

Abstract
Objective:
We sought to create a classification system for pediatric corpus callosal abnormalities (CCA) based upon midline sagittal brain MRI. We used the term CCA for patients with structural variants of the corpus callosum, excluding patients with interhemispheric cyst variant or pure dysplasia without hypoplasia. Currently, no system exists for nonsyndromic forms of CCA, and attempts to create such a system have been hampered by highly variable morphology in patients with sporadic CCA. We reasoned that any useful strategy should classify affected family members within the same type, and that phenotypic variability should be minimized in patients with recessive disease.
Methods:
We focused recruitment toward multiplex consanguineous families, ascertained 30 patients from 19 consanguineous families, and analyzed clinical features together with brain imaging.
Results:
We identified 3 major CCA classes, including hypoplasia, hypoplasia with dysplasia, and complete agenesis. Affected individuals within a given multiplex family usually displayed the same variant of the class of abnormality and they always displayed the same class of abnormality within each family, or they displayed complete agenesis. The system was validated among a second cohort of 10 sporadic patients with CCA.
Conclusions:
The data suggest that complete agenesis may be a common end-phenotype, and implicate multiple overlapping pathways in the etiology of CCA.
The corpus callosum (CC) structurally consists of 4 major anatomic features including the rostrum, genu, corpus, and splenium (figure 1). Corpus callosal abnormalities (CCA), a term encompassing agenesis of the corpus callosum (ACC), has an estimated prevalence of 0.3%–0.7% in patients undergoing brain imaging,1 but in Middle Eastern populations the prevalence is probably closer to 1%,2,3 due to the higher rates of inbreeding. The finding of CCA can be identified incidentally, or can be part of a developmental or cytogenetic disease.
Figure 1. Classification of morphologic spectrum of midline sagittal MRI appearance of the corpus callosum (CC).

Schematics depict the appearance of CC on midline sagittal MRI. Appearance of normal CC showing rostrum, genu, corpus, and splenium. CC abnormalities were divided into principal classes: HYPOPLASIA, DYSPLASIA WITHOUT HYPOPLASIA, HYPOPLASIA WITH DYSPLASIA, and COMPLETE AGENESIS. There are 3 subtypes of HYPOPLASIA: hypoplasia without dysplasia, apple core corpus callosal abnormalities (CCA), and anterior remnant CCA, based upon the physical appearance. DYSPLASIA WITHOUT HYPOPLASIA encompasses cases in which the CC is morphologically abnormal but has no evidence of hypoplasia, and is represented by a case with hump-shaped CC.25 In HYPOPLASIA WITH DYSPLASIA CCA, there are 2 subtypes, stripe CCA and kinked CCA. COMPLETE AGENESIS is characterized by absence of an evident CC at the level of MRI.
Previous studies have differentiated CCA into those with or without additional brain malformations3 or into partial or complete agenesis.4 However, in one study, roughly equal percentages of patients with either partial or complete agenesis displayed additional structural brain abnormalities, suggesting that this differentiation may not relate to causative etiology.5 The presence vs absence of Probst bundles (PBs) has also been used to define categories of disease,3 but has not been validated as a clinically predictive sign. CCA was previously divided into hypogenesis (incomplete formation) and dysgenesis (defective development),6 but further segregation based upon these initial findings has not been explored.
We aimed to refine this classification system, to account for similarities in CC appearance in affected siblings within a single family, where a common genetic etiology is most likely. Since recessive forms of disease (i.e., those observed in families with documented consanguinity and a recessive mode of inheritance) are the most likely to show consistent CC appearance, and are most often due to loss-of-function alleles (rather than environmental or dominant causes), the minimized intrafamilial variability could help determine the relationship between primary gene mutation and CCA morphology.
METHODS
Standard protocol approvals and patient consents.
This study was approved by an ethical standards committee on human subjects from UCSD and other collaborating institutions where applicable. Written informed consent was obtained from all families (patients or guardians) participating in this study.
We contacted several hundred clinicians including pediatric neurologists, radiologists, and clinical geneticists throughout the world in order to recruit patients with recessive developmental brain diseases. The resultant database is highly enriched for multiplex families that self-report consanguinity (i.e., parents are first- or second-degree cousins), and includes digital MRI and text descriptions of the MRI results, as reported by the attending neuroradiologist and one or more of the authors. We performed text queries of the dataset of 1,027 patients/pedigrees for CCA and variants, yielding 134 patients from 81 pedigrees. In order to limit this dataset to relatively pure forms of CCA, we excluded 53 patients in whom CCA was secondary to another diagnosis, 3 due to normal CC upon MRI re-evaluation, and 2 due to likely environmental causes. We excluded 46 patients who lacked adequate images. All patients were enrolled in this institutional review board–approved study according to institutional guidelines.
The final cohort included 30 individuals from 19 consanguineous families. There were 8 multiplex families with more than 2 affected siblings for whom MRI data were available. Because many of the images were from digital photographs of MRIs that were subsequently destroyed, image quality varied greatly. For analysis, midline sagittal MRI were cropped 2–4 cm anterior to the genu, posterior to the splenium, and inferior to IV ventricle to ensure uniform visualization of the CC and the adjacent structures.
RESULTS
Classification system.
We decided to adopt the term CCA to encompass all forms of CC abnormalities as well as complete agenesis. We decided against the term ACC because only a minority of patients showed complete agenesis. Corpus callosal dysplasia7 (figure 1) and ACC with interhemispheric cyst8 were not encountered and are thus not discussed further.
We identified 3 principal classes of CCA among our cohort (figures 1 and 2), all designated with upper case typeface. Within some classes, there were 2 or more variants, designated with italic typeface. HYPOPLASIA consisted of uniformly thin or partially underdeveloped CCA in the posterior region. Within the HYPOPLASIA class, we identified hypoplasia without dysplasia CCA, with all major anatomic features intact including a distinctive rostrum, genu, corpus, and splenium. More severe forms of HYPOPLASIA showed progressive loss of the more caudal aspects of the CC. This includes a form where the posterior CC is hypoplastic and foreshortened (i.e., corpus or splenium), taking on the appearance of an apple core CCA, and a form in which there is agenesis of the posterior CC (i.e., corpus and splenium), called anterior remnant CCA type.
Figure 2. Midline sagittal MRI representing the spectrum of corpus callosum (CC) abnormalities in 30 patients with corpus callosal abnormalities (CCA).

(A) Normal CC. (B–D) HYPOPLASIA class of CCA. (B) Hypoplasia without dysplasia CCA, (C) apple core CCA, (D) Anterior remnant CCA (black arrow). (E, F) HYPOPLASIA WITH DYSPLASIA class of CCA. (E) Stripe CCA, (F) kinked CCA. (G) COMPLETE AGENESIS. (H) Example of Probst bundles. Number of patients with each class of CCA, and each subtype of CCA, from a cohort of 29 patients.
The second class of CCA was defined as HYPOPLASIA WITH DYSPLASIA, with disturbance of the overall shape of the CC that was not limited to the posterior region, with 2 subtypes. The first consisted of a uniformly thin stripe CCA that lacked an anatomically distinct genu and splenium. The second was characterized by a thin CC that was hypoplastic as well as obviously kinked at one or more locations, either anteriorly or posteriorly, which we called kinked CCA.
The third class of CCA was characterized by absent CC at the resolution of MRI on a midline sagittal image. This class would thus be synonymous with COMPLETE AGENESIS or the previously used term ACC. In this class, it was important to distinguish the hippocampal commissure from the CC, the former retained in many COMPLETE AGENESIS cases.6
We also specifically commented on the appearance of the anterior commissure (AC), because recent data suggest a compensatory increase in the size of the AC (or other structures such as the internal capsule) in some cases of ACC.9 The AC was normal is 7, decreased in size in 14, absent in 8, and enlarged in 1 (table 1; see table e-1 on the Neurology® Web site at www.neurology.org for additional details). We did not utilize the appearance of the AC in our classification system due to unavoidable limitations in MRI quality in several cases.
Table 1.
Clinical features of patients with CCA in this study


Abbreviations: + = present; – = absent; +D = decreased size: referring to anterior commissure); +E = enlarged size: referring to anterior commissure); ABS = seizures (absence type); ANR = anterior remnant corpus callosum; APC = apple core corpus callosum; CAG = complete absence of corpus callosum; CBLND = cortical blindness; CCA = corpus callosum abnormality; Col = optic colobomas; CSP = cavum septum pellucidum; Ctrl = controlled (referring to seizures); HwoD = hypoplasia without dysplasia; KIN = kinked corpus callosum; MLD = mild; NCtrl = not controlled (referring to seizures); ON = optic nerve; ONA = optic nerve atrophy; PS = perisylvian; SEV = severe; STR = stripe corpus callosum; VEP = visual evoked potentials.
We categorized the 30 patients based upon this simple classification system (figure 2). We found 13 patients with HYPOPLASIA appearance, and of these, 6 showed hypoplasia without dysplasia CCA, 5 showed apple core CCA, and 2 showed anterior remnant CCA appearance. We found 12 patients with HYPOPLASIA WITH DYSPLASIA class, and of these, 6 showed stripe CCA and 6 showed a kinked CCA appearance. There were 5 patients with COMPLETE AGENESIS.
Validation of classification system in multiplex consanguineous families.
CCA-184, multiple epiphyseal dysplasia, and CCA.
CCA-184 is a consanguineous multiplex family with 3 affected branches from the United Arab Emirates (UAE, table 1 and figure 3). The affecteds within this family also displayed a mild form of multiple epiphyseal dysplasia, described previously,10,11 mild mental retardation, developmental delay, and ventriculomegaly. MRI was available in 4 of the affected members, and 2 (IV-I-2, IV-I-4) showed an intact splenium and genu, and were thus categorized as hypoplasia without dysplasia. In IV-II-7, the CCA was classified as the subtype apple core CCA because of foreshortening. In IV-III-1, there were fetal ultrasound studies that showed similar features, but MRI was not available, so he was excluded from further analysis. Interestingly, in IV-I-9, MRI showed no discernible CC, consistent with COMPLETE AGENESIS.
Figure 3. Imaging spectrum of HYPOPLASIA class.

(Top row) CCA-184: Affected children have multiple epiphyseal dysplasia, mild mental retardation, developmental delay, prominent ventricles, and corpus callosal abnormalities (CCA). (A, B) HYPOPLASIA, hypoplasia without dysplasia CCA. (D) HYPOPLASIA, apple core CCA subtype. (C) COMPLETE AGENESIS. CCA-385: Affected children have GnRH deficiency, respiratory abnormalities, septo-optic dysplasia, bilateral optic colobomas, pituitary hypoplasia, cerebellar vermis hypoplasia, mild polymicrogyria, and CCA. (E, F) Hypoplasia without dysplasia CCA. (Bottom row) CCA-542: Affected children have epilepsy, hypotonia and hyporeflexia, perisylvian polymicrogyria, and CCA. (G) HYPOPLASIA, apple core CCA. (H) HYPOPLASIA, Hypoplasia without dysplasia CCA. (I) COMPLETE AGENESIS. CCA-557: Affected children have cerebellar vermis hypoplasia and CCA. (J) Apple core CCA. (K) COMPLETE AGENESIS.
CCA-385, cerebellar vermis hypoplasia, and CCA.
Family CCA-385 displayed 2 affected branches and is from the UAE (figure 3). The clinical phenotype includes GnRH deficiency with pituitary hypoplasia, visual reduction, and mild ichthyosis.12 The affected members (IV-I-5, IV-II-7) showed mild ventriculomegaly, cerebellar vermis hypoplasia, and perisylvian polymicrogyria. The CC showed thinning of the entire structure, with most notable hypoplasia of the mid to posterior corpus, with an intact genu and splenium, consistent with hypoplasia without dysplasia CCA. MRI was not available on IV-II-5.
CCA-542, epilepsy, hypotonia, hyporeflexia, and CCA.
Family CCA-542 has one affected branch from a first-cousin marriage and is from Kuwait (figure 3). Their clinical phenotype included epilepsy, hypotonia, hyporeflexia, ventriculomegaly, and visual reduction. All 3 affected members showed perisylvian polymicrogyria. In III-3 the MRI showed posterior shortening of the CC with underdeveloped or absent splenium, consistent with apple core CCA. III-5 CC was classified as hypoplasia without dysplasia CCA. In III-9, MRI showed COMPLETE AGENESIS.
CCA-557, pontocerebellar hypoplasia, and CCA.
Family CCA-557 displays 2 affected members from a first-cousin marriage from Turkey (figure 3). The clinical phenotype suggested pontocerebellar hypoplasia. MRI in III-1 showed thinning and foreshortening of the CC, consistent with apple core CCA subtype. In III-2, MRI showed COMPLETE AGENESIS.
CCA-405, mental retardation, dysmorphic facies, and CCA.
Family CCA-405 displays 2 affected members from a first-cousin marriage from Oman (figure 4). Their clinical phenotype included mental retardation, dysmorphic facial features, and moderate spasticity in one of two children. MRI in III-1 and III-2 showed evidence of white matter disease with high signal on T2 and fluid-attenuated inversion recovery images and optic nerve atrophy. Lysosomal storage enzyme analysis, serum amino acids, urine organic acids, as well as lactate and pyruvate studies were unremarkable, and the clinical status improved over time. There was a uniformly thin appearance of the CC, consistent with stripe CCA, without clear anatomic features of a genu or splenium.
Figure 4. Imaging spectrum of HYPOPLASIA WITH DYSPLASIA class.

(Top row) CCA-405: Affected children have mental retardation, dysmorphic features, and corpus callosal abnormalities (CCA). (A, B) Stripe CCA. CCA-636: Affected children have spasticity, ataxia, white matter hypoplasia, cerebellar hypoplasia, and CCA. (C, D) Stripe CCA. (Bottom row) CCA-523: Affected children have spasticity, epilepsy, and CCA. (E, F) Kinked CCA. CCA-531: Affected children have cerebellar vermis hypoplasia, spasticity, and CCA. (G) Kinked CCA. (H) COMPLETE AGENESIS.
CCA-636, pontocerebellar hypoplasia, microcephaly, and CCA.
This family has 2 affected members from a first-cousin marriage from Egypt (figure 4). The clinical phenotype includes mild microcephaly, mild hypoplasia of the pons and cerebellum, and mild ventriculomegaly. MRI in III-1 and III-2 showed mild ventriculomegaly and a corpus callosum without a discernible genu, body, isthmus, and splenium that was classified as stripe CCA.
CCA-523, mental retardation, epilepsy, and CCA.
Family CCA-523 displays 2 affected members from a first-cousin (once removed) marriage, from Egypt (figure 4). Their clinical phenotype included mental retardation, spasticity, and epilepsy. The MRI showed similar appearance in the 2 affected members, the CC was severely thinned and kinked both in the anterior and posterior parts, most consistent with kinked CCA subtype. Additionally, there was some evidence of abnormal signal in the white matter, and was slightly more severe in IV-6.
CCA-531, cerebellar vermis hypoplasia, spasticity, and CCA.
Family CCA-531 displays 2 affected branches from Egypt (figure 4). There are 3 affected siblings that resulted from either a second-cousin or a first-cousin (once removed) marriage. MRI data were available from 2 affecteds. Their clinical phenotype included mild spasticity and microcephaly. In IV-I-1, MRI showed a thin CC that was kinked near the anterior region and the isthmus, consistent with kinked CCA subtype. In IV-II-2 MRI at 2 years showed complete absence of the CC, consistent with COMPLETE AGENESIS.
Evaluation of classification system in a cohort of CCA probands from consanguineous families.
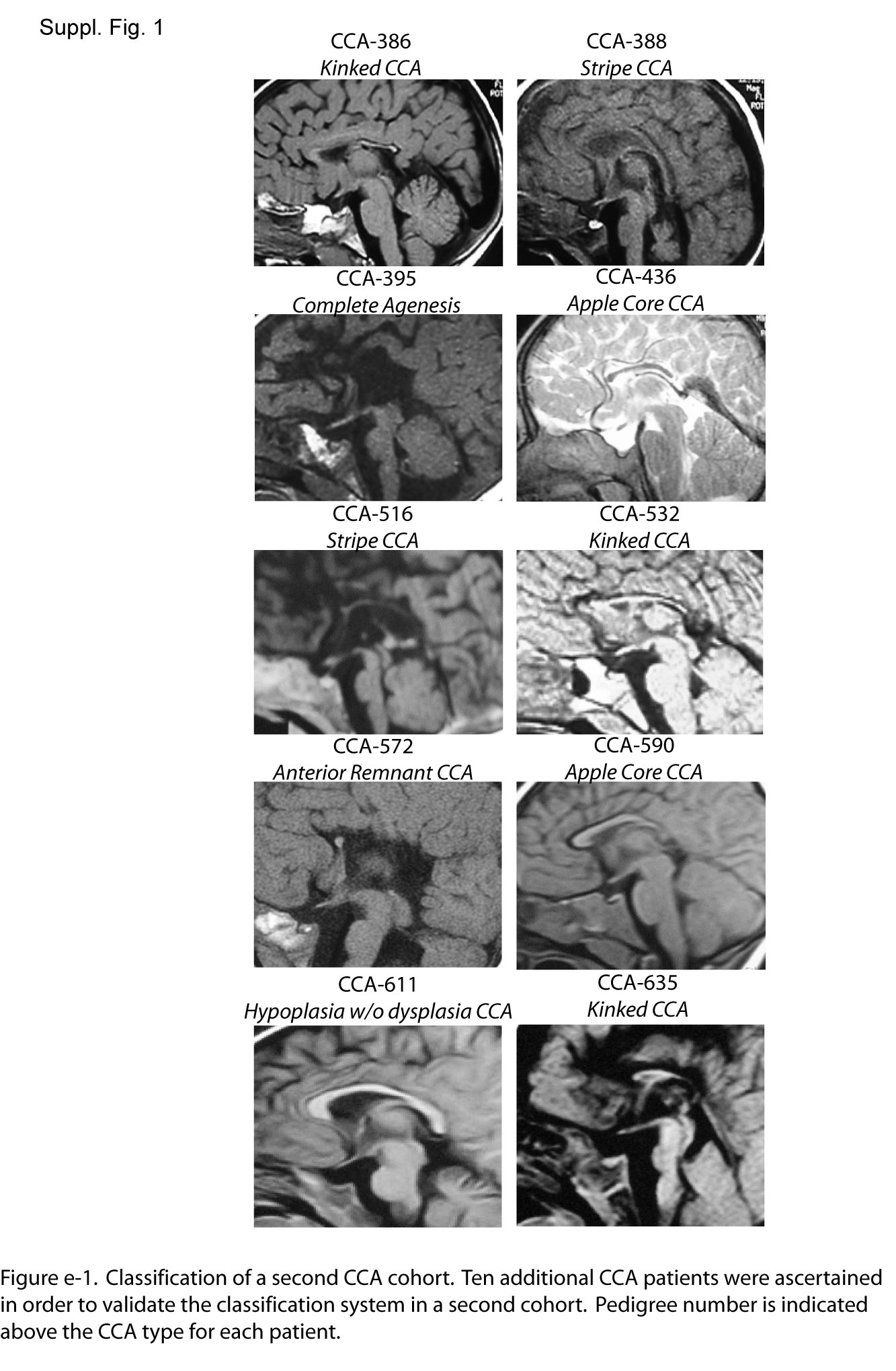
In order to determine if the proposed classification system could be applied to other cases with CCA, we used the system to classify an additional 10 families with CCA from our database (CCA-386, CCA-388, CCA-395, CCA-436, CCA-516, CCA-532, CCA-543, CCA-572, CCA-590, CCA-611, and CCA-635). All reported a history of consanguinity (typically first-cousin marriage), and 7 are multiplex (with more than one affected child), but in each case MRI was available from only a single affected within the family. There were 5 patients with some form of the HYPOPLASIA class, and of these, 2 patients showed hypoplasia without dysplasia CCA, 2 showed apple core CCA, and one showed anterior remnant CCA appearance (figure e-1). There were 5 patients with one of the HYPOPLASIA WITH DYSPLASIA class, and of these, 2 showed stripe CCA and 3 showed kinked CCA. There was one patient with COMPLETE AGENESIS. Therefore, the classification system appears to be applicable to patients with various forms of CCA, at least within the cohort tested.
Validation of the classification system by a blinded neuroradiologist and neurologist.
In order to validate the classification system proposed here, we presented the unlabeled images to both a blinded neuroradiologist and neurologist, each separately categorizing each patient according to the proposed classification system. As a result, there were 29 scans evaluated and concordance was established in 26 (89%) and 25 (86%), which is similar reliability to the 80%–90% concordance rate of other neuroimaging classification systems.13,14 The scans in which there was nonconcordance were at the interface of the hypoplasia without dysplasia and stripe CCA, because these 2 are along a continuum without an absolutely distinct separating line.
Clinical data.
Five of the 6 hypoplasia without dysplasia CCA cases were associated with mild to moderate ventriculomegaly (83%), whereas there was only one apple core CCA out of the 5, and 2 striped CCA out of the 6 that were associated with ventriculomegaly (table 1). Fifteen patients with CCA also had cerebellar affection of varying degrees (50%) and 7 (23%) had microcephaly (OFC <2%ile), but no correlation with type of CCA was apparent. There were 9 patients in our cohort with reduced white matter or evidence of abnormal white matter signal (30%), but this was observed in both HYPOPLASIA and HYPOPLASIA WITH DYSPLASIA classes. There were no obvious correlations between the presence or severity of extra-CNS or clinical features with the subtype or severity of CCA that we could detect. Statistical analysis was not attempted secondary to the small size and highly diverse phenotypic spectrum of the patient cohort.
DISCUSSION
The proposed CCA classification system can be applied when evaluating a single midline sagittal MRI from any patient, and does not require 3-dimensional reconstruction or volumetric analysis. Although future studies might utilize more advanced imaging technologies such as fMRI or fractional anisotropy for more highly precise anatomic subclassification, for most clinicians, a simple system based upon routine MRI is a necessary first step. We found that within a given family, the appearance of the CCA was remarkably similar, though in some instances there were different subtypes of a given class of abnormality seen within the same family. Clearly there is some interfamilial variability, although COMPLETE AGENESIS can be observed in family members as an “end phenotype” associated with most of the subtypes. Due to the relatively small cohort size, statistical analysis could not be applied, and we could not determine whether particular CNS or non-CNS clinical features consistently associated with specific CCA subtype.
The sequence of CC development begins with the differentiation of the commissural plate around 6 weeks gestation (GW), followed by crossing of pioneer axons, first in the rostrum and then proceeding more casually from 10 to 12 GW.9 CC myelination does not occur until after birth and proceeds until adolescence. Given this sequence, we might imagine that the insult timing of apple core and anterior remnant might be prior to midgestation, whereas hypoplasia without dysplasia might be later. Pinpointing genetic etiologies could better clarify precise mechanisms.
The clinical phenotype tended to be similar in siblings of a given family, but there was greater heterogeneity than for MRI findings. Some of the patients had evidence of a small brainstem/pons, which might represent deficient longitudinal pontine axons as part of an overall axonal deficiency in CCA, but a more comprehensive volumetric analysis of brainstem was not possible in this cohort due to limited image availability. Previous studies comparing neurodevelopmental outcome in relationship to partial or complete CCA have shown mixed results, some finding that complete CCA shows a poorer prognosis,15 whereas others have not validated this correlation.4,16 We found that the AC was present in most cases; however, in cases with COMPLETE AGENESIS, it tended to be absent or decreased in size.
Our observation fits well with the current understanding of CC development as a process that takes place along distinct anatomic axes, and a number of early events that are guided by independent signaling systems for each anatomic group of axons that cross the midline.17,18 Disturbance of an essential early event might therefore lead to hypoplasia or to complete agenesis, depending upon environmental or modifier gene influences. The predominant type of HYPOPLASIA WITHOUT DYSPLASIA showed some level of posterior CC hypoplasia, while only 2 cases showed near complete posterior CC hypoplasia (the anterior remnant CCA type). Presumptively in anterior remnant CCA patients, the genetic defect alters neurodevelopmental processes at an early time point, and involves the coordination of axons crossing in all but the most anterior regions of the CC.
A number of genetic disorders in humans have been associated with CCA, including several X-linked diseases, metabolic disorders, and contiguous gene deletion syndromes.17 However, in each of these, the CCA is usually secondary to a more defining characteristic (such as in X-linked lissencephaly with ambiguous genitalia syndrome19), or is just one of a number of defining characteristics (such as in Aicardi or Andermann syndromes).20,21 To date, no genes have been identified in which CCA has been the primary defining characteristic. A classification system such as the one that we propose may help lead to the identification of causes of this important neurologic disorder, by allowing patients with the same type of CCA to be grouped together for genetic analysis.
Investigations have identified several potential gene dose-dependent candidate loci for CCA.22–24 Additionally, cytogenetic abnormalities are detected in approximately 10% of patients with CCA.1 Analysis in mice has uncovered a large number of genes that when mutated lead to CCA, including guidance molecules, transcription factors, extracellular matrix molecules, signaling/cytoplasmic molecules, growth factors, as well as certain strains of mice that are particularly vulnerable to CCA.17 Thus, a large number of genes contribute to the development, morphology, and maintenance of the CC, some of which appear to play primary roles, while in others the effect on the CC may be secondary to a more notable structural CNS defect.
Our preliminary genetic mapping data from these consanguineous families suggest a multitude of new causative loci, which reflects the anatomic complexity that we describe here. Using a recessive disease model, it is possible to identify causative genes using homozygosity mapping and next-generation sequencing strategies in a straightforward manner. Therefore, the ability to select patients for evaluation at newly identified genes based upon similarities in the appearance of the CC will improve the ability to molecularly classify CCA patients.
Given this substantial proportion of sporadic patients with CCA and PBs, we felt our classification scheme should reflect their presence or absence, but rather than further subdivide the aforementioned types, we propose to indicate the presence of PBs by denoting + PBs next to the type. The explanation for the relatively low percentage of patients with PBs in our cohort compared with historic controls5 is not immediately apparent but may relate to the bias for large consanguineous families in this study.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the collaborators and families who participated in this study and A. James Barkovich for reviewing the text.
Supplemental data at www.neurology.org
- AC
- anterior commissure
- ACC
- agenesis of the corpus callosum
- CC
- corpus callosum
- CCA
- corpus callosal abnormalities
- GW
- weeks gestation
- PB
- Probst bundle
- UAE
- United Arab Emirates
DISCLOSURE
Dr. Hanna, Ms. Marsh, Ms. Swistun, Prof. Al-Gazali, Prof. Zaki, Dr. Abdel-Salam, Dr. Al-Tawari, and Dr. Bastaki report no disclosures. Dr. Kayserili serves on the editorial board of the European Journal of Medical Genetics; serves as a consultant in a clinical genetics outpatient division (10% effort) and conducts cranial MRI evaluations (20% effort); and receives research support from the Turkish Research Council, the EU, and the Istanbul University Research Fund. Dr. Rajab and Dr. Boglárka report no disclosures. Dr. Dietrich serves on the editorial board of Investigative Radiology. Dr. Dobyns serves on the editorial boards of the American Journal of Medical Genetics and Clinical Dysmorphology; and receives research support from the NIH. Dr. Truwit serves on the editorial board of Seminars in Roentgenology; is listed as an inventor on multiple patents, mostly related to medical device technologies; receives royalty payments for MR guided neurosurgery devices; receives research support from the NIH; and has served as an expert witness in the field of imaging of perinatal asphyxia. Dr. Sherr receives research support from Pfizer Inc, the NIH/NINDS, the March of Dimes, the Aicardi Syndrome Foundation, the Weston Havens Foundation, and the National Organization of Disorders of the Corpus Callosum. Dr. Gleeson reports no disclosures.
REFERENCES
- 1. Bedeschi MF, Bonaglia MC, Grasso R, et al. Agenesis of the corpus callosum: clinical and genetic study in 63 young patients. Pediatr Neurol 2006;34:186–193 [DOI] [PubMed] [Google Scholar]
- 2. Chacko A, Koul R, Sankhla DK. Corpus callosum agenesis. Saudi Med J 2001;22:22–25 [PubMed] [Google Scholar]
- 3. Sztriha L. Spectrum of corpus callosum agenesis. Pediatr Neurol 2005;32:94–101 [DOI] [PubMed] [Google Scholar]
- 4. Volpe P, Paladini D, Resta M, et al. Characteristics, associations and outcome of partial agenesis of the corpus callosum in the fetus. Ultrasound Obstet Gynecol 2006;27:509–516 [DOI] [PubMed] [Google Scholar]
- 5. Hetts SW, Sherr EH, Chao S, Gobuty S, Barkovich AJ. Anomalies of the corpus callosum: an MR analysis of the phenotypic spectrum of associated malformations. AJR Am J Roentgenol 2006;187:1343–1348 [DOI] [PubMed] [Google Scholar]
- 6. Barkovich AJ. Pediatric Neuroimaging. Philadelphia: Lippincott Williams & Wilkins; 2005 [Google Scholar]
- 7. Erdogmus B, Yazici B, Ozdere BA. Hump-shaped corpus callosum. Acta Radiol 2005;46:616–617 [DOI] [PubMed] [Google Scholar]
- 8. Barkovich AJ, Simon EM, Walsh CA. Callosal agenesis with cyst: a better understanding and new classification. Neurology 2001;56:220–227 [DOI] [PubMed] [Google Scholar]
- 9. Sarnat HB. Embryology and malformations of the forebrain commissure. In: Sarnat HB, Curatolo P, eds. Malformations of the Nervous System. St. Louis: Elsevier; 2008:67. [DOI] [PubMed] [Google Scholar]
- 10. Al-Gazali L, Bakalinova D. Autosomal recessive syndrome of macrocephaly, multiple epiphyseal dysplasia and distinctive facial appearance. Clin Dysmorphol 1998;7:177–184 [DOI] [PubMed] [Google Scholar]
- 11. Bayoumi R, Saar K, Lee YA, et al. Localisation of a gene for an autosomal recessive syndrome of macrocephaly, multiple epiphyseal dysplasia, and distinctive facies to chromosome 15q26. J Med Genet 2001;38:369–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Al-Gazali L, Hertecant J, Algawi K, El Teraifi H, Dattani M. A new autosomal recessive syndrome of ocular colobomas, ichthyosis, brain malformations and endocrine abnormalities in an inbred Emirati family. Am J Med Genet A 2008;146:813–819 [DOI] [PubMed] [Google Scholar]
- 13. Chawalparit O, Wonglaksanapimon S, Songsaeng D, Saenanarong V. Reliability study of white matter rating scale for the dementia and disability in Thai Elderly Project. J Med Assoc Thai 2009;92:543–547 [PubMed] [Google Scholar]
- 14. Ay H, Furie KL, Singhal A, Smith WS, Sorensen AG, Koroshetz WJ. An evidence-based causative classification system for acute ischemic stroke. Ann Neurol 2005;58:688–697 [DOI] [PubMed] [Google Scholar]
- 15. Goodyear PW, Bannister CM, Russell S, Rimmer S. Outcome in prenatally diagnosed fetal agenesis of the corpus callosum. Fetal Diagn Ther 2001;16:139–145 [DOI] [PubMed] [Google Scholar]
- 16. Moutard ML, Kieffer V, Feingold J, et al. Agenesis of corpus callosum: prenatal diagnosis and prognosis. Childs Nerv Syst 2003;19:471–476 [DOI] [PubMed] [Google Scholar]
- 17. Richards LJ, Plachez C, Ren T. Mechanisms regulating the development of the corpus callosum and its agenesis in mouse and human. Clin Genet 2004;66:276–289 [DOI] [PubMed] [Google Scholar]
- 18. Ren T, Anderson A, Shen WB, et al. Imaging, anatomical, and molecular analysis of callosal formation in the developing human fetal brain. Anat Rec A Discov Mol Cell Evol Biol 2006;288:191–204 [DOI] [PubMed] [Google Scholar]
- 19. Sherr EH. The ARX story (epilepsy, mental retardation, autism, and cerebral malformations): one gene leads to many phenotypes. Curr Opin Pediatr 2003;15:567–571 [DOI] [PubMed] [Google Scholar]
- 20. Larbrisseau A, Vanasse M, Brochu P, Jasmin G. The Andermann syndrome: agenesis of the corpus callosum associated with mental retardation and progressive sensorimotor neuronopathy. Can J Neurol Sci 1984;11:257–261 [DOI] [PubMed] [Google Scholar]
- 21. Aicardi J. Aicardi syndrome. Brain Dev 2005;27:164–171 [DOI] [PubMed] [Google Scholar]
- 22. Sherr EH, Owen R, Albertson DG, et al. Genomic microarray analysis identifies candidate loci in patients with corpus callosum anomalies. Neurology 2005;65:1496–1498 [DOI] [PubMed] [Google Scholar]
- 23. Boland E, Clayton-Smith J, Woo VG, et al. Mapping of deletion and translocation breakpoints in 1q44 implicates the serine/threonine kinase AKT3 in postnatal microcephaly and agenesis of the corpus callosum. Am J Hum Genet 2007;81:292–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Doco-Fenzy M, Mauran P, Lebrun JM, et al. Pure direct duplication (12) (q24.1–>q24.2) in a child with Marcus Gunn phenomenon and multiple congenital anomalies. Am J Med Genet A 2006;140:212–221 [DOI] [PubMed] [Google Scholar]
- 25. Davila-Gutierrez G. Agenesis and dysgenesis of the corpus callosum. Semin Pediatr Neurol 2002;9:292–301 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}