

Abstract
Oxidative stress is considered a major contributor in the pathology of multiple sclerosis (MS). Acrolein, a highly reactive aldehyde byproduct of lipid peroxidation, is thought to perpetuate oxidative stress. In this study, we aimed to determine the role of acrolein in an animal model of MS, experimental autoimmune enchephalomyelitis (EAE) mice. We have demonstrated a significant elevation of acrolein protein adduct levels in EAE mouse spinal cord. Hydralazine, a known acrolein scavenger, significantly improved behavioral outcomes and lessened myelin damage in spinal cord. We postulate that acrolein is an important pathological factor and likely a novel therapeutic target in MS.
Keywords: Multiple Sclerosis, EAE, Acrolein, Hydralazine, demyelination
Introduction
Multiple sclerosis (MS) is a severely debilitating neurodegenerative disease marked by progressive demyelination and functional loss in the central nervous system (CNS) (Gold et al., 2006; Compston and Coles, 2008). Oxidative stress resulting from inflammation is known to play a critical role in demyelination, a major pathology in MS (Smith et al., 1999; Gilgun-Sherki et al., 2004). However, conventional free radical scavengers have been unsuccessful at preventing disease development or progression (Smith et al., 1999; Gold et al., 2006; Compston and Coles, 2008). Hence, a priority in MS research is to improve understanding of the mechanisms of oxidative stress and thereby identify novel, more effective therapeutic targets.
Acrolein, a reactive α,β-unsaturated aldehyde, is a product of oxidative stress and lipid peroxidation (Esterbauer et al., 1991; Kehrer and Biswal, 2000). It is also a powerful toxin that has been shown to damage proteins, lipids, and DNA, as well as to generate more free radicals (Esterbauer et al., 1991; Uchida et al., 1998a; Kehrer and Biswal, 2000; Luo and Shi, 2004; Luo et al., 2005b; Luo and Shi, 2005). Acrolein is both a product and catalyst for lipid peroxidation that is capable of inducing a vicious cycle of oxidative stress, dramatically amplifying the effects (Esterbauer et al., 1991).
Furthermore, acrolein remains active in the body for several days (Ghilarducci and Tjeerdema, 1995) while more commonly studied oxidative species decay within seconds (Halliwell and Gutteridge, 1999). Therefore, we hypothesize that acrolein is a key factor in perpetuating oxidative stress, causes progressive myelin damage and functional loss, and that acrolein is a potential novel target for MS therapeutics.
The purpose of the current study is to examine the role of acrolein in the pathogenesis of MS using a well-established animal model of MS. Experimental autoimmune encephalomyelitis (EAE) was induced in mice, and acrolein levels were determined in control and control and experimental groups. Hydralazine, known to be an effective acrolein scavenger, was used to trap acrolein, which is significantly increased when the behavioral deficits emerge in EAE mice. Hydralazine treatment substantially alleviated MS motor deficits, accompanied by anatomical improvements, and tended to lower acrolein levels in spinal cord tissue.
Experimental Procedures
Animals
C57BL/6 female mice (8 weeks old) were purchased from Harlan Laboratories and were maintained in the lab animal housing facilities. These studies were performed in compliance with the Purdue Animal Care and Use Committee protocol guidelines at Purdue University, West Lafayette, Indiana.
Induction of EAE
Nine-twelve week old mice were subcutaneously injected with 0.1 mL MOG35–55/CFA emulsion (EK-0115, Hooke Laboratories) in the neck and lower back (total of 0.2 mL). Within two hours of the injection, 0.1 mL pertussis toxin (EK-0115, Hooke Laboratories) was administered intraperitoneally. A second dose of pertussis toxin of the same volume was given 22–26 hours later. The behavioral performance was assessed using a well established 5-point behavioral scoring system (Kalyvas and David, 2004). The animals were placed on a metal grate and their walking ability was recorded. The scoring system is as follows: 0 – no deficit; 1 – limp tail only; 2 – hind limb paresis but without leg dragging; 3 – partial hind limb weakness with one or both legs dragging; 4 – complete hind limb paralysis; 5 – moribund, paralysis in hind limbs and possibly in one forelimb. The animals were monitored and assessed three times during the first week and then assessed daily for the remainder of the study.
Hydralazine Treatment
A solution of hydralazine hydrochloride (Sigma) was prepared with phosphate buffered saline. The solution was then sterilized through a filter and stored at 4°C. Hydralazine (1 mg/kg) was administered through daily with intraperitoneal injections from the day the MOG/CFA emulsion was administered until the end of the 30 day study period. For sham treatments, mice were administered saline intraperitoneally rather than the hydralazine solution. Blood pressures were monitored using a CODA 2 system (Kent Scientific Corp.).
Detection of acrolein–Lysine adducts by immunoblotting
Acrolein–lysine adducts in the tissue homogenate was measured using a Bio-Dot SF Microfiltration Apparatus (Bio-Rad, Hercules, CA, USA), as previously described (Luo et al., 2005a; Shao et al., 2006; Hamann et al., 2008a). Briefly, the tissue was homogenized with TritonX-100 (3%), and the following anti-proteases were added: 2 mmol/L pefabloc, 15 lmol/L pepstatin A, 20 lg/mL aprotinin, and 25 lg/mL leupeptin. The solution was centrifuged to pellet large pieces of tissue and the supernatant was stored at −80°C until transferred to a nitrocellulose membrane. The membrane was blocked for 1 h in blocking buffer (0.2% casein and 0.1% Tween 20 in PBS) and transferred to 1: 1000 polyclonal rabbit anti-acrolein (in blocking buffer with 2% goat serum and 0.025% sodium azide) (Novus Biologicals) for 18 h at 4°C. Next, the membrane was washed in blocking buffer and then transferred to 1: 10000 alkaline phosphatase conjugated goat anti-rabbit IgG. It was then washed in blocking buffer followed by 0.1% Tween 20 in Tris-buffered saline. The membrane was exposed to Bio-Rad Immuno-Star Substrate (Bio-Rad) and visualized by chemiluminescence.
The optical density of bands was evaluated using Image J (NIH) and statistical comparison was performed with SAS 9.2 (SAS institute). Specifically, equal areas of each individual immunoblottied band of both anti-acrolein and anti-actin samples were selected and corresponding optical densities were obtained using Image J. The optical densities obtained from the anti-acrolein samples were standardized by their corresponding anti-actin samples before proceeding to statistical analysis. A Bicinchoninic acid (BCA) protein assay was also performed before the experiment to ensure equal loading of the samples.
Immunofluorescence imaging
Mice were perfusion fixed with 4% paraformaldehyde and the vertebral columns were removed and fixed in 4% paraformaldehyde overnight. Spinal cord tissues were extracted out of the vertebral column, cut into 1 cm pieces, and further fixed in 4% paraformaldhyde for 24–48 hours. The samples were cryoprotected by incubating for 24 h in a 10% sucrose solution, followed by 24 hours in a 20% sucrose solution. The samples were then imbedded in Tissue-Tek OCT compound (VWR, Batavia, IL) and frozen in liquid nitrogen. 15 μm sections were cut using a cryostat and mounted on gelatin coated slides. Sections were incubated with 5% goat serum and 0.5% Triton-X100 in phosphate buffered saline (PBS) for 30 minutes as blocking agents. After washing 3 times with PBS for 5 minutes, the sections were incubated in primary antibody for 1 hour at room temperature (RT). The sections were washed again for an additional 10 minutes and then incubated in secondary antibody for 1 hour at RT. After 15 minutes wash the sections were labeled by FluoroMyelin™ Red fluorescent myelin stain (Invitrogen, CA) for 30 minutes and washed. All sections were observed by fluorescence microscopy.
For quantification, the thoracic sections were imaged, and Adobe Photoshop was used to outline the demyelination area and total white matter area. Pixel area for each sample was calculated and the percentage of demyelination was obtained by dividing the total demyelinated area by total white matter area. Averages were obtained of the percent demyelination in 3 thoracic cross-sections for each animal. For each of the 3 groups, (control, EAE, EAE + HZ), 3 animals were used for immunofluorescence quantification.
Results
Acrolein protein adducts increased in EAE mice spinal cord
Whole spinal cord tissue from control mice (n = 3), EAE mice (n = 3), and EAE mice treated with hydralazine (HZ) (n = 3) were collected and examined for acrolein protein adducts using an immunoblotting assay (Fig. 1). Acrolein levels were determined at the conclusion of the experiment. The acrolein-lysine adduct levels were significantly increased in EAE mice spinal cord (20.27 ± 3.0 a.u.) compared to control healthy mice (12.30 ± 1.3 a.u., P < 0.05). The average acrolein-lysine adduct level in HZ treated EAE mice (15.14 ± 1.6 a.u.) was noticeably lower than that in EAE mice, though not significant (Fig. 1).
Figure 1.
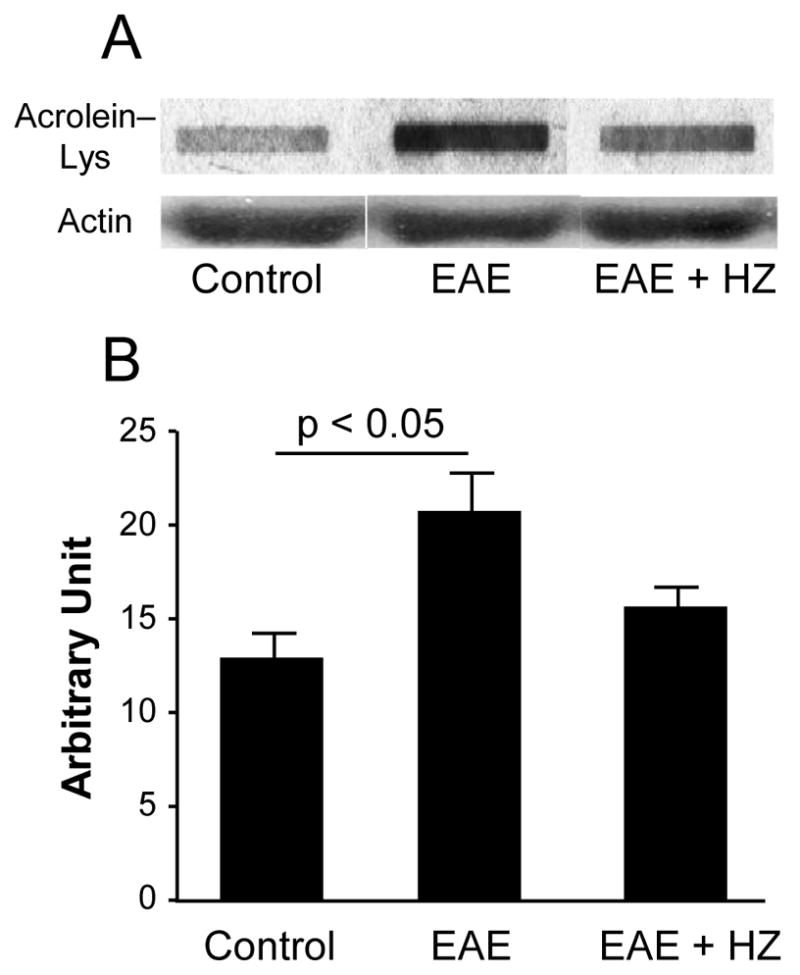
Immunoblotting showed increased acrolein-lysine adduct level in EAE mice spinal cord. The acrolein-lysine adducts in control (n = 3), EAE mice (n = 3) and EAE + HZ mice (n = 3) spinal cord were detected using Bio-Dot SF Microfiltration Apparatus. Loading controls were performed by Western blot using anti-actin antibody. Band intensities were analyzed using ImageJ (NIH) and are expressed in arbitrary units. Photograph (A) shows a representative blot for each condition. Bar graph (B) demonstrated that the acrolein-lysine adduct level in EAE mice spinal cord (20.27 ± 3.0 a.u.) was significantly higher than control (12.30 ± 1.3 a.u., p < 0.05). The acrolein-lysine adduct in EAE + HZ mice spinal cord was 15.14 ± 1.6 a.u. One way ANOVA and post hoc tests were used for statistical analysis. All data are expressed as mean ± SEM.
Hydralazine delayed the onset of EAE mice symptoms and reduced the severity of the paralysis
In this experiment, one group of EAE mice were injected with HZ (EAE + HZ) at the dose of 1 mg/kg daily starting from the day of induction (n=11). In the sham-treated group (EAE), equal amount of saline were injected daily (n=12). Both HZ and saline treatments were carried out for 30 days post induction. The behavioral scores of the two groups of mice were recorded daily (Fig. 2A). All the mice in EAE group showed symptoms of motor impairment as evaluated with the 5-point behavioral test. Nine of 11 mice in HZ-treated group developed behavioral deficits. However, the behavioral deficits in HZ-treated group emerged significantly later than in sham-treated group. Specifically, the average onset of symptoms for EAE + HZ group was 21.73 ± 2.1 days post induction, which was significantly longer than EAE group (15.42 ± 0.4 days post emulsion injection, p < 0.01, Fig. 2B). The onset of symptom for the 2 mice that did not develop behavioral deficits was considered to be at least 30 days post induction. In addition to onset, the severity of the symptoms in EAE + HZ group was significantly lower than the EAE group daily starting from 17 days post induction (p < 0.01, Fig. 2A). When the highest score for each mouse was taken and averaged within each group, EAE + HZ group had a significantly lower average behavioral score (1.72 ± 0.4) than the EAE group (3.33 ± 0.3, P < 0.05, Fig. 2C). No serious hypotension was detected following the treatment of hydralazine in both normal and EAE mice. Specifically, the average systolic pressure of normal mice are 125.6 ± 3.6 mmHg (no hydralazine) and 118.5 ± 6.4 mmHg (with hydralazine) (n= 4, p>0.05). The average systolic pressure of EAE mice are 153.9 ± 5.0 mmHg (no hydralazine) and 134.8 ± 4.5 mmHg (with hydralazine) (n= 3, p>0.05).
Figure 2.

(A) Comparison of behavioral score in each day between hydralazine and sham treated groups (* p < 0.01 for comparison between EAE + HZ and EAE after Day 17). (B) Comparison of the onset of symptoms between hydralazine (EAE + HZ) and sham treated (EAE) groups (p < 0.01). (C) Quantitative comparison of mean behavioral score of EAE + HZ and EAE groups (p< 0.05). The highest score for each mouse was taken and averaged within each group. All data are expressed as mean ± SEM.
Hydralazine treatment lessening the demyelination area on spinal cord cross section
The thoracic segment of spinal cord samples harvested from Control, HZ-treated (EAE + HZ), and Sham-treated mice (EAE) were cut in 15 μm cross sections and labeled with NF200 (green, denote axons) and fluoromyelin (red, denote myelin) (Fig. 3A). Total white matter area and demyelination area were measured and the percent of demyelination was estimated as outlined in Fig. 3B. Control sections showed no signs of demyelination or axonal loss. The percent of demyelination in white matter was compared between EAE and EAE + HZ groups. The HZ treatment significantly decreased demyelination area from 25.58 ± 3.8 % (sham-treated) to 5.10 ± 4.2 % (n = 3, p < 0.05, Fig. 3C).
Figure 3.

(A) A representative sample of spinal cord cross section that were stained with fluoromyelin (red) and anti-NF200 (green). Control sections showed no signs of demyelination or axonal loss. EAE sections showed demyelinated lesions and axonal loss. EAE + HZ treated sections also showed demyelinating lesions and some axonal loss, but noticeably less extensive than EAE. (B) Methodology used to quantify spinal cord lesions. Total white matter area and lesion areas were manually outlined and pixel area calculated. Percent demyelination was calculated by dividing total demyelinated area by total white matter area. (C) Histogram showed quantitative analysis of percent demyelination of EAE and EAE + HZ treated mice. EAE + HZ treated mice had significantly less demyelination compared to EAE mice (p < 0.05). Data are expressed as mean ± SEM. Scale bar = 500 μm.
Discussion
In previous studies we demonstrated that acrolein increased significantly in spinal cord injury (Luo et al., 2005a; Hamann et al., 2008b; Hamann and Shi, 2009) and that anti-acrolein treatment offers significant structural and functional benefits (Hamann et al., 2008a). In the current study, we show that acrolein is likely an important pathological factor in MS pathogenesis as well. First, acrolein-lysine adduct increased significantly in EAE mice when motor behavioral deficits emerge. Secondly, treatment with acrolein scavenger hydralazine alleviated behavioral deficits, reducing the severity and delaying the onset of the motor deficits in EAE mice. In addition to behavioral improvement, we also noted a significant reduction of demyelination in white matter of spinal cord. To the best of our knowledge, this is the first evidence indicating that acrolein is likely a critical factor in the pathogenesis of MS. Furthermore, anti-acrolein treatment appears to be an effective therapeutic strategy to curtail the progression of symptoms in EAE mice.
Acrolein’s ability to damage proteins, lipids, DNA, and to generate more free radicals is well established (Esterbauer et al., 1991; Adams and Klaidman, 1993; Uchida et al., 1998b; Kehrer and Biswal, 2000; Luo and Shi, 2004; Luo et al., 2005b; Luo and Shi, 2005; Luo et al., 2005a; Shao et al., 2005b; Shao et al., 2005a). Therefore, acrolein-mediated pathology in MS is likely mediated through multiple mechanisms. Primarily, acrolein likely breaks down the myelin sheath by attacking lipids and proteins, the main components of myelin (Morell and Quarles, 1999). However, since acrolein has been implicated in calpain activation, (Liu-Snyder et al., 2006), it is also possible that acrolein leads to enzymatic damage of myelin through (Shields and Banik, 1999; Shields et al., 1999). The reduction of EAE induced demyelination in spinal cord by anti-acrolein treatment is consistent with a causal role of acrolein in myelin damage.
Acrolein may also contribute directly to axonal degeneration, another major MS pathology (Trapp et al., 1998; Trapp et al., 1999; Trapp and Nave, 2008). This is due to acrolein compromising the axonal membrane and consequently triggering axonal degeneration (Shi et al., 2002; Luo and Shi, 2004). Such axonal damage can be further exacerbated through acrolein-mediated oxidative stress and mitochondrial dysfunction, known mechanisms of acrolein toxicity (Adams and Klaidman, 1993; Luo et al., 2005b; Luo and Shi, 2005). Therefore, anti-acrolein treatment may reduce both acrolein-mediated myelin damage as well as acrolein-mediated axonal damage. Taken together, acrolein is likely a major factor in MS that contributes to multiple mechanisms of demyelination, axonal degeneration, and functional loss.
Hydralazine is known to neutralize free acrolein (Burcham et al., 2000; Burcham et al., 2002; Kaminskas et al., 2004a) and acrolein-protein adducts (Burcham et al., 2004; Kaminskas et al., 2004b; Burcham and Pyke, 2006), both of which are cytotoxic. When injected at a concentration of 1 mg/kg body weight, a dosage known to be safe and effective in trapping acrolein (Kaminskas et al., 2004b), hydralazine significantly reduced behavioral deficits in EAE mice. The therapeutic effect of hydralazine in EAE is likely attributed to the trapping of acrolein. First, hydralazine is known to bind acrolein (Burcham et al., 2000; Burcham et al., 2002; Kaminskas et al., 2004a). Second, acrolein levels tended to decrease in the presence of hydralazine (Fig. 1). Third, it has been shown that hydralazine is not an effective superoxide scavenger (Hamann et al., 2008a). Thus, the neuroprotective effect of hydralazine demonstrated in this study is most likely due to scavenging acrolein rather than scavenging free radicals.
The effectiveness of hydralazine in the current study also suggests anti-acrolein treatment is a novel therapeutic regime to attenuate acrolein-mediated pathology in MS. The feasibility is further highlighted by the fact that there are multiple acrolein blocking agents, including hydralazine and phenelzine, which are FDA approved medications (Burcham et al., 2002; Burcham et al., 2004; Kaminskas et al., 2004a; Wood et al., 2006). Therefore, we predict that such a therapeutic strategy can be rapidly translated to the clinic for MS patients. Furthermore, other neurodegenerative diseases and trauma where oxidative stress has been implicated (Shibata et al., 2000; Lovell et al., 2001; Montine et al., 2002; Luo et al., 2005a; Hamann et al., 2008b; Hamann and Shi, 2009) may also benefit from such treatment.
Acknowledgments
We thank Melissa Tully and Sean Connell for their critical reading of the paper. We also appreciate technical assistance from Drs. Jianming Li and Amber Pond.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams JD, Jr, Klaidman LK. Acrolein-induced oxygen radical formation. Free Radical Biology & Medicine. 1993;15:187–193. doi: 10.1016/0891-5849(93)90058-3. [DOI] [PubMed] [Google Scholar]
- Burcham PC, Pyke SM. Hydralazine inhibits rapid acrolein-induced protein oligomerization: role of aldehyde scavenging and adduct trapping in cross-link blocking and cytoprotection. Mol Pharmacol. 2006;69:1056–1065. doi: 10.1124/mol.105.018168. [DOI] [PubMed] [Google Scholar]
- Burcham PC, Kerr PG, Fontaine F. The antihypertensive hydralazine is an efficient scavenger of acrolein. Redox Rep. 2000;5:47–49. doi: 10.1179/rer.2000.5.1.47. [DOI] [PubMed] [Google Scholar]
- Burcham PC, Kaminskas LM, Fontaine FR, Petersen DR, Pyke SM. Aldehyde-sequestering drugs: tools for studying protein damage by lipid peroxidation products. Toxicology. 2002:181–182. 229–236. doi: 10.1016/s0300-483x(02)00287-1. [DOI] [PubMed] [Google Scholar]
- Burcham PC, Fontaine FR, Kaminskas LM, Petersen DR, Pyke SM. Protein adduct-trapping by hydrazinophthalazine drugs: mechanisms of cytoprotection against acrolein-mediated toxicity. Mol Pharmacol. 2004;65:655–664. doi: 10.1124/mol.65.3.655. [DOI] [PubMed] [Google Scholar]
- Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372:1502–1517. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radical Biology & Medicine. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- Ghilarducci DP, Tjeerdema RS. Fate and effects of acrolein. Rev Environ Contam Toxicol. 1995;144:95–146. doi: 10.1007/978-1-4612-2550-8_2. [DOI] [PubMed] [Google Scholar]
- Gilgun-Sherki Y, Melamed E, Offen D. The role of oxidative stress in the pathogenesis of multiple sclerosis: the need for effective antioxidant therapy. J Neurol. 2004;251:261–268. doi: 10.1007/s00415-004-0348-9. [DOI] [PubMed] [Google Scholar]
- Gold R, Linington C, Lassmann H. Understanding pathogenesis and therapy of multiple sclerosis via animal models: 70 years of merits and culprits in experimental autoimmune encephalomyelitis research. Brain. 2006;129:1953–1971. doi: 10.1093/brain/awl075. [DOI] [PubMed] [Google Scholar]
- Halliwell B, Gutteridge JMC. Free radicals in biology and medicine. Oxford: Oxford University Press; 1999. [Google Scholar]
- Hamann K, Shi R. Acrolein scavenging: a potential novel mechanism of attenuating oxidative stress following spinal cord injury. J Neurochem. 2009;111:1348–1356. doi: 10.1111/j.1471-4159.2009.06395.x. [DOI] [PubMed] [Google Scholar]
- Hamann K, Nehrt G, Ouyang H, Duerstock B, Shi R. Hydralazine inhibits compression and acrolein-mediated injuries in ex vivo spinal cord. J Neurochem. 2008a;104:708–718. doi: 10.1111/j.1471-4159.2007.05002.x. [DOI] [PubMed] [Google Scholar]
- Hamann K, Durkes A, Ouyang H, Uchida K, Pond A, Shi R. Critical role of acrolein in secondary injury following ex vivo spinal cord trauma. J Neurochem. 2008b;107:712–721. doi: 10.1111/j.1471-4159.2008.05622.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyvas A, David S. Cytosolic phospholipase A2 plays a key role in the pathogenesis of multiple sclerosis-like disease. Neuron. 2004;41:323–335. doi: 10.1016/s0896-6273(04)00003-0. [DOI] [PubMed] [Google Scholar]
- Kaminskas LM, Pyke SM, Burcham PC. Reactivity of hydrazinophthalazine drugs with the lipid peroxidation products acrolein and crotonaldehyde. Org Biomol Chem. 2004a;2:2578–2584. doi: 10.1039/B408796H. [DOI] [PubMed] [Google Scholar]
- Kaminskas LM, Pyke SM, Burcham PC. Strong protein adduct trapping accompanies abolition of acrolein-mediated hepatotoxicity by hydralazine in mice. J Pharmacol Exp Ther. 2004b;310:1003–1010. doi: 10.1124/jpet.104.067330. [DOI] [PubMed] [Google Scholar]
- Kehrer JP, Biswal SS. The molecular effects of acrolein. Toxicological Sciences. 2000;57:6–15. doi: 10.1093/toxsci/57.1.6. [DOI] [PubMed] [Google Scholar]
- Liu-Snyder P, McNally H, Shi R, Borgens RB. Acrolein-mediated mechanisms of neuronal death. J Neurosci Res. 2006;84:209–218. doi: 10.1002/jnr.20863. [DOI] [PubMed] [Google Scholar]
- Lovell MA, Xie C, Markesbery WR. Acrolein is increased in Alzheimer’s disease brain and is toxic to primary hippocampal cultures. Neurobiology of Aging. 2001;22:187–194. doi: 10.1016/s0197-4580(00)00235-9. [DOI] [PubMed] [Google Scholar]
- Luo J, Shi R. Acrolein induces axolemmal disruption, oxidative stress, and mitochondrial impairment in spinal cord tissue. Neurochemsitry International. 2004;44:475–486. doi: 10.1016/j.neuint.2003.09.006. [DOI] [PubMed] [Google Scholar]
- Luo J, Shi R. Acrolein induces oxidative stress in brain mitochondria. Neurochem Int. 2005;46:243–252. doi: 10.1016/j.neuint.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Luo J, Uchida K, Shi R. Accumulation of acrolein-protein adducts after traumatic spinal cord injury. Neurochem Res. 2005a;30:291–295. doi: 10.1007/s11064-005-2602-7. [DOI] [PubMed] [Google Scholar]
- Luo J, Robinson JP, Shi R. Acrolein-induced cell death in PC12 cells: role of mitochondria-mediated oxidative stress. Neurochem Int. 2005b;47:449–457. doi: 10.1016/j.neuint.2005.07.002. [DOI] [PubMed] [Google Scholar]
- Montine TJ, Neely MD, Quinn JF, Beal MF, Markesbery WR, Roberts LJ, Morrow JD. Lipid peroxidation in aging brain and Alzheimer’s disease. Free Radic Biol Med. 2002;33:620–626. doi: 10.1016/s0891-5849(02)00807-9. [DOI] [PubMed] [Google Scholar]
- Morell P, Quarles RH. In: Basic Neurochemistry: Molecular, Cellular, and Medical Aspects. Siegel GJ, Agranoff BW, Alberts RW, Molinoff PB, editors. Philadelphia: Lippincott Williams & Wilkins; 1999. [Google Scholar]
- Shao B, O’Brien KD, McDonald TO, Fu X, Oram JF, Uchida K, Heinecke JW. Acrolein modifies apolipoprotein A-I in the human artery wall. Ann N Y Acad Sci. 2005a;1043:396–403. doi: 10.1196/annals.1333.046. [DOI] [PubMed] [Google Scholar]
- Shao B, Fu X, McDonald TO, Green PS, Uchida K, O’Brien KD, Oram JF, Heinecke JW. Acrolein impairs ATP binding cassette transporter A1-dependent cholesterol export from cells through site-specific modification of apolipoprotein A-I. J Biol Chem. 2005b;280:36386–36396. doi: 10.1074/jbc.M508169200. [DOI] [PubMed] [Google Scholar]
- Shao C, Roberts KN, Markesbery WR, Scheff SW, Lovell MA. Oxidative stress in head trauma in aging. Free Radic Biol Med. 2006;41:77–85. doi: 10.1016/j.freeradbiomed.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Shi R, Luo J, Peasley MA. Acrolein inflicts axonal membrane disruption and conduction loss in isolated guinea pig spinal cord. Neuroscience. 2002;115:337–340. doi: 10.1016/s0306-4522(02)00457-8. [DOI] [PubMed] [Google Scholar]
- Shibata N, Nagai R, Miyata S, Jono T, Horiuchi S, Hirano A, Kato S, Sasaki S, Asayama K, Kobayashi M. Nonoxidative protein glycation is implicated in familial amyotrophic lateral sclerosis with superoxide dismutase-1 mutation. Acta Neuropathol (Berl) 2000;100:275–284. doi: 10.1007/s004019900173. [DOI] [PubMed] [Google Scholar]
- Shields DC, Banik NL. Pathophysiological role of calpain in experimental demyelination. J Neurosci Res. 1999;55:533–541. doi: 10.1002/(SICI)1097-4547(19990301)55:5<533::AID-JNR1>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Shields DC, Schaecher KE, Saido TC, Banik NL. A putative mechanism of demyelination in multiple sclerosis by a proteolytic enzyme, calpain. Proc Natl Acad Sci U S A. 1999;96:11486–11491. doi: 10.1073/pnas.96.20.11486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KJ, Kapoor R, Felts PA. Demyelination: the role of reactive oxygen and nitrogen species. Brain Pathol. 1999;9:69–92. doi: 10.1111/j.1750-3639.1999.tb00212.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp BD, Nave KA. Multiple sclerosis: an immune or neurodegenerative disorder? Annu Rev Neurosci. 2008;31:247–269. doi: 10.1146/annurev.neuro.30.051606.094313. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Ransohoff R, Rudick R. Axonal pathology in multiple sclerosis: relationship to neurologic disability. Curr Opin Neurol. 1999;12:295–302. doi: 10.1097/00019052-199906000-00008. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338:278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- Uchida K, Kanematsu M, Morimitsu Y, Osawa T, Noguchi N, Niki E. Acrolein is a product of lipid peroxidation reaction. Formation of free acrolein and its conjugate with lysine residues in oxidized low density lipoproteins. J Biol Chem. 1998a;273:16058–16066. doi: 10.1074/jbc.273.26.16058. [DOI] [PubMed] [Google Scholar]
- Uchida K, Kanematsu M, Sakai K, Matsuda T, Hattori N, Mizuno Y, Suzuki D, Miyata T, Noguchi N, Niki E, Osawa T. Protein-bound acrolein: potential markers for oxidative stress. Proceedings of the National Academy of Sciences of the United States of America. 1998b;95:4882–4887. doi: 10.1073/pnas.95.9.4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood PL, Khan MA, Moskal JR, Todd KG, Tanay VA, Baker G. Aldehyde load in ischemia-reperfusion brain injury: neuroprotection by neutralization of reactive aldehydes with phenelzine. Brain Res. 2006;1122:184–190. doi: 10.1016/j.brainres.2006.09.003. [DOI] [PubMed] [Google Scholar]