingencies and to record data.
2.3 Chemicals
For in vivo studies, Δ9-THC (National Institute on Drug Abuse, Bethesda, MD), O-2050 and other sulfonamide Δ8-tetrahydrocannabinol analogs (Organix, Inc., Woburn, MA) and JWH-104 (Clemson University, Clemson, SC) were mixed in a vehicle of absolute ethanol, Emulphor-620 (Rhone-Poulenc, Inc., Princeton, NJ), and saline in a ratio of 1:1:18. All injections were administered at a volume of 0.1 ml/10 g. Route of administration for the antagonist tests in the ICR mice was i.v., except for the i.c.v. experiment described in the procedure section. For the drug discrimination experiments in C57/Bl6J mice and for the feeding experiment in ICR mice, the route of administration was s.c. The chemical names for CP55,940 and WIN55,212-2 are [(−)-cis-3-[2-hydroxy-4(1,1-dimethyl-heptyl)phenyl]-trans-4-(3-hydroxy-propyl)cyclohexanol] and [(R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl) pyrrolo-[1,2,3-d,e]-1,4-benzoxazin-6-yl]-1-naphthalenyl-methanone], respectively.
2.4 [3H]CP55,940 Binding
For cannabinoid CB1 receptor binding, rat cerebellar tissues, stored at −80°C, were thawed on the day of assay, placed in 20 volumes of cold Membrane Buffer (50 mM Tris–HCl, 3 mM MgCl2, 1 mM EGTA, pH 7.4), homogenized with a Polytron and centrifuged at 48,000 x g at 4°C for 10 min. The supernatants were discarded and the pellets were re-homogenized in Membrane Buffer, centrifuged at 48,000 x g and resuspended in Assay Buffer (50 mM Tris-HCl, 3mM MgCl2, 0.2 mM EGTA, 100 mM NaCl, pH 7.4). Adenosine deaminase (final concentration=0.004 units/ml) was added to the membrane homogenates, which were then preincubated for 10 min at 30°C. Total membrane protein was measured according to (Bradford, 1976). Binding was initiated by the addition of 150 μg of membrane to test tubes containing 1 nM of [3H] CP 55,940 (79 Ci/mmol) and a sufficient quantity of buffer to bring the total incubation volume to 1 ml. Nonspecific binding was determined by the addition of 1 μM unlabeled CP 55,940. The reaction was terminated by vacuum filtration though Whatman GF/B glass fiber filter that was pre-soaked in Tris buffer containing 5g/L BSA (Tris-BSA), followed by 3 washes with 4 C Tris-BSA. Bound radioactivity was determined by liquid scintillation spectrophotometry at 45% efficiency after extraction in ScinitSafe Econo 1 scintillation fluid.
For cannabinoid CB2 receptor binding, cell lines stably expressing mouse cannabinoid CB2 receptors (CB2-CHO cells) were cultured in a humidified atmosphere of 5% CO2/95% air at 37°C, in a 1:1 mixture of DMEM and Nutrient Mixture F12 containing 5% fetal bovine serum, 100 units/ml each of penicillin and streptomycin and 0.4 mg/ml hygromycin B. Cells were harvested by replacing the media with PBS + 0.4% EDTA and collected by centrifugation at 1,000 x g for 15 min at 4°C. Cells were homogenized in 20 vol. ice-cold membrane buffer. The homogenate was centrifuged at 48,000 x g for 10 min at 4°C, the supernatant discarded and the pellet resuspended in assay buffer A, centrifuged, and the final pellet resuspended in assay buffer. CB2-CHO cell membranes (12 μg) were incubated at 37°C for 90 min with 1 nM [3H]CP55,940 and the test compound in assay buffer containing 0.5% BSA. Binding was initiated as described above for cannabinoid CB1 receptors.
2.5 Agonist-Stimulated [35S]GTPγS Binding
Rat cerebellar membranes, prepared and assayed for protein content as for [3H]CP55,940 binding assays, were used for [35S]GTPγS binding assays. Concentration-effect curves for O-2050 and inhibition of CP55,940 activity by O-2050 were generated by incubating membrane protein (11-16 μg) in Assay Buffer with 1.4g/L bovine serum albumin (BSA) (Assay Buffer + BSA) in the presence of 30μM GDP and 0.1nM [35S]GTPγS in 0.5mL total volume for 2 hours at 30°C with 0.1-10,000nM O-2050 or 10-100,000nM O-2050 in the presence of 1000nM CP55,940, respectively. Basal binding was measured in the absence of agonist and non-specific binding was measured in the presence of 20μM unlabeled GTPγS. The reaction was terminated by vacuum filtration though Whatman GF/B glass fiber filters, followed by three washes with 4°C Tris buffer (50 mM Tris-HCl, pH 7.4). Bound radioactivity was determined by liquid scintillation spectrophotometry at 95% efficiency after 10-hour extraction in ScintiSafe Econo 1 scintillation fluid.
Specific [35S]GTPγS binding was determined by subtracting non-specific binding values from total binding values, and net-agonist stimulated binding was determined by subtracting specific basal binding values (obtained in the absence of agonist) from specific binding values obtained in the presence of agonist(s). Percent stimulation of [35S]GTPγS binding was calculated as net-agonist stimulated binding values divided by specific basal binding. Emax and EC50 or IC50 and Imax values for O-2050 were determined by non-linear fitting of % stimulation data to one-site models using Prism (GraphPad software). The KB value of O-2050 was calculated as IC50/(([L]/EC50)-1), where [L] = the concentration of CP55,940 and EC50 is that obtained for CP 55,940 in [35S]GTPγS binding assays in rat cerebellum.
2.6 Inhibition of forskolin-stimulated cyclic AMP production
Adherent Chinese hamster ovary (CHO) cells stably transfected with human cannabinoid CB1 receptors were washed once with Dulbecco’s phosphate buffered saline (PBS) and detached using a non-enzymatic cell dissociation solution. After centrifugation, the cells were resuspended (2 × 106 cells ml−1) in buffer containing PBS (calcium and magnesium free), 1% bovine serum albumin and 10 μM rolipram and then incubated for 30 minutes at 37°C with CP55940 or O-2050. A further 30 min incubation was performed with 10 μM forskolin in a total volume of 500 μl. The reaction was terminated by addition of 0.1 M HCl, followed by centrifugation to remove cell debris. The pH was then adjusted to between 8 and 9 by addition of 1 M of NaOH and cyclic AMP content was measured using a radioimmunoassay kit (Biotrak, Amersham). Forskolin and rolipram were dissolved in DMSO and stored at −20°C as 10 mM stock solutions.
2.7 Mouse Vas Deferens Procedure
Vasa deferentia were obtained from male MF1 outbred albino mice (Harlan UK Ltd., Blackthorn, UK) weighing 34 to 46 g. Each mouse vas deferens was mounted in a 4 ml organ bath at an initial tension of 0.5 g as described previously (Pertwee et al., 2002). The baths contained Mg2+-free Krebs solution which was kept at 35 to 36°C and bubbled with 95% O2 and 5% CO2. The composition of the Krebs solution was (mM): NaCl 118.2, KCl 4.75, KH2PO4 1.19, NaHCO3 25.0, glucose 11.0 and CaCl2.6H2O 2.54. Isometric contractions were evoked by applying 0.5 s trains of pulses of 110% maximal voltage (train frequency 0.1 Hz; pulse frequency 5 Hz; pulse duration 0.5 ms) through a platinum electrode attached to the upper end and a stainless steel electrode attached to the lower end of each bath. Stimuli were generated by a Grass S48 stimulator, then amplified (channel attenuator; MedLab Instruments) and divided to yield separate outputs to eight organ baths (StimuSplitter; MedLab Instruments). Contractions were monitored by computer (Apple Macintosh) using a data recording and analysis system (MacLab; ADInstruments, Chalgrove, England) that was linked via preamplifiers (Macbridge; ADInstruments, Chalgrove, England) to either Pioden UF1 transducers (Harvard Apparatus Ltd, Edenbridge, England) or MLT1030 transducers (ADInstruments, Chalgrove, England). After placement in an organ bath, each tissue was subjected to electrical stimulation of progressively greater intensity, followed by an equilibration procedure in which it was exposed to alternate periods of stimulation (2 min) and rest (10 min) until consistent twitch amplitudes were obtained. The stimulator was now switched off and O-2050 or DMSO added. Thirty minutes later, the first addition of WIN55212-2 was made. Additions of WIN55212-2 were made cumulatively at 15 min intervals without washout, the tissues being stimulated for the final two minutes of the 15 min exposure to each concentration of this agonist. Only one concentration-response curve was constructed per tissue. O-2050 was dissolved in DMSO and WIN55212-2 in a solution consisting of 50% DMSO and 50% saline. By themselves, these vehicles did not inhibit the twitch response. Drug additions were made in a volume of 10 μl.
For mouse vas deferens experiments, values are expressed as means and variability as 95% confidence limits or S.E.M. The degree of inhibition of evoked contractions induced by WIN55212-2 was calculated in percentage terms by comparing the amplitude of the twitch response after each addition of WIN55212-2 with its amplitude immediately before the first addition of this agonist. The dissociation constant (KB) of O-2050 was calculated from the slope of the best-fit straight line of a plot of (x-1) against B, constructed by linear regression analysis and constrained to pass through the origin (GraphPad Prism). The equation for this graph is (x-1) =B/KB, where x (the “concentration ratio”) is the concentration of a twitch inhibitor that produces a particular degree of inhibition in the presence of a competitive reversible antagonist at a concentration, B, divided by the concentration of the same twitch inhibitor that produces an identical degree of inhibition in the absence of the antagonist. The Schild slope for the interaction between WIN55212-2 and O-2050 was obtained from the best-fit straight line of a plot of log (x-1) against log B (GraphPad Prism). The equation for this graph, log (x-1) = log B-log KB, predicts a slope of unity for all receptor-mediated interactions between agonists and antagonists that are competitive and reversible. Values of the concentration ratio and its 95% confidence limits were determined by symmetrical (2 + 2) dose parallel line assays. This method was also used to establish whether 2-point log concentration-response plots deviated significantly from parallelism. Some mean values have been compared with zero using the 1-sample t test (GraphPad Prism).
2.8 Mouse In Vivo Screening Test Battery
Prior to testing in the behavioral procedures, mice were acclimated to the experimental setting (ambient temperature 22-24°C) overnight. Pre-injection control values were determined for rectal temperature and tail-flick latency (in sec). For agonist tests, mice were injected i.v. with an experimental compound or vehicle 5 min before being placed in individual activity chambers. For antagonist tests, mice were injected i.v. with the antagonist compound 10 min before the i.v. agonist injection. Five min later they were placed in individual activity chambers and spontaneous activity was measured for 10 min. Activity was measured as total number of interruptions of 16 photocell beams per chamber during the 10-min test and expressed as % inhibition of activity of the vehicle group. Tail-flick latency was measured at 20 min post-injection. Maximum latency of 10 s was used. Antinociception was calculated as percent of maximum possible effect {%MPE = [(test - control latency)/ (10-control)] X 100}. Average (± S.E.M.) control latency was 2.5 (0.08) sec. At 30 min post-injection, rectal temperature was measured. This value was expressed as the difference between control temperature (before injection) and temperatures following drug administration (Δ°C). Different mice (n=5-6 per dose) were tested for each dose of each compound. Each mouse was tested in each of the 3 procedures.
Introcerebroventricular (i.c.v.) injections were given as described previously (Welch et al., 1998; Wiley et al., 2000). Briefly, mice were prepared under light anesthesia and an incision was made to expose bregma. Injections were given with a 26-gauge needle to a site 2-mm lateral and 2-mm caudal to bregma at a depth of 2 mm. Each mouse received an i.c.v. injection volume of 5 μl of a given concentration of O-2050 dissolved in dimethyl sulfoxide, regardless of weight. Ten min later the mice were injected i.v. with 3 mg/kg Δ9-tetrahydrocannabinol or with dimethyl sulfoxide with subsequent placement into the locomotor chambers for the start of the test battery.
Data analysis for the structure-activity experiment was based on a scheme we have used in numerous previous studies with cannabinoids, with maximal cannabinoid effects in each procedure estimated as follows: 90% inhibition of spontaneous activity, 100% MPE in the tail flick procedure, and −6 °C change in rectal temperature. ED50 was defined as the dose at which half maximal effect occurred. For compounds that produced one or more cannabinoid effect, ED50 was calculated separately using least-squares linear regression on the linear part of the dose-effect curve for each measure in the mouse tetrad, plotted against log10 transformation of the dose. Data collected during combination tests with O-2050 and 3 mg/kg Δ9-tetrahydrocannabinol were converted to percentage antagonism [(mean score of group that received vehicle and 3 mg/kg Δ9-tetrahydrocannabinol minus score obtained with each O-2050 dose and 3 mg/kg Δ9-tetrahydrocannabinol)/(mean score of group that received vehicle and 3 mg/kg Δ9-tetrahydrocannabinol) X 100]. Data for each measure in the i.c.v. experiments were analyzed with two-way ANOVA (i.c.v. injection X i.v. injection). Tukey post hoc tests (α=0.05) were used to specify differences in the case of significant main or interactive effects of the ANOVA.
2.9 Mouse Feeding Experiment
The effects of O-2050 on feeding were assessed in separate group of mice. Twenty-four hours before the start of a feeding trial, all food was removed from the home cages of mice to be tested. The next day mice were transported to the laboratory at least one h before the beginning of the feeding trial. Thirty min before the feeding session they were injected i.p. with O-2050. Subsequently, they were placed in a clear plastic cage with thick brown paper lining the bottom and allowed access to a pre-measured amount of their regular lab chow. At the end of one hour, mice were removed from the test cage and placed back into their home cage. The amount of food left in the test cage, including crumbs was measured, and amount consumed was calculated. Mice in the feeding study received all doses of O-2050 presented in randomized Latin square order. Mice received no more than two feeding trials per week, separated by at least 72 hours.
Locomotor trials associated with the feeding experiment occurred in a different laboratory. For these tests, mice were injected i.p. with vehicle or O-2050 thirty min before being placed in individual activity chambers and spontaneous activity was measured for 10 min. Activity was measured as total number of interruptions of 16 photocell beams per chamber during the entire test session and expressed as % inhibition of activity of the vehicle group.
Separate one-way ANOVAs were used to analyze amount eaten during the feeding trials and % inhibition of activity in the locomotor tests. Tukey post hoc tests (α=0.05) were used to specify differences between individual means.
2.10 Drug Discrimination
Mice were trained to insert their snout into the nose poke apertures to receive food reinforcement as described in (Vann et al., 2009). Following acquisition of the nose poke response, mice were trained to respond on one nose poke following administration of 5.6 mg/kg Δ9-tetrahydrocannabinol and to respond on the opposite nose poke following vehicle injection according to a fixed ratio 10 schedule of reinforcement wherein they received a food pellet for every 10 consecutive nose pokes in the injection-appropriate aperture. Each response in the incorrect aperture reset the response requirement on the correct aperture. Daily injections were administered on a double alternation sequence of Δ9-tetrahydrocannabinol and vehicle (e.g., drug, drug, vehicle, vehicle). Daily 15-min training sessions were held Monday-Friday until the mice had met three criteria during 7 of 8 consecutive sessions: (1) the first completed fixed ratio-10 (e.g., consecutive correct responses ≥ 10) on the correct aperture, (2) ≥ 80% of the total responding occurred on the correct aperture and (3) average response rate ≥ 0.17 responses per second. When these three criteria were met, acquisition of the discrimination was established and substitution testing began.
Following successful acquisition of the discrimination, stimulus substitution tests were conducted on Tuesdays and Fridays during 15-min test sessions. Training continued on Mondays, Wednesdays, and Thursdays. During training sessions, only nose pokes in the injection-appropriate aperture were reinforced under a fixed ratio-10 schedule. During test sessions, responses on either aperture delivered reinforcement according to a fixed ratio-10 schedule. To be tested, mice must have completed the first ten responses on the correct aperture and ≥ 80% of the total responding must have occurred on the correct aperture on the preceding day. In addition, the mouse must have met these same criteria during previous training sessions with the alternate training condition (drug or vehicle). Prior to substitution tests with O-2050 and JWH-104, a generalization curve for Δ9-tetrahydrocannabinol was generated for each mouse.
For each drug discrimination test session, percentage of responses on the drug lever and response rate (responses/s) were calculated. Since mice that responded less than 10 times during a test session did not press either lever a sufficient number of times to earn a reinforcer, their data were excluded from analysis of drug lever selection, but their response rate data were included. Response-rate suppression (relative to rates after vehicle administration) was determined by separate analyses of variance (ANOVA) using GBSTAT statistical software (GB-STAT software; Dynamic Microsystems, Silver Spring, MD). Significant ANOVAs were further analyzed with Tukey post hoc tests (α= 0.05) to specify differences between means.
3.0 Results
3.1 Results Cannabinoid Receptor Affinities and In Vivo Assessment of O-2050
Table 1 shows results of tests with sulfonamide analogs of Δ8-tetrahydrocannabinol, in which the pentyl side chain at position C3 is replaced by an acetylene moiety or a dimethylpentyl group with a sulfonamide substituent and terminal carbon chain of varying length. Methyl substitution at the terminal end (O-2050) resulted in the best cannabinoid CB1 and CB2 receptor affinities of compounds with an acetylene substituent, with affinities for both cannabinoid receptors progressively decreasing as the chain was lengthened to ethyl (O-1991) and butyl (O-1993). In vivo potencies of O-1993 were correspondingly decreased by 4- to 14-fold as compared to O-1991; however, O-2050, the compound with the best cannabinoid CB1 receptor affinity of the three acetylene analogs, was not active in any of the three in vivo tests. For the final compound (O-2113), a dimethylpentyl group was substituted for the acetylene group of O-1991. O-2113 showed the best affinities for cannabinoid CB1 and CB2 receptors of all of the compounds presented in Table 1, with corresponding increases in potencies for all three in vivo tests (1.6- to 4.5-fold as compared to those of O-1991).
O-2050 had good cannabinoid CB1 receptor affinity; yet, it was not fully active when tested in vivo (up to doses of 30 mg/kg, i.v.) Consequently, further evaluation of its pharmacological effects was undertaken. First, O-2050 was tested for antagonist effects in mice. At doses up to 30 mg/kg i.v., O-2050 did not antagonize the in vivo pharmacological effects of a 3 mg/kg dose of Δ9-tetrahydrocannabinol (Fig. 1). Doses of up to 3 and 10 mg/kg O-2050 i.v. also did not antagonize the in vivo effects of 1 mg/kg WIN 55,212-2 (data not shown). Given previous reports of delayed action of some nitrogen containing Δ8-tetrahydrocannabinol analogs, 30 mg/kg O-2050 was injected 24 hr prior to injection with 3 mg/kg Δ9-tetrahydrocannabinol and subsequent in vivo testing. O-2050 did not have delayed antagonist effects (data not shown). When injected i.c.v., 10 μg O-2050 produced a 2-fold increase in antinociception as compared to vehicle (Fig. 2, middle left panel), but neither dose of O-2050 (3 or 10 μg) antagonized any of the in vivo effects of 3 mg/kg Δ9-tetrahydrocannabinol (Fig. 2, right panels), suggesting that failure to cross the blood-brain barrier was not a likely explanation of its lack of antagonism.
Fig. 1.
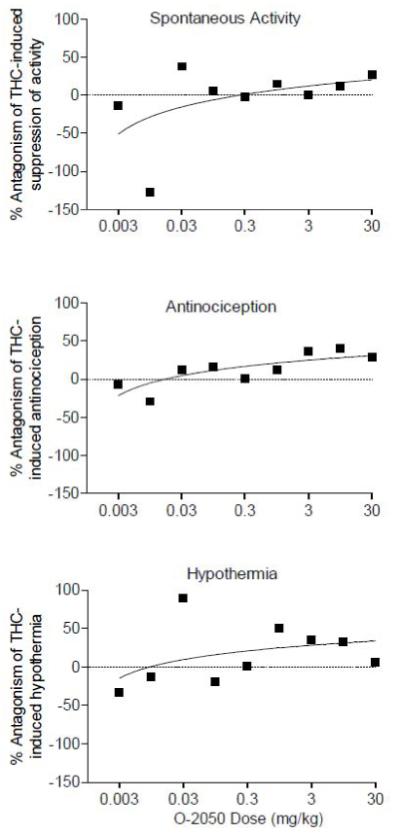
Effects of O-2050 (i.v.) across a wide dose range on percentage antagonism of hypomobility (top panel), antinociception (middle panel) and hypothermia (bottom panel) produced by 3 mg/kg (i.v.) Δ9-tetrahydrocannabinol in mice (n=5-6 mice/dose combination).
Fig. 2.
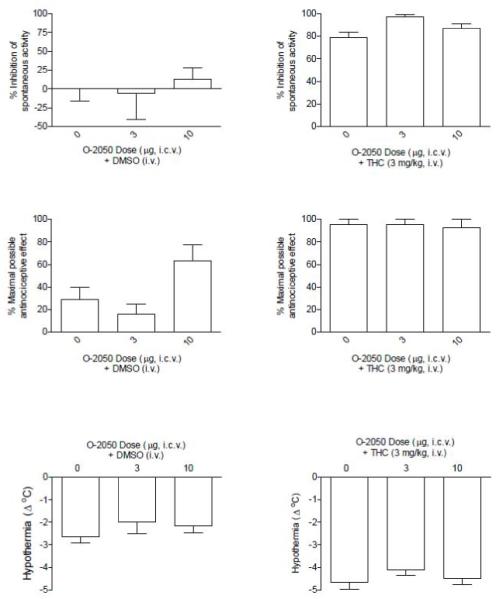
Effects of 3 and 10 μg O-2050 (i.c.v. at a volume of 5 μl) in combination with dimethyl sulfoxide (i.v.) [left panels] or with 3 mg/kg Δ9-tetrahydrocannabinol (i.v.) [right panels] on suppression of spontaneous activity (top panels), antinociception (middle panels) and hypothermia (bottom panels). Each bar represents the mean (± S.E.M.) of data from 4-5 mice.
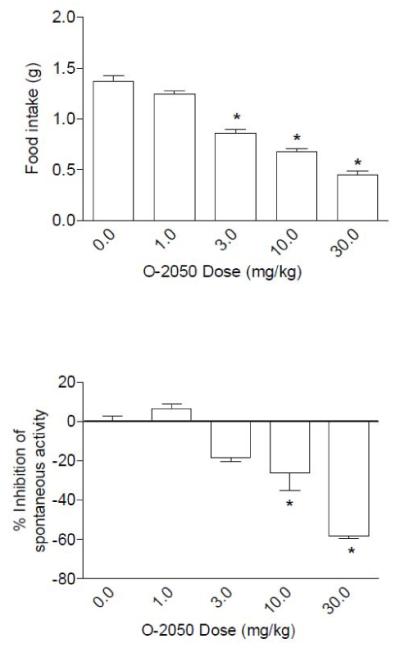
Fig. 3 shows the results of O-2050 on food intake in mice that had been food deprived for 24 hr. In this feeding model, O-2050 produced significant and dose-dependent decreases in food intake (Fig. 3, top panel). In addition, significant locomotor stimulation was observed at the 10 and 30 mg/kg doses (Fig. 3, bottom panel).
Fig. 3.
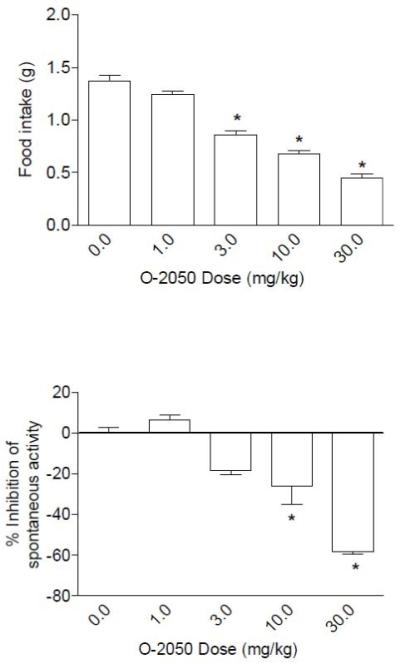
Effects of O-2050 on food intake (top panel) and suppression of locomotor activity (bottom panel). Bars represent the mean (± S.E.M.) of data from the same 10 mice at each dose for food intake and data from 5 separate mice at each dose for assessment of locomotor activity (% inhibition compared to vehicle group). * indicates significant difference from vehicle control (P < 0.05). (Note: On all graphs that illustrate % inhibition, negative numbers represent stimulation of locomotor activity.)
In mice trained to discriminate 5.6 mg/kg Δ9-tetrahydrocannabinol from vehicle, O-2050 produced discriminative stimulus effects that were similar to those of Δ9-tetrahydrocannabinol (Fig. 4). In contrast, JWH-104, a Δ8-THC analog with poor affinity and low efficacy, failed to produce Δ9-tetrahydrocannabinol-like discriminative stimulus effects. All three compounds significantly decreased response rates at higher doses (Fig. 4, bottom panel).
Fig. 4.
Effects of Δ9-tetrahydrocannabinol, O-2050 and JWH-104 on percentage of Δ9-tetrahydrocannabinol-appropriate responding (top panel) and response rates (bottom panel) in mice trained to discriminate 5.6 mg/kg Δ9-tetrahydrocannabinol from vehicle. Points above VEH and THC represent the results of control tests with vehicle and 5.6 mg/kg Δ9-tetrahydrocannabinol conducted before each dose-effect determination. Asterisks (*) represent significant decreases or increases in rates of responding compared to vehicle (P < 0.05). For each dose-effect curve determination, values represent the mean (±S.E.M.) of 5-11 mice.
3.2 [35S]GTPγS Binding, Cyclic AMP, and Mouse Vas Deferens Assays
Fig. 5 shows that O-2050 alone produced maximum stimulation of 11 ± 3% in the [35S]GTPγS binding assay. This magnitude of stimulation was only about half that produced by Δ9-THC (Breivogel et al., 1998). O-2050 appeared to exhibit an EC50 very close to the lowest concentration of O-2050 assayed (0.09 ± 0.01 nM). In addition to its weak stimulatory effects, O-2050 produced concentration-dependent inhibition of [35S]GTPγS binding stimulated by 1000 nM CP55,940. It blocked all but 11 ± 6 % of the stimulation by CP 55,940 with a KB of 22 ± 8 nM.
Fig. 5.
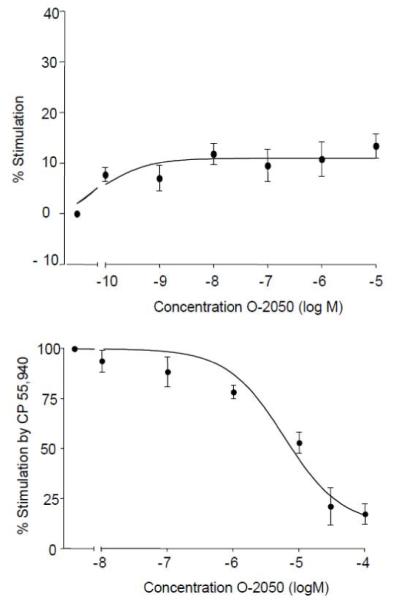
To evaluate agonism in the [35S]GTPγS binding assay, the effects of various concentrations of O-2050 alone were tested (top panel). To evaluate antagonism, O-2050 concentrations were tested in combination with 1000nM CP55,940 (bottom panel) in the assay. [35S]GTPγS binding was conducted in membranes prepared from rat cerebellum.
In a cyclic AMP assay, O-2050 acted as a partial agonist, producing dose-dependent inhibition of forskolin-induced stimulation of cyclic AMP (Fig. 6, top panel). The magnitude of this inhibition was approximately half of that observed with the full agonist CP55,940. Emax values were 37.4% (26.8 and 48.1%) for O-2050 and 74.4% (67.0 and 81.9%) for CP55,940. The corresponding EC50 values with 95% confidence intervals shown in parentheses were 40.4 nM (3.7 and 441 nM) and 12.2 nM (5.4 and 27.3 nM), respectively.
Fig. 6.
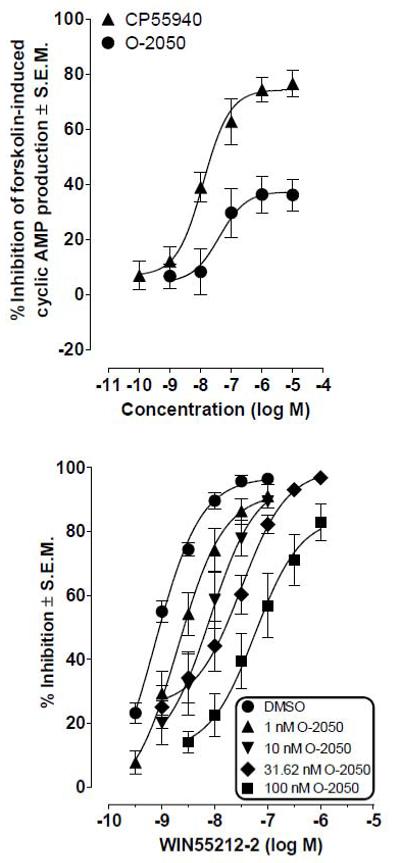
Top panel: Effects of O-2050 and CP55,940 on forskolin-induced stimulation of cyclic AMP production in human CB1 transfected CHO cells. Symbols represent mean values ± S.E.M. (n=5 for O-2050 and n=4 for CP55,940). Bottom panel: Effects of pretreatment with O-2050 on the mean log concentration-response curve of WIN55,212-2 in the mouse isolated vas deferens. Each symbol represents the mean value (± S.E.M.) for inhibition of electrically-evoked contractions expressed as a percentage of the amplitude of the twitch response measured immediately before the first addition of WIN55,212-2 to the organ bath. O-2050 or dimethyl sulfoxide was added 30 min before the first addition of WIN55,212-2, further additions of which were made at 15 min intervals. Each log concentration-response curve was constructed cumulatively (n=6 or 11).
In contrast to its partial agonist effects in the cyclic AMP assay, O-2050 acted only as an antagonist in the mouse vas deferens assay (Fig. 6, bottom panel). In this assay, it attenuated the ability of WIN 55,212-2 to inhibit electrically-evoked contractions, producing concentration-related dextral shifts in the log concentration response curve of WIN 55,212-2 that did not deviate significantly from parallelism. Dextral shifts after 1, 10, 31.62 and 100 nM O-2050 with 95% confidence limits in parentheses were 2.8 (1.7 - 4.4), 7.5 (4.1 - 13), 15.0 (9.9 - 21.9) and 103.9 (60.0 - 180.7), respectively (n=6 or 11). Data analysis yielded a Schild plot with a slope that was close to unity (0.83±0.18) and a mean KB value for O-2050 of 1.0±0.1 nM. O-2050 had little effect on the twitch response when administered by itself at concentrations of 1 nM to 1 μM. More specifically, mean changes in twitch height 30 min after 1, 10, 31.62, 100, or 1000 nM O-2050 were 6.03±6.21%, −0.45±5.9%, 3.39±9.4%, 3.8±8.1% and 6.19±16.4% respectively (n=6 to 11) and none of them was significantly different from zero (1-sample t test; P>0.05).
4.0 Discussion
Traditional receptor theory posits that ligand-receptor interactions may be characterized at minimum by affinity and efficacy, with the property of affinity shared by both agonists and antagonists whereas efficacy for receptor activation is specific for agonists. In the case of inverse agonists, the functional consequences of the resulting receptor activation are the reverse of those observed with direct agonists (i.e., negative modulation). At cannabinoid CB1 receptors, the prototypic antagonist rimonabant has good affinity without efficacy at lower concentrations; however, at higher concentrations, it has been purported to possess inverse agonist effects (Landsman et al., 1997). Consequently, synthesis efforts in this area have emphasized development of a neutral cannabinoid CB1 receptor antagonist. O-2050, one promising candidate, was the focus of investigation in this study.
O-2050 was originally synthesized as one of a series of Δ8-tetrahydrocannabinol derivatives with carboxamide or sulfonamide substituents at the terminal end of the C3 side chain. Previous research had shown that incorporation of a nitrogen substituent into the C3 side chain resulted in a series of compounds with agonist-antagonist properties (Martin et al., 1999; Wiley et al., 1996). In particular, terminal cyano substitution was shown to produce irregular effects on in vivo potency (enhancement or no effect; Martin et al., 1999; Singer et al., 1998; Wiley et al., 1996), but, in certain cases, to produce antagonism in vitro (Crocker et al., 1999; Griffin et al., 1999; Pertwee et al., 1996). In addition, various carboxamide and sulfonamide derivatives exhibited a pattern of no effect or partial agonist effects in vivo despite good binding affinity (Singer et al., 1998). These latter derivatives also showed potent antagonist activity for the peptidoleukotrienes, a class of eicosanoids (Jacobs et al., 1993; Matassa et al., 1990).
In contrast with the findings reported for other sulfonamide series, the four compounds in the abbreviated series of sulfonamide cannabinoids presented here exhibited good to modest cannabinoid CB1 receptor affinity, with three of the four showing even better affinity for cannabinoid CB2 receptors (12-fold for O-2050 and 21-fold for O-1991 and O-2113). With the exception of O-2050, all compounds acted as agonists in a battery of in vivo tests in mice. The flexibility of the proximal end of the C3 side chain was restricted through inclusion of an acetylene moiety or a dimethyl group in all four compounds. Notably, however, the portion of the C3 substituent that was terminal to the sulfonamide group was shortest for O-2050 and presumably had the least flexibility as a consequence. Previous structure-activity relationship analysis of a series of 1′,1′-dimethylalkyl-Δ8-tetrahydrocannabinol analogs demonstrated that optimum affinity and potency of tetrahydrocannabinols were achieved when the terminal end of the side chain retained the ability to loop back in proximity to the phenolic ring (Huffman et al., 2003). These results suggest that the acetylene moiety of O-2050 may force the side chain into a different angle than in alkyl substituted tetrahydrocannabinols, thereby allowing the sulfonamide moiety to interact with the cannabinoid CB1 receptor at a site which results in high recognition but very poor activation. Sulfonamides with longer terminal side chains may have sufficient flexibility to overcome the restriction imposed by the acetylene moiety.
In functional assays, O-2050 alone failed to alter the mouse vas deferens twitch response and produced only minor stimulation of [35S]GTPγS binding. It also partially inhibited forskolin-stimulated cyclic AMP production. Interestingly, the magnitude of O-2050 efficacy in the latter assay was greater than for [35S]GTPγS binding, as is consistent with previous research showing that partial agonists exhibit greater efficacy in cyclic AMP signaling as compared to upstream signaling mechanisms (Breivogel and Childers, 2000). These results also offer some support for the idea that O-2050 may have poor efficacy at cannabinoid CB1 receptors, despite its good binding affinity. Also consistent with this idea that O-2050 has little efficacy at cannabinoid CB1 receptors is the fact that it acted as an antagonist in the mouse vas deferens assay. Further, and in contrast to rimonabant (Pertwee et al., 1996), O-2050 did not enhance electrically-evoked contractions of the vas deferens even at concentrations well above its KB value, suggesting that O-2050 is not an inverse agonist and may be a neutral cannabinoid CB1 antagonist. Results from in vivo evaluation, however, are not consistent with this hypothesis. While O-2050 does not produce the Δ9-tetrahydrocannabinol-like effects of hypomobility, antinociception and hypothermia, it also does not antagonize these effects (or those of WIN 55,212-2). This lack of antagonism is not due to failure to reach the brain or to delayed action, as has been reported for some nitrogen-containing cannabinoids (Compton et al., 1990).
When administered by itself to mice, the pharmacological profile of O-2050 is even more complex. Some of its effects resemble those seen with rimonabant. Namely, both O-2050 and rimonabant decrease eating (present study; Gardner and Mallet, 2006) and increase locomotion (present study). Yet, O-2050 also produces full, dose-dependent Δ9-tetrahydrocannabinol-like discriminative stimulus effects, which is typically only observed with psychoactive cannabinoid agonists (Balster and Prescott, 1992) and is antagonized by rimonabant (Wiley et al., 1995). These results are discordant with the lack of agonist activity in locomotor, nociceptive and temperature assays and are without a clear explanatory mechanism; however, it is worth noting that the partial agonist effects of O-2050 to inhibit cyclic AMP and stimulate G-protein signaling is consistent with its Δ9-tetrahydrocannabinol-like effects in drug discrimination. While cannabinoid discrimination in mice is relatively novel (most previous research in this area was done in rats), it has been used successfully to distinguish cannabinoids from non-cannabinoids (McMahon et al., 2007; Vann et al., 2009). Further, in the present study, JWH-104, a Δ8-tetrahydrocannabinol analog from another series (Wiley et al., 2002), did not substitute for Δ9-tetrahydrocannabinol, suggesting that substitution was somewhat selective in that it did not occur across all cannabinoids within this class. Several other factors may have contributed to the disconnection between the agonist-like substitution for Δ9-tetrahydrocannabinol in a drug discrimination procedure and the absence of O-2050 effects in the triad of in vivo assays. First, ICR mice were tested in the triad assays whereas C57/Bl6 mice were used in the drug discrimination experiments. Route of administration also differed (i.v. and i.c.v. in triad and s.c. in drug discrimination), as did the degree of lifelong cannabinoid exposure (acute in triad and chronic in drug discrimination). Finally, the degree to which interaction of O-2050 with cannabinoid CB2 receptors may have modulated responses in the different procedures is unknown. Although a direct role of cannabinoid CB2 receptors alone in mediation of effects in the triad and discriminative stimulus effects of cannabinoid agonists has been ruled out (Järbe et al., 2006; Wiley et al., 2002), possible effects of interactions among cannabinoid CB1 and CB2 receptor mechanisms in these behaviors have not been extensively explored.
In summary, O-2050 presents a complex pharmacological profile that includes some aspects which resembled cannabinoid CB1 receptor antagonism. Similar to rimonabant, O-2050 exhibited excellent affinity for cannabinoid CB1 receptors and lacked efficacy in some functional in vitro assays or in a battery of in vivo tests in mice. Further, O-2050 antagonized the in vitro effects of cannabinoid agonists. By itself, it stimulated locomotor activity and decreased feeding. Other aspects of the pharmacological profile of O-2050, however, resembled cannabinoid agonism. Although O-2050 failed to produce characteristic cannabinoid effects in a battery of in vivo tests, it also did not reverse those produced by cannabinoid agonists. The Δ9-tetrahydrocannabinol-like discriminative stimulus effects produced by O-2050 are even more intriguing, as is its ability to serve as a partial agonist in assays measuring inhibition of cyclic AMP signaling and stimulation of G-protein binding. Hence, O-2050 produces myriad pharmacological in vivo and in vitro effects, only some of which are consistent with antagonism at CB1 receptors. Further, its good affinity for cannabinoid CB2 receptors complicates its use as a tool to evaluate the unique contribution of cannabinoid CB1 receptor mediation of specific pharmacological effects. Together, these results suggest that overall classification of O-2050 as a neutral cannabinoid CB1 antagonist is inaccurate.
Acknowledgements
This work was supported by National Institutes of Health, National Institute on Drug Abuse Grants DA-03672, DA-05488, and DA-03590.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Balster RL, Prescott WR. Δ9-Tetrahydrocannabinol discrimination in rats as a model for cannabis intoxication. Neurosci. Biobehav. Rev. 1992;16:55–62. doi: 10.1016/s0149-7634(05)80051-x. [DOI] [PubMed] [Google Scholar]
- Bass CE, Griffin G, Grier M, Mahadevan A, Razdan RK, Martin BR. SR-141716A-induced stimulation of locomotor activity. A structure-activity relationship study. Pharmacol. Biochem. Behav. 2002;74:31–40. doi: 10.1016/s0091-3057(02)00945-0. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Breivogel CS, Selley DE, Childers SR. Cannabinoid receptor agonist efficacy for stimulating [35S]GTPγS binding to rat cerebellar membranes correlates with agonist-induced decreases in GDP affinity. J. Biol. Chem. 1998;273:16865–16873. doi: 10.1074/jbc.273.27.16865. [DOI] [PubMed] [Google Scholar]
- Chambers AP, Vemuri VK, Peng Y, Wood JT, Olszewska T, Pittman QJ, Makriyannis A, Sharkey KA. A neutral CB1 receptor antagonist reduces weight gain in rat. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007;293:R2185–2193. doi: 10.1152/ajpregu.00663.2007. [DOI] [PubMed] [Google Scholar]
- Compton DR, Aceto MD, Lowe J, Martin BR. In vivo characterization of a specific cannabinoid receptor antagonist (SR141716A): inhibition of delta 9-tetrahydrocannabinol-induced responses and apparent agonist activity. J. Pharmacol. Exp. Ther. 1996;277:586–594. [PubMed] [Google Scholar]
- Compton DR, Little PJ, Martin BR, Gilman JW, Saha JK, Jorapur VS, Sard HP, Razdan RK. Synthesis and pharmacological evaluation of amino, azido, and nitrogen mustard analogues of 10-substituted cannabidiol and 11- or 12- substituted Δ8-tetrahydrocannabinol. J. Med. Chem. 1990;33:1437–1443. doi: 10.1021/jm00167a025. [DOI] [PubMed] [Google Scholar]
- Cota D, Marsicano G, Lutz B, Vicennati V, Stalla GK, Pasquali R, Pagotto U. Endogenous cannabinoid system as a modulator of food intake. Int. J. Obes. Relat. Metab. Disord. 2003;27:289–301. doi: 10.1038/sj.ijo.0802250. [DOI] [PubMed] [Google Scholar]
- Coutts AA, Brewster N, Ingram T, Razdan RK, Pertwee RG. Comparison of novel cannabinoid partial agonists and SR141716A in the guinea-pig small intestine. Br. J. Pharmacol. 2000;129:645–652. doi: 10.1038/sj.bjp.0703094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crocker PJ, Saha B, Ryan WJ, Wiley JL, Martin BR, Ross RA, Pertwee RG, Razdan RK. Development of agonists, partial agonists and antagonists in the Δ-tetrahydrocannabinol series. Tetrahedron. 1999;55:13907–13926. [Google Scholar]
- Gardner A, Mallet PE. Suppression of feeding, drinking, and locomotion by a putative cannabinoid receptor ‘silent antagonist’. Eur. J. Pharmacol. 2006;530:103–106. doi: 10.1016/j.ejphar.2005.11.032. [DOI] [PubMed] [Google Scholar]
- Griffin G, Wray EJ, Rorrer WK, Crocker PJ, Ryan WJ, Saha B, Razdan RK, Martin BR, Abood ME. An investigation into the structural determinants of cannabinoid receptor ligand efficacy. Br. J. Pharmacol. 1999;126:1575–1584. doi: 10.1038/sj.bjp.0702469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi S, Irie K, Mishima S, Araki M, Ohji M, Shirakawa A, Akitake Y, Matsuyama K, Mishima K, Mishima K, Iwasaki K, Fujiwara M. The cannabinoid 1-receptor silent antagonist O-2050 attenuates preference for high-fat diet and activated astrocytes in mice. J. Pharmacol. Sci. 2010;112:369–372. doi: 10.1254/jphs.09326sc. [DOI] [PubMed] [Google Scholar]
- Huffman JW, Miller JR, Liddle J, Yu S, Thomas BF, Wiley JL, Martin BR. Structure-activity relationships for 1′,1′-dimethylalkyl-Delta8-tetrahydrocannabinols. Bioorg. Med. Chem. 2003;11:1397–1410. doi: 10.1016/s0968-0896(02)00649-1. [DOI] [PubMed] [Google Scholar]
- Jacobs RT, Brown FJ, Cronk LA, Aharony D, Buckner CK, Kusner EJ, Kirkland KM, Neilson KL. Substituted 3-(phenylmethyl)-1H-indole-5-carboxamides and 1-(phenylmethyl)indole-6-carboxamides as potent, selective, orally active antagonists of the peptidoleukotrienes. J. Med. Chem. 1993;36:394–409. doi: 10.1021/jm00055a011. [DOI] [PubMed] [Google Scholar]
- Järbe TU, Liu Q, Makriyannis A. Antagonism of discriminative stimulus effects of delta(9)-THC and (R)-methanandamide in rats. Psychopharmacology (Berl) 2006;184:36–45. doi: 10.1007/s00213-005-0225-y. [DOI] [PubMed] [Google Scholar]
- Landsman RS, Burkey TH, Consroe P, Roeske WR, Yamamura HI. SR141716A is an inverse agonist at the human cannabinoid CB1 receptor. Eur. J. Pharmacol. 1997;334:R1–2. doi: 10.1016/s0014-2999(97)01160-6. [DOI] [PubMed] [Google Scholar]
- Lichtman AH, Varvel SA, Martin BR. Endocannabinoids in cognition and dependence. Prostaglandins Leukot. Essent. Fatty Acids. 2002;66:269–285. doi: 10.1054/plef.2001.0351. [DOI] [PubMed] [Google Scholar]
- Martin BR, Jefferson R, Winckler R, Wiley JL, Huffman JW, Crocker PJ, Saha B, Razdan RK. Manipulation of the tetrahydrocannabinol side chain delineates agonists, partial agonists, and antagonists. J. Pharmacol. Exp. Ther. 1999;290:1065–1079. [PubMed] [Google Scholar]
- Matassa VG, Maduskuie TP, Jr., Shapiro HS, Hesp B, Snyder DW, Aharony D, Krell RD, Keith RA. Evolution of a series of peptidoleukotriene antagonists: synthesis and structure/activity relationships of 1,3,5-substituted indoles and indazoles. J. Med. Chem. 1990;33:1781–1790. doi: 10.1021/jm00168a037. [DOI] [PubMed] [Google Scholar]
- Mato S, Pazos A, Valdizan EM. Cannabinoid receptor antagonism and inverse agonism in response to SR141716A on cAMP production in human and rat brain. Eur. J. Pharmacol. 2002;443:43–46. doi: 10.1016/s0014-2999(02)01575-3. [DOI] [PubMed] [Google Scholar]
- McMahon LR, Ginsburg BC, Lamb RJ. Cannabinoid agonists differentially substitute for the discriminative stimulus effects of Delta(9)-tetrahydrocannabinol in C57BL/6J mice. Psychopharmacology (Berl) 2008;198:487–495. doi: 10.1007/s00213-007-0900-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Research Council . Guide for the Care and Use of Laboratory Animals. National Academy Press; Washington, D.C.: 1996. [Google Scholar]
- Pertwee RG. Inverse agonism and neutral antagonism at cannabinoid CB1 receptors. Life Sci. 2005;76:1307–1324. doi: 10.1016/j.lfs.2004.10.025. [DOI] [PubMed] [Google Scholar]
- Pertwee RG, Fernando SR, Griffin G, Ryan W, Razdan RK, Compton DR, Martin BR. Agonist-antagonist characterization of 6′-cyanohex-2′-yne-delta-8-tetrahydrocannabinol in two isolated tissue preparations. Eur. J. Pharmacol. 1996;315:195–201. doi: 10.1016/s0014-2999(96)00631-0. [DOI] [PubMed] [Google Scholar]
- Pertwee RG, Ross RA, Craib SJ, Thomas A. (−)-Cannabidiol antagonizes cannabinoid receptor agonists and noradrenaline in the mouse vas deferens. Eur. J. Pharmacol. 2002;456:99–106. doi: 10.1016/s0014-2999(02)02624-9. [DOI] [PubMed] [Google Scholar]
- Rinaldi-Carmona M, Barth F, Heaulme M, Alonso R, Shire D, Congy C, Soubrie P, Breliere JC, Le Fur G. Biochemical and pharmacological characterisation of SR141716A, the first potent and selective brain cannabinoid receptor antagonist. Life Sci. 1995;56:1941–1947. doi: 10.1016/0024-3205(95)00174-5. [DOI] [PubMed] [Google Scholar]
- Rinaldi-Carmona M, Barth F, Héaulme M, Shire D, Calandra B, Congy C, Martinez S, Maruani J, Néliat G, Caput D, Ferrara P, Soubrié P, Brelière JC, Le Fur G. SR141716A, a potent and selective antagonist of the brain cannabinoid receptor. FEBS Lett. 1994;350:240–244. doi: 10.1016/0014-5793(94)00773-x. [DOI] [PubMed] [Google Scholar]
- Sano K, Koushi E, Irie K, Higuchi S, Tsuchihashi R, Kinjo J, Egashira N, Oishi R, Uchida N, Nagai H, Nishimura R, Tanaka H, Morimoto S, Mishima K, Iwasaki K, Fujiwara M. Delta(9)-tetrahydrocannabinol enhances an increase of plasma corticosterone levels induced by forced swim-stress. Biol. Pharm. Bull. 2009;32:2065–2067. doi: 10.1248/bpb.32.2065. [DOI] [PubMed] [Google Scholar]
- Selley DE, Stark S, Sim LJ, Childers SR. Cannabinoid receptor stimulation of guanosine-5′-O-(3-[35S]thio)triphosphate binding in rat brain membranes. Life Sci. 1996;59:659–668. doi: 10.1016/0024-3205(96)00347-5. [DOI] [PubMed] [Google Scholar]
- Singer M, Ryan WJ, Saha B, Martin BR, Razdan RK. Potent cyano and carboxamide side-chain analogus of 1′,1′-dimethyl-Δ8-tetrahydrocannabinol. J. Med. Chem. 1998;41:4400–4407. doi: 10.1021/jm9803875. [DOI] [PubMed] [Google Scholar]
- Solinas M, Goldberg SR, Piomelli D. The endocannabinoid system in brain reward processes. Br. J. Pharmacol. 2008;154:369–383. doi: 10.1038/bjp.2008.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas A, Ross RA, Saha B, Mahadevan A, Razdan RK, Pertwee RG. 6″-Azidohex-2″-yne-cannabidiol: a potential neutral, competitive cannabinoid CB1 receptor antagonist. Eur. J. Pharmacol. 2004;487:213–221. doi: 10.1016/j.ejphar.2004.01.023. [DOI] [PubMed] [Google Scholar]
- Vann RE, Warner JA, Bushell K, Huffman JW, Martin BR, Wiley JL. Discriminative stimulus properties of Delta9-tetrahydrocannabinol (THC) in C57Bl/6J mice. Eur. J. Pharmacol. 2009;615:102–107. doi: 10.1016/j.ejphar.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch SP, Huffman JW, Lowe J. Differential blockade of the antinociceptive effects of centrally administered cannabinoids by SR141716A. J. Pharmacol. Exp. Ther. 1998;286:1301–1308. [PubMed] [Google Scholar]
- Wiley JL, Burston JJ, Leggett DC, Alekseeva OO, Razdan RK, Mahadevan A, Martin BR. CB1 cannabinoid receptor-mediated modulation of food intake in mice. Br. J. Pharmacol. 2005;145:293–300. doi: 10.1038/sj.bjp.0706157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley JL, Compton DR, Gordon PM, Siegel C, Singer M, Dutta A, Lichtman AH, Balster RL, Razdan RK, Martin BR. Evaluation of agonist-antagonist properties of nitrogen mustard and cyano derivatives of delta 8-tetrahydrocannabinol. Neuropharmacology. 1996;35:1793–1804. doi: 10.1016/s0028-3908(96)00120-7. [DOI] [PubMed] [Google Scholar]
- Wiley JL, Jefferson RG, Griffin G, Liddle J, Yu S, Huffman JW, Martin BR. Paradoxical pharmacological effects of deoxy-tetrahydrocannabinol analogs lacking high CB1 receptor affinity. Pharmacology. 2002;66:89–99. doi: 10.1159/000065631. [DOI] [PubMed] [Google Scholar]
- Wiley JL, Lowe JA, Balster RL, Martin BR. Antagonism of the discriminative stimulus effects of delta 9-tetrahydrocannabinol in rats and rhesus monkeys. J. Pharmacol. Exp. Ther. 1995;275:1–6. [PubMed] [Google Scholar]
- Wiley JL, Patrick GS, Crocker PC, Saha B, Razdan RK, Martin BR. Antinociceptive effects of tetrahydrocannabinol side chain analogs: dependence upon route of administration. Eur. J. Pharmacol. 2000;397:319–326. doi: 10.1016/s0014-2999(00)00259-4. [DOI] [PubMed] [Google Scholar]