

Abstract
Recently, we revealed the architecture of the clamp–loader–helicase (τ-DnaB) complex in Bacillus by atomic force microscopy imaging and constructed a structural model, whereby a pentameric clamp–loader interacts with the hexameric helicase. Crucial to this model is the assumption that the clamp–loader forms a pentamer in the absence of other components of the clamp–loader complex such as δδ′. Here, we show that the Bacillus subtilis τ protein, even in the absence of δδ′, interacts as a pentamer with the hexameric DnaB and that the L381 of τ is critical for the integrity of the τ oligomer and interaction with DnaB. The effects of the L381A mutation were confirmed by gel filtration, ultracentrifugation, circular dichroism, cross-linking studies, and genetic replacement of the dnaX gene with a mutant L381A dnaX gene in vivo. The L381A protein is able to support growth in vivo only when expressed in high quantities. Finally, despite the fact that a mutation at P465 has been reported to result in a thermosensitive gene in vivo, a P465L mutant protein interacts with DnaB in vitro suggesting that this defect is not a result of a defective τ-DnaB interaction.
DNA polymerase III holoenzyme (DNA pol. III HE)1 is a 10-polypeptide complex that forms part of the replisome (1). It consists of three functionally distinct but interconnected subassemblies (2). The pol. III core is formed by three different polypeptides, α, ε, and θ. The sliding clamp (or processivity factor β) and the clamp–loader complex (also known as the DnaX or γ complex), are DNA-dependent ATPases that consist of six different polypeptides, τ, γ, δ, δ′, χ, and ψ. Functional homologues exist in eukaryotes where the proliferating cell nuclear antigen (PCNA) and the replication factor C (RFC) complex play the roles of the sliding clamp processivity factor and the clamp–loader, respectively (3, 4). Likewise, in bacteriophage T4, the functional homologues of the sliding clamp and the clamp loader are the gp45 and gp44/62 proteins, respectively (5). The τ and γ polypeptides are coded by the same gene (dnaX), but γ lacks the C-terminal domain (Cτ) of τ because of premature translational termination (6, 7). Both τ and γ (to avoid confusion, DnaX refers to both τ and γ) form the central structural core to which δ, δ′, χ, and ψ bind to form the complete clamp–loader complex that binds and delivers the processivity factor, β, onto primed DNA (8). The DnaX complex is also involved in interactions with the pol. III core (9), DnaB (10), and the single-strand DNA binding protein, SSB (11). Its role is not limited to a structural one, by acting as a connector at the center of the replisome, or simply delivering the sliding clamp processivity factor β, as the τ-mediated stimulation of DnaB helicase activity is crucial for the fast progression of the DNA replication fork (10). Mutations in the large subunit of RFC or in the C-terminal portion of τ result in elevated mutation rates during DNA replication (12, 13). Therefore, clamp–loaders are also crucial for maintaining high fidelity during DNA replication. Domain mapping analysis located both the α and the DnaB interaction interfaces within the Cτ domain of τ (14, 15). Although the τ interaction interface of DnaB is still unknown, the equivalent interaction interface of pol. α has been identified to the last 48 amino acid residues at the C-terminus of α (9).
Structural information from the crystal structures of the Escherichia coli clamp–loader complex, containing the γ polypeptide bound to δδ′ (16), the δβ complex (17), and the δ′ (18) and β subunits (19), has revealed important insights into the mechanism of the clamp–loading reaction (reviewed in ref 8). A surprising finding of the γ complex structure was the stoichiometry of the complex, δ′γ3δ. It was previously assumed that the correct stoichiometry of the complex was τ4δδ′χψ (20, 21). Recent biochemical data also provided evidence for the DnaX3δδ′χψ stoichiometry (22). Domain III binds δδ′ and χψ (23), and although chemical cross-linking and immunoblotting experiments showed that both γ and τ can bind δ′ in reconstituted in vitro complexes of purified DNA pol. III HE, only γ cross-links to δ′ (24). Clearly, domain III is important for oligomerization and cooperative interactions between the subunits in the DnaX complex. The γ polypeptide in the crystal structure is a truncated version (residues 1–373) lacking the last nine residues of domain III (16, 23). Furthermore, at least one of the subunits in the DNA pol. III HE must be the τ polypeptide, which is absent from the crystal structure, and its effect on the overall conformation of the complex is unknown. The Cτ domain mediates the interaction with DnaB, but the molecular details of this interaction are poorly understood at present. On the basis of atomic force microscopy (AFM) images, we were able to construct a structural model for the τ-DnaB interaction whereby a τ pentamer interacts with the hexameric DnaB. The model is based upon the assumption that τ forms pentamers that interact with DnaB. In this work, we tested this assumption using biochemical and biophysical methodology. Since DnaB from Bacillus stearothermophilus strain NCA1503 (ATCC 29609) is a stable hexamer under a variety of conditions, including low or high salt and the presence or absence of Mg2+, nucleotides, and DNA (25), we decided to take advantage of these properties in this study and also in our previous structural studies. This strain is related more closely to B. subtilis than other stearothermophilus strains (26), with the subtilis and stearothermophilus DnaB proteins sharing 82% identity and 92% similarity. Studying the B. subtilis τ protein also allows us to compare our results with the more extensively studied τ protein from the Gram -ve E. coli.
We present in vitro biochemical and biophysical evidence to show that, even in the absence of δδ′, τ forms pentamers that interact with DnaB hexamers. This is distinctly different than the E. coli system where τ has been reported to exist as a tetramer. Furthermore, a conserved leucine residue (L381) is critical for pentamerization and also for interaction with DnaB. A mutant L381A protein forms a lower oligomer and is capable of supporting growth in vivo only when expressed in large quantities consistent with a somewhat defective interaction with DnaB. In contrast, a mutation at another position (P465) does not affect binding to DnaB despite the fact that a ts B. subtilis strain (ts dnaX51) has been previously reported to carry a mutation at this position. The structural significance of the τ-DnaB (pentamer:hexamer) complex has already been revealed by AFM.
EXPERIMENTAL PROCEDURES
Cloning the B. subtilis dnaX Gene
The B. subtilis (strain 168 EMG50) dnaX gene was cloned by PCR using the XNcoI (5′-GGGCAAACCCATGGTGAGTTACCAAGCTTTA-3′) and XBam (5′-TCATTTTGGGATCCTTAGTCTTTTATTTCA-3′) primers that introduce unique NcoI and BamHI sites at the beginning and end of the gene, respectively. The starting codon of the dnaX gene is GTG, and the XNcoI primer is inserted the sequence ATGGGG in front of the GTG codon. This modification introduced a methionine–glycine dipeptide in front of the gene that improved expression of the τ protein. The inserted dipeptide is unlikely to have resulted in structural changes, as the first two residues M and A of the E. coli γ protein are not visible in the crystal structure of the γ complex because they are highly mobile and free of interactions with other residues (16). Furthermore, an N-terminally tagged E. coli τ behaves like the untagged protein, implying that these N-terminal modifications have no adverse structural effect (14, 23). The amplified dnaX gene was cloned as an NcoI–BamHI fragment into the pET28a (Novagen) expression vector to construct the pET28adnaX plasmid. A glutamate was found at position 533 instead of an alanine (data not shown). The same sequence was also reported in the B. subtilis YB886 dnaX gene (27), raising the possibility that the sequence deposited (NCBI-GB NC000964) is incorrect.
Protein Purifications
BstDnaB was purified as described before (25, 28). Full-length τ was purified using a combination of medium substitution Blue Sepharose and MonoQ and Superdex S200 chromatography (Amersham Pharmacia Biotech). Typically, 4 L of BL21(DE3) E. coli cells were grown at 37 °C, in the presence of kanamycin (30 μg/mL) until the OD600 was 0.6. Expression was induced by IPTG (1 mM) for 3 h. The cell pellet was suspended in buffer A (50 mM Tris pH 7.5, 2 mM EDTA, 1 mM DTT), and cells were disrupted by sonication in the presence of PMSF (20 μM). The clarified supernatant was loaded onto a medium substitution Blue Sepharose column, maintaining the conductivity below 10 mS, and eluted with 2 M NaCl in buffer A. After precipitation with (NH4)2SO4, the protein was suspended in buffer B (50 mM glycine pH 9.5, 2 mM EDTA, 1 mM DTT) and loaded onto a MonoQ column maintaining the conductivity below 10 mS and eluted with a NaCl gradient (0–600 mM). Finally, the protein was applied onto a gel filtration Superdex S200 column equilibrated in buffer A plus 100 mM NaCl. Fractions containing τ were pooled, made up to 10% v/v glycerol, aliquoted, frozen in liquid nitrogen, and stored at −80 °C. Typical yields of 10–12 mg of τ per liter were obtained. The protein was >98% pure as assessed by SDS–PAGE and Coomassie blue staining (data not shown). Mutant proteins were purified as described for the wt protein.
Gel Filtration
The formation of a stable τ-DnaB complex was monitored by gel filtration using Superose 6 HR 10/30 or Superdex S200 HR 10/30 size exclusion columns (Amersham Pharmacia Biotech), equilibrated with TED/100 buffer (buffer A, 100 mM NaCl). The complex was formed by mixing 1.37 nM DnaB with 4.1 nM τ (referring to monomers) in a total volume of 0.5 mL of TED/100 buffer. The mixture was incubated on ice for 15 min and then loaded onto the appropriate column, at 0.5 mL/min, collecting 0.5 mL fractions. Control experiments with DnaB and τ proteins separately were carried out in an identical manner. The elution profile was monitored at 280 nm, and fractions containing the proteins were analyzed by SDS–PAGE and Coomassie blue staining. Similar experiments were carried out for the mutant τ proteins.
Gel filtration with the Superdex S200 and S75 HR 10/30 columns was also used to estimate the MW of the various DnaB-τ and τ complexes. Both columns were calibrated using proteins of known MW purified previously in our lab, P16 the 16 kDa C-terminal fragment of B. stearothermophilus DnaG, the 36 kDa DnaI and 44 kDa YxaL proteins from B. subtilis, the 82.5 kDa PcrA helicase from B. stearothermophilus, and the 303 kDa hexameric DnaB helicase from B. stearothermophilus (data not shown).
Site-Directed Mutagenesis
Site-directed mutagenesis was carried out using the two-step splicing by overlap method, as described elsewhere (29). The Q380A and L381A mutations were constructed by mutagenic forward and reverse oligonucleotides as follows: Q380AF (5′-GAAAAAAATCCAGGCGCTCGAACAGGA-3′), Q380AR (5′-TCCTGTTCGAGCGCCTGGATTTTTTC-3′), L381AF (5′-AATCCAGCAGGCGGAACAGGAAGTAG-3′), and L381-AR (5′-CTACTTCCTGTTCCGCCTGCTGGATT-3′).
The dnaXL381PP465L mutant gene was isolated during PCR cloning of the wt dnaX gene. The L381P and P465L single mutants were constructed by rearranging restriction endonuclease fragments within dnaX. The wt and L381PP465L dnaX genes were digested with EcoRI and BamHI restriction endonucleases. EcoRI restriction endonuclease cuts at a unique site between the two mutations. The EcoRI/BamHI fragment from the wt dnaX gene was ligated into the EcoRI/BamHI-cut dnaXL381PP465L gene to produce the dnaXL381P single mutant. The EcoRI/BamHI-cut fragment from the dnaXL381PP465L gene was ligated into the EcoRI/BamHI-cut wt dnaX gene to produce the dnaXP465L single mutant. All mutations as well as the absence of spurious mutations were verified by sequencing.
Sedimentation Velocity Ultracentrifugation
Sedimentation velocity ultracentrifugation experiments of wt τ (5 μM), L381A (16 μM), DnaB (11 μM), the DnaB:τ (3:2 molar ratio), and DnaB:L381A (2:1 molar ratio) complexes, in buffer A supplemented with 100 mM NaCl and 10% v/v glycerol, were carried out at 40 000 rpm for 6 h at 20 °C in a Beckman Optima XL-A analytical ultracentrifuge, at the National Centre of Molecular Hydrodynamics (NCMH), University of Nottingham. Sedimenting species were monitored by absorbance at 280 nm. Data were captured at 3 min intervals and analyzed as described in ref 30, using partial specific volumes of 0.736 and 0.742 for DnaB and τ proteins, respectively, and plotted as a Gaussian distribution of the apparent sedimentation coefficient, s*(20,w), uncorrected for density, viscosity, or solute concentration and expressed in Svedbergs (Sv). All concentrations above refer to monomers.
Sedimentation Equilibrium Ultracentrifugation
DnaB (10.56 μM), τ (7.2 μM), L381A (6.4 μM), and DnaB-τ complex (formed by mixing τ, 0.99 μM and DnaB, 0.79 μM and then purified by gel filtration) were loaded into 2× single-channel and 1 × 3-channel Beckman XL-A AUC cells, and centrifugation was performed initially at 7000 rpm for 28 h at 20 °C. Absorption scans were performed at 280 nm and logged. The data were analyzed using the program WinNONLIN, floating σ, offset, and initial y value (30). The program SEDNTERP (30) was employed to assign σ values into MW values, employing partial specific volumes of 0.736 and 0.742 for DnaB and τ proteins, respectively. All concentrations above refer to monomers. The L381A protein was analyzed further by additional experiments at 16 000 rpm, using two different samples.
Circular Dichroism (CD) Analysis
CD experiments were carried out using τ and L381A proteins in 20 mM phosphate buffer, pH 7.4 and three different concentrations of each protein, 350, 250, and 150 nM (referring to monomers), in a final volume of 2.5 mL for each sample. Spectra were run on a Pi. Star 180 Applied Photophysics system using a 1 cm cell at 293 K, over a wavelength range of 200–300 nm. Triplicate experiments for each sample were carried out with the baseline corrected by solvent subtraction. Mean residue ellipticity (θ) was calculated for each experiment and expressed in cm2 dmol−1.
Chemical Cross-Linking
Protein cross-linking using glutaraldehyde was carried out at 0.002% v/v glutaraldehyde and 1.6–1.9 nM (referring to monomers) τ poteins (either wt or L381A mutant) in 20 mM phosphate buffer pH 7.4. Reaction mixtures were incubated at room temperature, and samples were removed at appropriate time intervals and quenched with 50 mM Tris-HCl pH 7.5. Proteins were resolved in an 8% SDS–PAGE gel.
Construction of Plasmids for in Vivo Gene Replacement
Gene replacement in vivo was carried out using a xylose-dependent conditional expression system for modulated expression in B. subtilis (31, 32). The expression system is contained on plasmid pSWEET for integration at the amyE locus of B. subtilis. To create the pSWEET-dnaX and pSWEET-dnaXLA plasmids, the dnaX and dnaXLA genes were excised out of the pET28a plasmid as XbaI–NotI fragments (the XbaI end was blunted) and inserted into the PacI (blunted)–NotI sites of the pSWEET-bgaB plasmid. The cloning was directional and resulted in the replacement of the bgaB gene with the dnaX (pSWEET-dnaX) and dnaXLA (pSWEET-dnaXLA) genes.
To precisely replace the dnaX gene, approximately 700 bp of sequence flanking either side of dnaX was amplified via a splicing by overlap method (33) by using primer pairs MS1/MS1int (MS1, 5′-GTGCAGTCGTACTTTCTCGAGTGGAAAAAGTG-3′ and MS1int, 5′-GTTATAAATTTGGCCCGGGAGGTTATTGCGTGGGGTTTGCCCTCCTCCGTTATTCATC-3′) and MS2/MS2int (MS2, 5′-AGAAATAGCGCCGTGAAGAACGTGATACTGTC-3′ and MS2int, 5′-CACGCAATAACCTGCCCGGGCCAAATTTATAACCA--AAATGAAAGAGAGTGAATGCTATG-3′) for sequence upstream and downstream of dnaX, respectively. The flanking sequence, reamplified with primers MS1/MS2 to generate a single product with a unique SrfI site in the middle, was cloned as a blunt-ended fragment into the EcoRV site of pBluescript SK-, generating the pBS-FLANK plasmid. The spectinomycin (SPC) resistance cassette was amplified from pUS19 (32) with primers AB14/AB15 (AB14, 5′-GGTTTACACTTACTTTAGTTTTATGGAAAT-GAAAGATC-3′ and AB15, 5′-TTATAATTTTTTTAATCTGTTATTTAAATAGTTTATAG-3′) and subsequently incorporated in the middle of the dnaX flanking sequence (at the SrfI site) in the pBS-FLANK plasmid to generate the pFLANKspc plasmid. All sequences obtained by PCR were sequenced to verify the absence of spurious mutations.
Manipulation of B. subtilis
Strains encoding the wt and L381A dnaX alleles were constructed as described elsewhere (31, 32). The pSWEET plasmid was kindly provided by Dr. Eric Brown. The pSWEET-dnaX and pSWEET-dnaXLA plasmids were linearized with ScaI and introduced into the B. subtilis 168 derivative EMG50 competent cells by transformation (34). Transformants were selected on YT plates containing chloramphenicol (10 μg/mL) and tested for amylase activity using starch plates and flooding with iodine. One of the amy– derivatives containing wt dnaX, RT1, and one amy– derivative containing the L381A dnaX allele, RT2, were used to prepare competent cells. The plasmid pFLANK-spc was linearized with ScaI and introduced into RT1 and RT2 competent cells plating on YT + 2% w/v xylose, chloramphenicol (10 μg/mL), and spectinomycin (100 μg/mL) to form RT10 and RT20, respectively. PCR was used to ensure that the dnaX gene had been replaced by a double crossover between the flanking DNA dnaX and the homologous DNA in the plasmid in both RT10 and RT20. Primers that bound to the dnaX gene were used to amplify part of the dnaX alleles in RT10 and RT20 to verify by DNA sequencing that they contained the wt and L381A alleles, respectively.
Leaky expression was observed in a variety of media tested (data not shown), but tight xylose-dependent growth without leaky expression was achieved in Schaeffers sporulation medium (34). Growth curves were performed in Schaeffers medium containing chloramphenicol (10 μg/mL), spectinomycin (100 μg/mL), 0.2% w/v glucose, and varying amounts of xylose. Overnight cultures of RT10 and RT20 in Schaeffers plus antibiotics and xylose were washed twice in medium lacking xylose and used to inoculate cultures with 2, 0.2, 0.02, and 0% w/v xylose. Cultures were grown with shaking at 37 °C, and samples were taken at various time points to measure the OD600. Three independent colonies for each of the two strains were used to repeat the growth curves in triplicate results.
RESULTS
Stable Interaction between the B. stearothermophilus DnaB and the B. subtilis τ Protein Detected by Gel Filtration
A stable interaction between the E. coli τ and the DnaB proteins at μM concentrations has been reported previously (10). Under the conditions of these experiments, E. coli DnaB was a trimer, and the true stoichiometry of this interaction could not be established although the assumption is that a τ tetramer interacts with a DnaB hexamer. We employed a similar procedure to demonstrate a stable interaction between the BstDnaB helicase and the B. subtilis τ protein. A stable DnaB-τ complex was formed at low nM concentrations (Figure 1A). The individual elution peaks of the two proteins are close to each other, but in a mixture of both proteins together, an earlier peak appears in the elution profile. The presence of both proteins in this peak was confirmed by SDS–PAGE analysis suggesting a stable interaction between DnaB and τ under our experimental conditions (Figure 1A). This interaction is salt dependent as at 500 mM NaCl no interaction was detected, suggesting that it is mainly ionic (data not shown).
Figure 1.

Mutations of τ that affect binding to DnaB. (A) Formation of a stable complex between the BstDnaB and the B. subtilis τ proteins. The DnaB-τ complex was resolved by gel filtration through a Superose 6 column. Samples from peaks (indicated by numbers) were analyzed by SDS–PAGE. The numbering of lanes corresponds to the numbering shown in gel filtration profiles. Lane 5 is a control lane, showing a mixture of purified τ and DnaB proteins in a 2:3 molar ratio (referring to monomers), for comparison. (B) A summary of the apparent oligomerization and DnaB-binding properties of τ mutants. A plus sign indicates apparent oligomerization and DnaB-binding properties similar to wt τ, whereas a negative sign indicates a defect. (C) Examining the L381P–BstDnaB interaction by gel filtration. A mixture of the L381P mutant protein and DnaB was resolved by gel filtration through a Superdex S-200 column. Samples, indicated by numbers, were analyzed by SDS–PAGE. The numbering of lanes corresponds to the numbers shown in gel filtration profiles. L381P and DnaB proteins are indicated by arrows. No stable interaction was detected. (D) Examining the L381A–BstDnaB interaction by gel filtration. The same experiment using the L381A mutant. Again, no stable interaction was detected.
Gel Filtration Shows that L381 Is a Critical Amino Acid Residue in τ that Affects Oligomerization and Interaction with DnaB
During cloning of the dnaX gene, we isolated a mutant gene with two mutations, L381P and P465L. Subsequent overexpression, purification, and biochemical analysis of the L381PP465L mutant protein revealed two defects: its oligomerization was different to wt τ and failed to interact with DnaB (Figure 1B and data not shown). To investigate which of these mutations is responsible for these defects, we constructed the corresponding single mutants (L381P and P465L) and investigated their oligomerization states and DnaB-binding ability. Using analytical gel filtration, we showed that the P465L mutant behaves comparably to wt τ (Figure 1B and data not shown), whereas the L381P mutant has different oligomerization properties and fails to form a stable complex with DnaB (Figure 1B,C). We therefore concentrated on investigating further the importance of L381.
The replacement of leucine by a proline at position 381 is likely to have caused local alterations in the structure that may affect the ability of the protein to form correct oligomers and interact with DnaB. We constructed the single L381A and also the adjacent Q380A mutants to study the individual contributions of these residues to the oligomerization and DnaB-binding abilities of τ. L381A formed a different oligomer in solution, as determined by its elution profile in gel filtration studies, and failed to form a stable complex with DnaB (Figure 1B,D). These results suggest that L381 is a critical residue for correct oligomer formation and DnaB interaction. However, our data cannot differentiate whether L381 is involved directly in interactions with DnaB or whether its defective interaction with DnaB is the indirect effect of its altered oligomeric state. Mutating the neighboring glutamine to an alanine (Q380A) had no apparent effect on either oligomerization or interaction with DnaB (Figure 1B and data not shown).
Gel Filtration Shows that P465 Is Not Critical for Oligomerization and Interaction with DnaB
Interestingly, a ts dnaX51 B. subtilis strain has been reported to carry the P465S mutation. It grows at the permissive (30 °C) but not at the nonpermissive (50 °C) temperature (27). This mutation is within the Cτ domain shown to interact with DnaB and DNA polymerase α in E. coli. Our data show that the P465L mutant behaves comparably to wt τ protein. It forms similar oligomers that interact stably with DnaB in vitro, as shown by gel filtration (data not shown). It is therefore likely that the thermosensitive defect of ts dnaX51 is not because of a defective interaction with DnaB. Instead, it may be attributed to defective interactions with either (or both) DnaE or PolC polymerases at the nonpermissive temperature, although currently there is no such evidence available.
Sedimentation Velocity Ultracentrifugation Also Supports the Importance of L381 for Oligomerization and DnaB Binding
The significance of L381 for oligomerization and interaction with DnaB was also confirmed with sedimentation velocity ultracentrifugation experiments. DnaB, τ, and L381A proteins sedimented with apparent s*(20,w) values of 10.229 ± 0.0017 Sv, 5.813 ± 0.046 Sv, and 2.739 ± 0.025 Sv, respectively (Figure 2A). A mixture of DnaB and τ, in a 3:2 molar ratio, sedimented as major species with s*(20,w) = 12.031 ± 0.161 Sv and a minor species with s*(20,w) = 9.811 ± 0.350 Sv, corresponding to the DnaB-τ complex and traces of free DnaB, respectively (Figure 2B). By comparison, a mixture of DnaB and L381A sedimented as two distinct major species with s*(20,w) = 2.850 ± 0.075 Sv and s*(20,w) = 8.224 ± 0.017 Sv, corresponding to free L381A and DnaB, respectively (Figure 2B). These data are consistent with the analytical gel filtration results, showing that the L381A mutant fails to form a stable complex with DnaB. We proceeded to investigate the molecular masses and thus the stoichiometries of the DnaB, τ, and L381A proteins as well as the DnaB-τ complex by sedimentation equilibrium analytical centrifugation.
Figure 2.

Sedimentation velocity ultracentrifugation confirms the importance of L381. Experiments for DnaB, L381A, and τ proteins (A), as well as the DnaB-τ and DnaB–L381A mixtures (B) are shown. Data are presented as Gaussian distribution of apparent sedimentation coefficients and show an interaction between DnaB and τ but not between DnaB and L381A.
Sedimentation Equilibrium Ultracentrifugation Reveals that a Hexamer of DnaB Interacts with a Pentamer of τ and that L381A Forms a Variety of Complexes
Sedimentation equilibrium provides shape-independent estimations for the molecular mass of proteins. We verified the oligomeric species of our proteins (DnaB, τ, L381A, and DnaB-τ complex) using this method. The molecular masses obtained for DnaB and τ were 303 (hexamer) and 290 kDa (closer to a pentamer than a tetramer), respectively, with very good fits and σ values of 1.7342 and 1.7006, respectively (Figure 3A). For the DnaB-τ complex, the residuals were elevated in the middle and reduced on either end (Figure 3B). When we analyzed the data from near the meniscus (upper region) and further down the tube (lower region), the data fit into two species, a major one with molecular mass of 616 kDa (lower region) and a minor one with molecular mass of 286 kDa (upper region) with σ values 3.539 and 1.675, respectively (Figure 3C). These represent the major species, DnaB-τ complex (hexamer–pentamer), and the minor species, τ (pentamer), and are compatible with the slight excess of τ over DnaB used to make the complex, assuming a τ pentamer and a DnaB hexamer.
Figure 3.

Stoichiometry of the τ-DnaB complex. Equilibrium ultracentrifugation data for DnaB and τ proteins (A) and for the DnaB-τ complex treated as monodisperse (B) with residuals clearly reduced on either side and elevated in the middle. (C) The same data for the DnaB-τ complex analyzed near the meniscus (representing traces of τ) and near the base (representing the true DnaB-τ complex). The sizes of the complexes indicate that DnaB and τ are hexameric and pentameric, respectively, and one hexamer of DnaB interacts with one pentamer of τ.
The initial data collected for two different samples of the L381A mutant indicated that its profile is significantly different than the wt. Although the average molecular weight of the oligomeric species in the two samples was 247 kDa for sample 1 (Figure 4A) and 274 kDa for sample 2 (data not shown), there seemed to be a degree of polydispersity and some caution is needed in the interpretation of these data. Our initial analysis of sample 1 showed that a single-species fit was not a particularly good fit, as judged by the residuals (Figure 4A). The σ value, 8.773, was intermediate between a tetramer and a pentamer. Fits to data from both samples were attempted based on the hypothesis that the system consisted of tetramers with varying degree of association (K2). The σ value was therefore fixed at 7.5, with ln(K2) floated. Data from both samples could be fit well on this basis (Figure 4B shows sample 1) yielding a molecular mass of 250 kDa. It is therefore likely that L381A exists as a tetramer.
Figure 4.
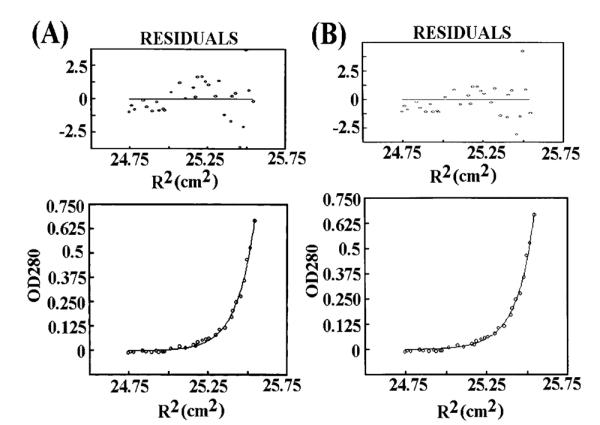
L381A forms a tetramer of varying degrees of association. (A) Equilibrium ultracentrifugation data for L381A (sample 1) from an initial analysis based upon a single stable tetrameric species. The residuals reveal a poor fit. (B) Analysis of the same data based upon the assumption of tetramers of varying degrees of association (see Results). The residuals reveal a good fit suggesting a tetramer.
CD Analysis of the L381A Mutant
Large conformational changes affecting the folding of the L381A protein were eliminated by CD analysis. The CD spectra of wt τ and L381A proteins at three different concentrations (350, 250, and 150 nM) were superimposable, displaying the characteristics of predominantly α helical proteins with negative transitions near 208 and 222 nm and a positive transition near 192 nm (data not shown). The data revealed that there were no gross changes in the overall fold of the L381A mutant protein. The different oligomeric states of wt and L381A τ proteins were also verified by a comparison of their glutaraldehyde-mediated cross-linking patterns.
Glutaraldehyde Cross-Linking
Exposure to glutaraldehyde for up to 30 min resulted in the formation of lower mobility complexes I and II for the wt protein and only complex I for the L381A protein (Figure 5). Although it is difficult to determine unequivocally the stoichiometry of these complexes, it appears that complex I represents a dimer (~135 kDa), whereas complex II represents a higher oligomer, either a tetramer of ~250 kDa or a pentamer of ~312 kDa. Extended exposure of L381A to glutaraldehyde for more than 60 min resulted in the formation of some complex II (data not shown). These data show that there is a clear difference between the wt and the L381A oligomers but cannot determine whether this is the result of different oligomeric states (different stoichiometry), different types of oligomers (different shape), or even differences in the stability of the oligomers.
Figure 5.
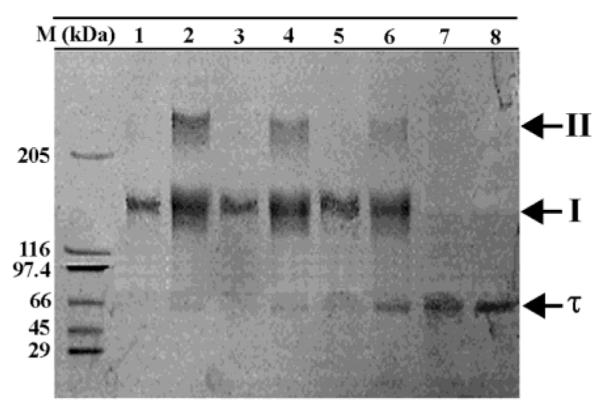
L381A forms lower oligomers as compared to wt τ. Glutaraldehyde cross-linking of wt τ and L381A proteins. An 8% SDS–PAGE gel showing complexes I and II obtained by glutaraldehyde cross-linking of wt τ for 0, 10, 20, and 30 min (lanes 8, 6, 4, and 2, respectively). The equivalent experiment for the L381A protein is shown in lanes 7, 5, 3, and 1. Molecular weight markers are as indicated.
Replacement of the wt dnax Gene with the Mutant L381A dnax Gene in Vivo
The dnaX alleles were placed under the control of the xylose inducible promoter and introduced ectopically, disrupting the amy gene. These merodiploids grew normally in the presence or absence of 2% xylose indicating that the mutant dnaX did not have a dominant negative phenotype. Strains in which the dnaX alleles are conditional on xylose were constructed by deletion of the endogenous dnaX gene. RT10 and RT20 contain the wt and mutant dnaX alleles, respectively, both under the control of the xylose promoter. In this way, the expression of both genes can be controlled under identical conditions to ensure comparable expression. On Schaeffers medium with glucose, neither strain could grow in the absence of xylose (data not shown). When 2% xylose was added to this medium, both strains grew equally well, suggesting that under these conditions the L381A mutant protein was able to support growth, but at lower xylose concentrations the growth defect was more pronounced for the RT20 as compared to the RT10 strain (data not shown).
We tested the activity of the L381A protein as the xylose levels become limiting. Growth curves were performed on RT10 and RT20 in Schaeffers medium containing glucose and increasing amounts xylose (Figure 6). These conditions were chosen to increase the basal repression of the xylose promoter by glucose catabolite repression in the absence of xylose. Both strains grew well with glucose and xylose at 2%, but with lower levels of xylose, growth was affected. With RT10 there was a long lag phase, and then surprisingly all four cultures grew at the same rate, even the culture containing no xylose (see later for the explanation). With RT20, the rate of growth was dependent on the amount of xylose added. After inoculation, the RT10 cells may still contain xylose, despite being washed twice in an equal volume of medium without xylose, and/or they may have a high level of τ that even with dilution caused by cell division can still support growth. Thus, RT10, even without inducer, can still undergo several doublings when the xylose concentration is less than 2%. With RT20, growth is much more dependent on the level of xylose in the medium with only a small increase in the OD600 apparent with 0 or 0.02% xylose. These data suggest that it is the endogenous L381A present in the cells on inoculation that limits growth. We interpret these data to indicate that the mutant L381A is defective as compared to wt τ, but this defect can be suppressed by a high level of expression.
Figure 6.
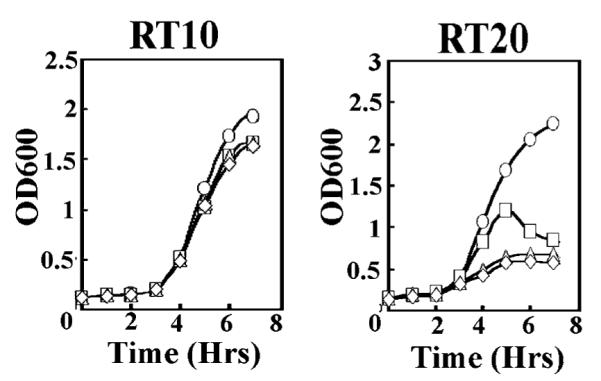
Effect of the L381A mutation in vivo. Growth curves of RT10 and RT20 strains. All cultures were grown at 37 °C with shaking in the presence of chloramphenicol (10 μg/mL), spectinomycin (100 μL/ml), 0.2%w/v glucose, and 2, 0.2, 0.02, and 0% w/v xylose (circles, squares, triangles, and diamonds, respectively). RT10 exhibited no growth defect, whereas RT20 exhibited growth inhibition at low xylose levels.
DISCUSSION
τ-DnaB Interaction
Studies for the E. coli τ protein using Surface Plasmon Resonance (SPR) with truncated versions of the protein revealed that the DnaB interaction interface includes 66 amino acid residues (431–496) in domain IV (15). Domain IV includes the C-terminal 17 residues of γ, but these were reported not to contribute significantly to DnaB binding (ref 15 and Figure 7). Although the DnaB interaction interface is in the Cτ domain, no stable DnaB-Cτ interaction has been demonstrated to date, despite the fact that a stable complex between the full-length τ and DnaB has been isolated by gel filtration (10). Instead, the interaction has been detected indirectly by SPR (15) and cross-linking followed by immunodetection (28). However, Cτ has been shown to substitute full-length τ in the presence of exogenous γ in reconstituted E. coli replication forks, suggesting that this domain by itself is functionally competent (28). Our studies show that L381, which is also conserved in E. coli τ (L419), is critical for oligomerization and DnaB binding. By comparison, the neighboring Q380 residue is not required for oligomerization or interaction with DnaB. Although L419 is in domain IV of the E. coli τ protein, it is within the N-terminal 17 amino acid residues of domain IV that are not part of Cτ and are instead the last C-terminal 17 residues of the shorter γ protein (ref 15 and Figure 7). An extended version of Cτ that includes these 17 residues was reported to bind to DnaB with an apparent Kd similar to that observed for the interaction of DnaB with Cτ, suggesting that these residues do not contribute significantly to DnaB interactions (15). Similar results were obtained with our protein (unpublished work). However, our gel filtration and ultracentrifugation data show that this region is crucial for oligomerization and stable DnaB binding. Unfortunately, the crystal structure of the E. coli γ complex shows a truncated γ protein (residues 1–373) missing L419 and does not give us an insight into the structure of this particular region of the protein (16). The importance of L381 is also highlighted by its good conservation among different τ proteins. It is conserved not only between E. coli and B. subtilis proteins but also in S. pyogenes (L384), B. halodurans (L387), B. stearothermophilus strain 10 (L382) while a valine (V394) is found in the equivalent position in the L. lactis τ protein. The neighboring glutamine residue (Q380) is not as well conserved, as there are threonine and asparagine residues in the equivalent positions in the B. stearothermophilus strain 10 and L. lactis τ proteins, respectively.
Figure 7.

CLUSTAL W sequence alignment of the E. coli and B. subtilis τ proteins. Only the C-terminal ends are shown with identical amino acids labeled by asterisks. An arrow indicates the position that defines the shorter E. coli γ polypeptide. The smallest box indicates Q and L residues that have been mutated, whereas the other boxes indicate the 66 amino acid residue region of the E. coli τ that has been implicated in DnaB binding (15).
Is Oligomerization of τ Essential for Functional DnaB Binding?
Our ultracentrifugation data show that, even in the absence of δδ′,a τ pentamer interacts with a DnaB hexamer in vitro. The structural significance of this interaction becomes apparent in a later paper. It is more likely that L381 does not contribute directly to DnaB binding, but because of its importance to the integrity of the correct pentameric conformation, its removal in the L381A mutant affects binding to DnaB indirectly. It appears that the integrity of the correct pentameric conformation is important for stable interaction with DnaB, although less stable (perhaps transient) interactions are also possible with lower oligomeric forms of τ in the presence of other replisomal proteins, as the unique Cτ domain has been shown to partially substitute the function of the full-length protein in reconstituted E. coli replication forks (35), and our genetic data show that L381A can sustain bacterial growth in vivo provided it is expressed in high amounts. Interconversions of the γ complex between DnaB stably interacting and transiently interacting states may also be functionally significant, with the latter perhaps serving roles in β recycling from completed Okazaki fragments (ref 36) or in mismatch repair (37). Such an idea has been suggested previously for γ complexes with different τ,γ compositions (35).
Significance of the L381A Mutation in Vivo
The functional significance of the L381A mutation was verified by in vivo gene replacement experiments. The data suggest that the L381A protein in vivo can still interact with the helicase when produced in relatively large quantities and in the presence of other replisomal proteins (normal growth in the presence of xylose) but cannot do so when produced in trace amounts. The simplest explanation is that L381A interacts with DnaB in a transient manner as compared to wt τ. Alternatively, it is likely that τ will form hetero-oligomers with the δδ′ proteins to function in the loading of the β clamp. It may also be possible that the L381A mutation affects the correct assembly of the τ–δ–δ′ hetero-oligomer and thus also the loading of the β clamp. Whatever the precise functional defect of the L381A mutant, it is clear that it becomes apparent in vivo only when the expression of the mutant protein is limited under low concentrations of xylose. Under high concentrations of xylose when expression of the mutant protein is high, there is no observable functional defect.
Oligomeric States of the wt τ and L381A Proteins
Our data show an apparent disparity between the molecular masses of the wt τ and mutant L381A proteins determined by gel filtration (this paper and ref 27) and those obtained by analytical ultracentrifugation (this paper). The former method suggests an apparent tetramer and trimer for wt τ and L381A/P proteins, respectively, whereas the second method suggests a pentamer and tetramer for wt τ and L381A, respectively. This disparity highlights the dangers of relying solely on gel filtration for molecular mass estimations and may be explained because of the dependence of the elution volume of a protein on its shape or by nonspecific adsorption of the protein onto the column matrix. The frictional coefficient, f0, for a nonhydrated, spherical, and compact particle in solution is given by the following expression:
where η is the viscosity of the solution, r(0) = (3MV)/4πN)0.333, M is the molecular mass of the particle, V is its partial specific volume, and N is Avogadros’ number. However, proteins are hydrated and sometimes asymmetric, and the frictional coefficient, f, is often greater than f0. Then
where r(H) is the Stokes’ radius. The ratio f/f0 is the frictional ratio. If a gel filtration column is calibrated with a set of proteins of approximately the same frictional ratios, then the elution position is related to the molecular weight. If, however, a protein with a different frictional ratio is run through the same calibrated column, an erroneous estimate will be yielded for its molecular weight. However, the equilibrium ultracentrifugation technique gives molecular mass estimations that are independent of shape and represent the true oligomeric state of a protein.
Summarizing, in this study we present evidence that, even in the absence of δδ′, a pentamer of τ interacts with a hexamer of DnaB in vitro, and we identified a leucine residue that is crucial for the integrity of the τ pentamer and its interaction with DnaB. The fact that the L381A dnaX gene at low expression levels fails to support bacterial growth in vivo suggests that the integrity of L381 is important for the function of τ. Finally, we showed that the P465L mutant behaves in a manner comparable to the wt protein in terms of its oligomeric state and its interaction with DnaB; therefore, the growth defects observed with the ts dnaX51 strain carrying the P465S mutation are unlikely to be due to a defective interaction with DnaB. The architecture of a pentameric τ interacting with a hexameric DnaB has already been revealed by AFM (38).
ACKNOWLEDGMENT
We thank Ingrid Davies for DNA sequencing; Dr. Neil Errington and Prof. Arthur J. Rowe for assistance with the analytical ultracentrifugation experiments, useful discussions, and analysis of the data; and Dr. Eric Brown for the kind gift of the pSWEET-bgaB and pUS19 plasmids.
Footnotes
This work was supported by a BBSRC grant (42/B15519) to P.S.A.H. is a postgraduate student supported by a University of Nottingham Research Scholarship.
- AFM
- atomic force microscopy
- DNA
- deoxyri-bonucleic acid
- RFC
- replication factor C
- SSB
- single-strand DNA binding protein
- DTT
- dithiothreitol
- PMSF
- phenyl methyl sulfonyl fluoride
- EDTA
- ethylene diaminotetraacetic acid
- SDS–PAGE
- sodium dodecyl sulfate–polyacrylamide gel electrophoresis
- HE
- holoenzyme
- MW
- molecular weight
- NCMH
- National Centre for Molecular Hydrodynamics
REFERENCES
- 1.Kornberg A, Baker TA. DNA replication. 2nd ed W. H. Freeman; New York: 1991. [Google Scholar]
- 2.Kelman Z, O’Donnell M. DNA polymerase III holoenzyme: Structure and function of a chromosomal replicating machine. Annu. Rev. Biochem. 1995;64:171–200. doi: 10.1146/annurev.bi.64.070195.001131. [DOI] [PubMed] [Google Scholar]
- 3.Stillman B. Smart machines at the DNA replication fork. Cell. 1994;78:725–728. doi: 10.1016/s0092-8674(94)90362-x. [DOI] [PubMed] [Google Scholar]
- 4.Waga S, Stillman B. The DNA replication fork in eukaryotic cells. Annu. Rev. Biochem. 1998;67:721–751. doi: 10.1146/annurev.biochem.67.1.721. [DOI] [PubMed] [Google Scholar]
- 5.Young MC, Reddy MK, von Hippel PH. Structure and function of the bacteriophage T4 DNA polymerase holoenzyme. Biochemistry. 1992;31:8675–8690. doi: 10.1021/bi00152a001. [DOI] [PubMed] [Google Scholar]
- 6.Blinkowa AL, Walker JR. Programed ribosomal frameshifting generates the Escherichisa coli DNA polymerase III γ subunit from within the τ subunit reading frame. Nucleic Acids Res. 1990;18:1725–1729. doi: 10.1093/nar/18.7.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsuchihashi Z. Translational frameshifting in the Escherichia coli dnaX gene in vitro. Nucleic Acids Res. 1991;19:2457–2462. doi: 10.1093/nar/19.9.2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ellison V, Stillman B. Opening of the clamp: An intimate view of an ATP-driven biological machine. Cell. 2001;106:655–660. doi: 10.1016/s0092-8674(01)00498-6. [DOI] [PubMed] [Google Scholar]
- 9.Kim DR, McHenry CS. Biotin tagging deletion analysis of domain limits involved in protein–macromolecular interactions. Mapping the τ binding domain of the DNA polymerase III α subunit. J. Biol. Chem. 1996;271:20690–20698. doi: 10.1074/jbc.271.34.20690. [DOI] [PubMed] [Google Scholar]
- 10.Kim S, Dallmann HG, McHenry CS, Marians KJ. Coupling of a replicative polymerase and helicase: A τ–DnaB interaction mediates rapid replication fork movement. Cell. 1996;84:643–650. doi: 10.1016/s0092-8674(00)81039-9. [DOI] [PubMed] [Google Scholar]
- 11.Glover BP, McHenry CS. The χψ subunits of DNA polymerase III holoenzyme bind to single-stranded DNA-binding protein (SSB) and facilitate replication of an SSB-coated template. J. Biol. Chem. 1998;273:23476–23484. doi: 10.1074/jbc.273.36.23476. [DOI] [PubMed] [Google Scholar]
- 12.McAlear MA, Tuffo KM, Holm C. The large subunit of replication factor C (Rfc1p/Cdc44p) is required for DNA replication and DNA repair in Saccharomyces cerevisiae. Genetics. 1996;142:65–78. doi: 10.1093/genetics/142.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kunkel TA, Bebenek K. DNA replication fidelity. Annu. Rev. Biochem. 2000;69:497–529. doi: 10.1146/annurev.biochem.69.1.497. [DOI] [PubMed] [Google Scholar]
- 14.Gao D, McHenry CS. τ binds and organizes Escherichia coli replication proteins through distinct domains. Partial proteolysis of terminally tagged τ to determine candidate domains and assign domain V as the α binding domain. J. Biol. Chem. 2001;276:4433–4440. doi: 10.1074/jbc.M009828200. [DOI] [PubMed] [Google Scholar]
- 15.Gao D, McHenry CS. τ binds and organizes Escherichia coli replication proteins through distinct domains. Domain IV, located within the unique C terminus of τ, binds the replication fork helicase. J. Biol. Chem. 2001;276:4441–4446. doi: 10.1074/jbc.M009830200. [DOI] [PubMed] [Google Scholar]
- 16.Jeruzalmi D, O’Donnell M, Kuriyan J. Crystal structure of the processivity clamp loader γ complex of Escherichia coli DNA polymerase III. Cell. 2001;106:429–441. doi: 10.1016/s0092-8674(01)00463-9. [DOI] [PubMed] [Google Scholar]
- 17.Jeruzalmi D, Yurieva O, Zhao Y, Young M, Stewart J, Hingorani M, O’Donnell M, Kuriyan J. Mechanism of processivity clamp opening by the δ subunit wrench of the clamp loader complex of Escherichia coli DNA polymerase III. Cell. 2001;106:417–428. [PubMed] [Google Scholar]
- 18.Guenther BD, Onrust R, Sali A, O’Donnell M, Kuriyan J. Crystal structure of the δ′ subunit of the clamp–loader complex of Escherichia coli DNA polymerase III. Cell. 1996;91:335–345. doi: 10.1016/s0092-8674(00)80417-1. [DOI] [PubMed] [Google Scholar]
- 19.Kong XP, Onrust R, O’Donnell M, Kuriyan J. Three-dimensional structure of the β subunit of Escherichia coli DNA polymerase III holoenzyme: a sliding DNA clamp. Cell. 1992;69:425–437. doi: 10.1016/0092-8674(92)90445-i. [DOI] [PubMed] [Google Scholar]
- 20.Onrust R, Finkelstein J, Naktinis V, Turner J, Fang L, O’Donnell M. Assembly of a chromosomal replication machine: two polymerases, a clamp loader, and sliding clamps in one holoenzyme particle. I. Organization of the clamp loader. J. Biol. Chem. 1995;270:13348–13357. doi: 10.1074/jbc.270.22.13348. [DOI] [PubMed] [Google Scholar]
- 21.Dallmann HG, McHenry CS. DnaX complex of Escherichia coli DNA polymerase III holoenzyme. Physical characterization of the DnaX subunits and complexes. J. Biol. Chem. 1995;270:29563–29569. [PubMed] [Google Scholar]
- 22.Pritchard AE, Dallmann GH, Glover BP, McHenry CS. A novel assembly mechanism for the DNA polymerase III holoenzyme DnaX complex: association of δδ′ with DnaX4 forms DnaX3δδ′. EMBO J. 2000;19:6536–6545. doi: 10.1093/emboj/19.23.6536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao D, McHenry CS. τ binds and organizes Escherichia coli replication proteins through distinct domains. Domain III, shared by γ and τ, binds δδ′. J. Biol. Chem. 2001;276:4447–4453. doi: 10.1074/jbc.M009827200. [DOI] [PubMed] [Google Scholar]
- 24.Glover BP, McHenry CS. The DnaX-binding subunits δ′ and ψ are bound to γ and not τ in the DNA polymerase III holoenzyme. J. Biol. Chem. 2000;275:3017–3020. doi: 10.1074/jbc.275.5.3017. [DOI] [PubMed] [Google Scholar]
- 25.Bird LE, Pan H, Soultanas P, Wigley DB. Mapping protein–protein interactions within a stable complex of DNA primase and DnaB helicase from Bacillus stearothermophilus. Biochemistry. 2000;39:171–182. doi: 10.1021/bi9918801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baker DG. Cloning and amplified expression of the tyrosyl-tRNA synthetase genes of Bacillus stearothermophilus and Escherichia coli. Eur. J. Biochem. 1982;125:357–360. doi: 10.1111/j.1432-1033.1982.tb06691.x. [DOI] [PubMed] [Google Scholar]
- 27.Martinez-Jimenez MI, Mesa P, Alonso JC. Bacillus subtilis τ subunit of DNA polymerase III interacts with bacteriophage SPP1 replicative DNA helicase G40P. Nucleic Acids Res. 2002;30:5056–5064. doi: 10.1093/nar/gkf650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bird LE, Wigley DB. The Bacillus stearothermophilus replicative helicase: cloning overexpression and activity. Biochem. Biophys. Acta. 1999;1444:424–428. doi: 10.1016/s0167-4781(99)00024-x. [DOI] [PubMed] [Google Scholar]
- 29.McPherson MJ, Quirke P, Taylor GR. PCR, a practical approach. IRL Press; Oxford: 1992. pp. 207–209. [Google Scholar]
- 30.Harding SE, Rowe AJ, Horton JC. Analytical ultracentrifugation in biochemistry and polymer science. The Royal Society of Chemistry; Letchworth, UK: 1992. Ch. 7; pp. 909–126. [Google Scholar]
- 31.Bhavsar AP, Zhao X, Brown ED. Development and characterisation of a xylose-dependent system for expression of cloned genes in Bacillus subtilis: conditional complementation of a teichoic acid mutant. Appl. Environ. Microbiol. 2001;67:403–410. doi: 10.1128/AEM.67.1.403-410.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bhavsar AP, Beveridge TJ, Brown ED. Precise deletion of tagD and controlled depletion of its product glycerol 3-phosphate cytidylyltransferase leads to irregular morphology and lysis of Bacillus subtilis grown at physiological temperature. J. Bacteriol. 2001;183:6688–6693. doi: 10.1128/JB.183.22.6688-6693.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Link AJ, Phillips D, Church GM. Methods for generating precise deletions and insertions in the genome of wild-type Escherichia coli: application to open reading frame characterization. J. Bacteriol. 1997;179:6228–6237. doi: 10.1128/jb.179.20.6228-6237.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harwood CR, Cutting SM. Molecular Biological Methods for Bacillus. John Wiley and Sons; New York: 1990. [Google Scholar]
- 35.Dallmann HG, Kim S, Pritchard AE, Marians KJ, McHenry CS. Characterization of the unique C terminus of the Escherichia coli τ protein: Monomeric C-τ binds α and DnaB and can partially replace τ in reconstituted replication forks. J. Biol. Chem. 2000;275:15512–15519. doi: 10.1074/jbc.M909257199. [DOI] [PubMed] [Google Scholar]
- 36.Stukenberg PT, Turner J, O’Donnell M. An explanation for lagging strand replication: polymerase hopping among DNA sliding clamps. Cell. 1994;78:877–887. doi: 10.1016/s0092-8674(94)90662-9. [DOI] [PubMed] [Google Scholar]
- 37.Lahue RS, Au KG, Modrich P. DNA mismatch correction in a defined system. Science. 1989;245:160–164. doi: 10.1126/science.2665076. [DOI] [PubMed] [Google Scholar]
- 38.Haroniti A, Anderson C, Doddridge Z, Gardiner L, Roberts CJ, Allen S, Soultanas P. The clamp–loader–helicase interaction in Bacillus. Atomic force microscopy reveals the structural organisation of the DnaB-tau complex in Bacillus. Structure. 2003 doi: 10.1016/j.jmb.2003.12.043. (submitted for publication) [DOI] [PMC free article] [PubMed] [Google Scholar]