

Abstract
Several pathovars of Pseudomonas syringae produce the phytotoxin coronatine (COR), which contains an unusual amino acid, the 1-amino-2-ethylcyclopropane carboxylic acid called coronamic acid (CMA), which is covalently linked to a polyketide-derived carboxylic acid, coronafacic acid, by an amide bond. The region of the COR biosynthetic gene cluster proposed to be responsible for CMA biosynthesis was resequenced, and errors in previously deposited cmaA sequences were corrected. These efforts allowed overproduction of P. syringae pv. glycinea PG4180 CmaA in P. syringae pv. syringae FF5 as a FLAG-tagged protein and overproduction of P. syringae pv. tomato CmaA in Escherichia coli as a His-tagged protein; both proteins were in an enzymatically active form. Sequence analysis of CmaA indicated that there were two domains, an adenylation domain (A domain) and a thiolation domain (T domain). ATP-32PPi exchange assays showed that the A domain of CmaA catalyzes the conversion of branched-chain l-amino acids and ATP into the corresponding aminoacyl-AMP derivatives, with a kinetic preference for l-allo-isoleucine. Additional experiments demonstrated that the T domain of CmaA, which is posttranslationally modified with a 4′-phosphopantetheinyl group, reacts with the AMP derivative of l-allo-isoleucine to produce an aminoacyl thiolester intermediate. This covalent species was detected by incubating CmaA with ATP and l-[G-3H]allo-isoleucine, followed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis analysis. It is postulated that the l-allo-isoleucine covalently tethered to CmaA serves as the substrate for additional enzymes in the CMA biosynthetic pathway that catalyze cyclopropane ring formation, which is followed by thiolester hydrolysis, yielding free CMA. The availability of catalytically active CmaA should facilitate elucidation of the details of the subsequent steps in the formation of this novel cyclopropyl amino acid.
Coronatine (COR) (Fig. 1) is a novel phytotoxin that is produced by five distinct pathovars of Pseudomonas syringae, including P. syringae pv. atropurpurea, P. syringae pv. glycinea, P. syringae pv. maculicola, P. syringae pv. morsprunorum, and P. syringae pv. tomato, which infect ryegrass, soybean, crucifers, Prunus spp., and tomato, respectively. COR contributes to virulence in several host-pathogen interactions and elicits diffuse chlorosis in a wide variety of plant species (3, 13). COR also induces hypertrophy, inhibits root elongation, and stimulates ethylene production (11, 14, 17, 39). In several reports workers have noted the striking structural and functional homologies among COR, jasmonic acid, and 12-oxophytodienoic acid, suggesting that COR may function as a molecular mimic of the octadecanoid signaling molecules of higher plants (11, 14, 17, 39). COR consists of a bicyclic polyketide moiety, coronafacic acid (CFA), that is linked to an ethylcyclopropyl amino acid moiety, coronamic acid (CMA) (Fig. 1). These two moieties are produced by different biosynthetic pathways (26). Three acetate units, one butyrate unit, and a four-carbon unit that is derived from glutamic acid are combined to form CFA (27), while CMA is derived from l-isoleucine via l-allo-isoleucine which is oxidatively cyclized to form the cyclopropane ring (25). A ligase is then presumed to join CFA to CMA and produce COR via formation of an amide bond (2, 18). In the fermentation broth of P. syringae pv. glycinea, COR is accompanied by small amounts of norcoronatine, which contains norcoronamic acid (20) (Fig. 1), and by other congeners in which CMA is replaced by leucine, valine, isoleucine, or allo-isoleucine (20, 21).
FIG. 1.

Structures of COR, norcoronatine, CFA, CMA, and norcoronamic acid (norCMA).
The genes required for COR biosynthesis were first identified in P. syringae pv. glycinea PG4180, in which the 32.8-kb COR gene cluster is borne on a 90-kb plasmid designated p4180A (3). Investigations have shown that the structural genes for CFA and CMA biosynthesis are located at opposite ends of the gene cluster (40). An intermediate region, located between the biosynthetic regions, encodes the three regulatory proteins involved in transcriptional control of the other two regions (29, 31, 38). Nucleotide sequence analysis of each of the biosynthetic regions revealed open reading frames which indicate that CFA is biosynthesized by monofunctional and multifunctional polyketide synthase proteins (30, 32), whereas CMA appears to be biosynthesized by a thiotemplate mechanism that resembles nonribosomal peptide synthetases (37). Protein overexpression and function assays are required to confirm these predictions based on in silico data.
To begin to decipher the mechanism of CMA biosynthesis, we report here the initial characterization of CmaA, a protein encoded by the CMA region that appears to be a didomain protein containing an adenylation domain (A domain) and a thiolation domain (T domain). We found that CmaA catalyzes the adenylation of l-allo-isoleucine and the attachment of l-allo-isoleucine to the CmaA T domain. We postulate that the enzyme-bound l-allo-isoleucine serves as the substrate for the later stages of CMA biosynthesis.
MATERIALS AND METHODS
Bacterial strains and plasmids.
Pseudomonas strains were routinely cultured on King's medium B (16) at 28 or 18°C, while Escherichia coli cultures were grown on Luria-Bertani medium at 37°C (33). Ampicillin (100 μg/ml) and kanamycin (30 μg/ml) were used for plasmid selection in both organisms.
Reagents.
Unless indicated otherwise, all chemicals were purchased from Sigma Chemical Co. (St. Louis, Mo.). Restriction enzymes were obtained from NEB Life Technologies (Beverly, Mass.), and Pfu Turbo polymerase was purchased from Stratagene (La Jolla, Calif.). Protein concentrations were determined with the Advanced protein assay reagent from Cytoskeleton Inc. (Denver, Colo.) or with Bio-Rad protein assay reagent (Bio-Rad Laboratories, Hercules, Calif.).
DNA manipulations.
Agarose gel electrophoresis, restriction enzyme digestion, and purification of DNA fragments were performed by standard procedures (33). Plasmids were prepared by using a Qiaprep Spin miniprep kit (Qiagen, Chatsworth, Calif.). Electrocompetent P. syringae FF5 cells were prepared as described previously (12), and electroporation was conducted by using a Bio-Rad Gene Pulser II at 200 Ω, 2.5 kV, and 25 μF. Creation of the Pseudomonas expression vectors pSFFLAG-CTC and pSFFLAG-MAC has been described previously (9). P. syringae strains harboring the FLAG-tagged expression vectors were selected by ampicillin resistance. The nucleotide sequences of all expression constructs were confirmed by sequencing to verify the absence of errors. Resequencing of the CMA region of PG4180 was carried out by primer walking by using pSAY10 (40) as the template and was performed by a commercial sequencing facility. Polyacrylamide gel electrophoresis (PAGE)-purified sequencing and PCR primers were synthesized by Sigma Genosys (The Woodlands, Tex.).
Protein manipulations.
Native PAGE and sodium dodecyl sulfate (SDS)-PAGE were performed by using the separation and development units of the PhastSystem (Amersham Biosciences, Piscataway, N.J.) or by using the Mini-Protean II system (Bio-Rad Laboratories). SDS-PAGE protein molecular weight standards (broad range) were obtained from Bio-Rad Laboratories. Native PAGE molecular weight standards were created by combining chymotrypsinogen A (molecular mass, 25 kDa), albumin (67 kDa), aldolase (158 kDa), catalase (232 kDa), ferritin (440 kDa), and thyroglobulin (669 kDa), all of which were purchased from Amersham Biosciences.
Computational analyses.
Primary sequence alignment was performed by using the Gene Inspector 1.5 software (Textco, Inc., West Lebanon, N.H.) run on a Macintosh computer. Basic local alignment search tool (BLAST) analyses were performed at the National Center for Biotechnology Information web site (http://www.ncbi.nlm.nih.gov/). The ScanProsite program, available at the Expasy web site (http://us.expasy.org/tools/scanprosite/), was used to scan the CmaA amino acid sequence for the presence of signature motifs. GraphPad Prism 3.0 (GraphPad Software Inc., San Diego, Calif.) was used for nonlinear regression analysis of all enzyme assay data. Sequencher, version 4.1 (Gene Codes Corporation, Ann Arbor, Mich.), was used to compile DNA sequence data.
Cloning of CmaA. (i) FLAG-tagged proteins.
Two sets of PCR primer pairs were used to amplify the P. syringae pv. glycinea cmaA gene from plasmid pSAY10 (40). The first set (primer pair 1) consisted of CmaA-F-EcoRI (5′-TATGAATTCCATGACCTCCTACCATTCACAT-3′; restriction site is underlined) and CmaA-R-SmaI (5′-AACCCCGGGTCTCAGTCATTTCCATGTTGGCTCC-3′), and the second set (primer pair 2) consisted of CmaA-F-EcoRI and CmaA-RNS-SmaI (5′-AACCCCGGGTCGTCATTTCCATGTTGGCTCC-3′). The two sets of primer pairs resulted in a PCR product containing unique EcoRI and SmaI restriction sites (underlined) at the 5′ and 3′ ends, respectively. After high-fidelity PCR amplification with Pfu Turbo DNA polymerase and either primer pair 1 or primer pair 2, the ∼1.8-kb PCR product was purified with a Qiaquick PCR purification kit (Qiagen), digested with EcoRI and SmaI, and ligated into the multiple cloning site of EcoRI-SmaI-digested pSFFLAG-MAC (for the product of primer pair 1) or pSFFLAG-CTC (for the product of primer pair 2). The resulting constructs were electroporated into E. coli by using a Gene Pulser II electroporator (Bio-Rad Laboratories) according to the manufacturer's instructions. The transformants were confirmed by ampicillin selection, restriction mapping, and DNA sequencing and were designated pSFFLAG-MACcmaA and pSFFLAG-CTCcmaA. For overproduction of CmaA in Pseudomonas, the constructs were electroporated into P. syringae pv. syringae FF5, a COR nonproducer (36), and protein expression and purification were carried out as previously described (9).
(ii) His-tagged proteins.
The P. syringae pv. tomato DC3000 cmaA gene was amplified from the genomic DNA of this strain, which is a known COR producer. DC3000 genomic DNA was prepared by using a Bactozol kit (Molecular Research Center, Inc., Cincinnati, Ohio). PCR amplification with primers cmaA-NdeI (5′-GGAATTCCATATGACCTCCTACCATTCA-3′) and cmaA-EcoRI (5′-AAAAAAGAATTCTCAGTCATTTCCATGTTG-3′) resulted in a product containing unique NdeI and EcoRI sites (underlined). The PCR product was purified as described above and ligated into the multiple cloning site of NdeI-EcoRI-digested vector pET28b (Novagen, Madison, Wis.). The constructs were transformed into E. coli as described above and were selected by kanamycin resistance. DNA sequencing confirmed the identity of the insert. For overproduction of CmaA in E. coli, this construct was transformed into E. coli BL21(DE3) along with plasmid pSU20-Sfp containing the gene encoding Sfp, a Bacillus subtilis phosphopantetheinyl transferase with broad substrate specificity (22). pSU20-Sfp was constructed by removal of the sfp gene, along with its promoter and ribosome binding site, from plasmid pUC8-Sfp (24) as an EcoRI-BamHI fragment and ligation of the fragment into plasmid pSU20 (1) digested with EcoRI and BamHI.
For overproduction of the His-tagged CmaA protein (His-CmaA), BL21(DE3) cells harboring the desired plasmids were grown in Luria-Bertani medium supplemented with 30 μg of kanamycin per ml and 34 μg of chloramphenicol per ml. One liter of medium was inoculated with 10 ml of an overnight starter culture and incubated at 25°C until an optical density at 595 nm of 0.55 was reached. Protein expression was then induced by addition of 100 μM isopropyl-β-d-thiogalactopyranoside (IPTG), and cells were allowed to grow for an additional 15 h at 15°C. Cells were harvested by centrifugation (10 min at 6,000 × g) and resuspended in lysis buffer (25 mM Tris [pH 8], 500 mM NaCl, 10% glycerol) and lysed by two passages through a French press at 10,000 lb/in2. Cell extracts were clarified by centrifugation (30 min at 15,000 × g) and applied to nickel-nitrilotriacetic acid resin (1 ml of resin for 3 liters of culture) (Qiagen). Cell lysate was allowed to bind in batch to the resin for 2 h at 4°C and then was decanted into a column. The resin was washed with 15 column volumes of lysis buffer and then eluted with a step gradient of lysis buffer containing increasing amounts of imidazole (5, 30, 60, 100, and 500 mM). CmaA eluted in the 100 and 500 mM imidazole fractions. Fractions containing the desired protein were dialyzed against 25 mM Tris (pH 7.5)-200 mM NaCl-10% glycerol and frozen at −80°C.
A C-terminal His-tagged version of CmaA was also constructed by amplification of the gene with primers cmaA-NcoI (5′-AAAAAACCATGGATGACCTCCTACCATTCA-3′; restriction site is underlined) and cmaA-XhoI (5′-AAAAAACTCGAGCTCATTTCCATGTTGGCT-3′). The resulting product was ligated into NcoI-XhoI-digested pET28b. Selection, transformation, and protein expression were performed as described above.
ATP-32PPi exchange assays.
ATP-PPi exchange reactions were carried out at 25°C in 100-μl mixtures that contained 75 mM Tris-HCl (pH 7.5), 10 mM MgCl2, 1 mM tris-(2-carboxyethyl)phosphine (Molecular Probes, Eugene, Oreg.), 1 mM [32P]sodium pyrophosphate (5 Ci/mol; Dupont NEN, Boston, Mass.), 5 mM ATP, 0.2 nmol of FLAG-tagged CmaA protein (CmaA-FLAG) or 0.14 nmol of His-CmaA, and various concentrations of amino acid substrate. The reactions were initiated by addition of enzyme, were allowed to proceed for 10 min, and then were quenched by addition of a 1.6% activated charcoal-4.46% tetrasodium pyrophosphate-3.5% perchloric acid mixture in water. The charcoal was collected by either centrifugation or filtration, washed twice with a 4.46% tetrasodium pyrophosphate-3.5% perchloric acid solution, and then, if collected by centrifugation, resuspended in 0.5 ml of water, combined with liquid scintillation cocktail (ScintiVerse; Fisher Scientific, Pittsburgh, Pa.), and placed in a liquid scintillation counter. If filtration was used to collect the charcoal, the entire filter paper was placed directly into the scintillation vial after the wash step, mixed vigorously with scintillation cocktail, and counted. Each reaction was performed at least in duplicate. The amount of charcoal-bound radioactivity was converted into reaction velocity by using the specific activity of the 32PPi. A nonlinear regression fit of the plot of velocity versus substrate concentration provided the Vmax and Km values.
PPi release assay.
PPi levels were measured by monitoring the appearance of NADH (at 340 nm) generated by a coupled, continuous spectrophotometric assay (8). Reactions were carried out at 25°C, and the reaction mixtures (500 μl) contained 1 M Tris-HCl (pH 8), 50 mM UDP-glucose (Sigma-Aldrich, St. Louis, Mo.), 50 mM glucose 1,6-bisphosphate (Sigma-Aldrich), 10 mM NAD+, 100 mM dithiothreitol, 5 mM EDTA, 100 mM ATP, 1 M MgCl2, 1 U of UDP-glucose pyrophosphorylase (Sigma-Aldrich), 5 U of phosphoglucomutase (Sigma-Aldrich), 8 U of glucose-6-phosphate dehydrogenase (Sigma-Aldrich), 0.2 nmol of CmaA, and various concentrations of amino acid substrate. The reactions were initiated by addition of CmaA and were monitored for 30 to 45 min with a Hewlett-Packard 8453 diode array spectrophotometer (Agilent Technologies, Palo Alto, Calif.). The spectrophotometer recorded data points every 20 s. Each assay was performed in duplicate. A linear reaction velocity was obtained by using a minimum of 30 colinear data points and an extinction coefficient of 6,220 M−1 cm−1 for NADH.
Aminoacylation of the T domain of CmaA by l-allo-isoleucine.
Radioautographic studies were performed to examine the reaction of enzymatically generated l-allo-isoleucyl-AMP with the free thiol of the 4′-phosphopantetheine arm located within the T domain of CmaA. Reactions were performed at 25°C, and the reaction mixtures (40 μl) contained 100 mM Tris-HCl (pH 8), 10 mM MgCl2, 1 mM TCEP [tris-(2-carboxyethyl)phosphine], 5 mM ATP, 800 μM l-[G-3H]allo-isoleucine (generally tritiated; 320 Ci/mol; Moravek Biochemicals, Brea, Calif.), and 0.3 nmol of FLAG-CmaA or 0.14 nmol of His-CmaA. A control reaction, from which ATP was excluded, was also carried out. The reaction was initiated by addition of CmaA and was allowed to proceed for 30 min. The reaction was terminated by addition of 8 ml of 6× SDS sample buffer (280 mM Tris-HCl [pH 6.8], 10% SDS, 30% glycerol, 0.6 M dithiothreitol, 0.01% bromophenol blue) and boiling for 5 min. Samples were analyzed by electrophoresis with a SDS-10% PAGE gel. After electrophoresis, the gel was soaked in fixing solution (isopropanol-water-acetic acid, 25:65:10) for 30 min, soaked in Amplify reagent (Amersham Biosciences) for 30 min, vacuum dried (60°C for 1 h), and exposed to Kodak Biomax XR X-ray film (Eastman Kodak Company, Rochester, N.Y.) for 3 days at −80°C before the film was developed.
RESULTS
Sequence revisions.
In 1998, a corrected version of the P. syringae pv. glycinea PG4180 cmaA nucleotide sequence was deposited in the GenBank database (4). In order to characterize the CmaA protein, it was critical to confirm the accuracy of this sequence. Therefore, the cmaA region of P. syringae pv. glycinea PG4180 was resequenced. A comparison of the new sequence, which has at least twofold coverage in the forward and reverse directions of the entire gene, with the 1998 sequence revealed a number of differences between the two sequences. To clarify these differences, the P. syringae pv. tomato DC3000 cmaA gene sequence was retrieved from The Institute for Genomic Research (TIGR) web site (www.tigr.org) and compared with the other two cmaA gene sequences. The TIGR P. syringae pv. tomato sequence exhibited 97% identity with the newly obtained cmaA nucleotide sequence but only 93% identity with the 1998 cmaA gene sequence. At the protein level, the TIGR P. syringae pv. tomato amino acid sequence exhibited 96% identity with the new PG4180 CmaA sequence (Fig. 2) but only 79% identity with the 1998 CmaA sequence (data not shown). The differences are due to reading frame changes caused by inserted or deleted bases in the 1998 cmaA sequence. Because of the repeated coverage of the resequenced cmaA region and the high degree of similarity between the new cmaA sequence and the TIGR sequence, we assumed that the correct sequence for the PG4180 cmaA gene has been obtained (GenBank accession number AY386681). This sequence was used as a basis for overproduction of CmaA in P. syringae FF5.
FIG. 2.

Alignment of CmaA amino acid sequences. A comparison of the CmaA sequence from P. syringae pv. glycinea with the CmaA sequence from P. syringae pv. tomato DC3000 indicates that these two sequences are 96% identical. The residues enclosed in the rectangle create an AMP-binding domain signature sequence, whereas the residues enclosed in the ellipses are common to a T domain, as determined by using the ScanProsite program at the Expasy web site. The arrow indicates the putative 4′-phosphopantetheine attachment site. The underlined residues are the residues that contribute to the specificity-conferring code of A domains in nonribosomal peptide synthetases. See the text for further discussion.
Because of the errors discovered in the previously deposited PG4180 cmaA sequence, the entire CMA region of PG4180 was also resequenced, and the resequenced region was compared with the sequence reported for P. syringae pv. tomato. A significant number of errors were discovered in the original PG4180 sequence. Furthermore, both the new PG4180 sequence (GenBank accession number AY391839) and the P. syringae pv. tomato sequence appeared to contain at least three previously unrecognized open reading frames, which we designated cmaC, cmaD, and cmaE (Fig. 3).
FIG. 3.

Gene organization in the CMA region of the COR biosynthetic gene cluster of P. syringae pv. glycinea PG4180.
CmaA sequence analysis.
An analysis of either the P. syringae pv. glycinea or P. syringae pv. tomato cmaA nucleotide sequence revealed a 1,788-bp open reading frame encoding a 595-amino-acid CmaA protein. A BLAST analysis of each CmaA protein revealed that the greatest similarities were with adenylation activation enzymes and domains that play a role in nonribosomal peptide biosynthesis (results not shown). Furthermore, screening the CmaA amino acid sequence for the presence of signature motifs by using the Expasy ScanProsite algorithm resulted in identification of both an AMP-binding domain signature sequence motif (A domain) and several residues that are characteristic of a T domain (Fig. 2). Both of these domains are common in adenylation activation proteins. Thus, based upon these sequence analyses, it appeared reasonable to propose that CmaA is an adenylation activation enzyme that covalently loads its substrate by first adenylating it (via the A domain) and then transferring it onto the 4′-phosphopantetheine arm located within the T domain (7, 19). The presence of A and T domains in CmaA was recognized previously, despite the errors present in the cmaA nucleotide sequence (37).
In an attempt to identify the substrate specificity of CmaA, key residues that comprise the A domain specificity-conferring code (5, 35) were examined (underlined residues in Fig. 2). Of the 10 residues that make up the specificity-conferring code, 7 are found in A domains that are specific for l-isoleucine, l-leucine, and l-valine. Both l-isoleucine and l-allo-isoleucine have been shown to be incorporated into CMA, but l-allo-isoleucine, whose specificity-conferring code has not been defined yet, is a much more efficient precursor (25). The timing of the epimerization of l-isoleucine to l-allo-isoleucine is currently unknown. By analogy with the conversion of l-allo-isoleucine into CMA, it appears that norcoronamic acid should be derived from l-valine. For these reasons, l-isoleucine, l-allo-isoleucine, and l-valine each appeared to be a potential substrate for CmaA.
Protein overexpression and visualization.
Two complementary approaches were taken to overexpress CmaA for substrate specificity assays. In one approach, the P. syringae pv. glycinea CmaA protein was expressed in P. syringae pv. syringae FF5, a Pseudomonas strain that lacks the COR gene cluster (36). Additionally, the P. syringae pv. tomato CmaA protein was overexpressed in E. coli. To generate the appropriate constructs, the cmaA gene was PCR amplified from P. syringae pv. glycinea and P. syringae pv. tomato, cloned into expression vectors that produced FLAG-tagged and His-tagged proteins, respectively, and transformed into the appropriate hosts. The soluble protein yields were compared with N-terminal and C-terminal tag locations, and a C-terminal FLAG-tagged protein and an N-terminal His-tagged protein were selected for subsequent functional assays. Figure 4A shows the SDS-PAGE results for the N-terminal His-CmaA and the C-terminal CmaA-FLAG. As shown in the Fig. 4, denatured CmaA-FLAG electrophoresed faster than expected, since the protein migrated at a molecular weight <66,000, whereas the predicted molecular weight is actually ∼68,000. Denatured His-CmaA electrophoresed at approximately the same position as CmaA-FLAG. Native PAGE of CmaA-FLAG indicated that the protein exists as a dimer (Fig. 4B).
FIG. 4.

PAGE analysis of affinity-tagged CmaA. (A) SDS-PAGE analysis. Denatured CmaA migrated at a molecular weight (MW) slightly less than the molecular weight (68,500) predicted from its amino acid sequence. (B) Native PAGE analysis. CmaA-FLAG appeared to migrate as a dimer.
PPi exchange assay.
The substrate specificity of CmaA was assayed by using the amino acid-dependent exchange of radiolabel from 32PPi into ATP (10). This assay measures the reversible formation of the aminoacyl-AMP derivative and allows determination of amino acid selectivity. His-CmaA and FLAG-CmaA were each assayed with variable concentrations of substrate in a buffered solution containing ATP and 32PPi. After a brief incubation period, newly formed, radiolabeled ATP was collected by using activated charcoal and subsequently was quantified by liquid scintillation counting. A nonlinear regression analysis of reaction velocity versus substrate concentration provided the Km and Vmax values, the latter of which was used to calculate kcat.
l-Leucine, l-valine, and l-isoleucine were suggested to be possible substrates by examining the specificity-conferring code of CmaA. A series of qualitative assays with His-CmaA indicated that l-allo-isoleucine is the preferred substrate, and a small amount of activation was also observed with l-leucine, l-valine, or l-isoleucine (Fig. 5). Kinetic parameters for the four isoleucine diastereomers were measured (Table 1). A more detailed kinetic analysis was performed with the more active CmaA-FLAG.
FIG. 5.

Relative amino acid substrate activities as determined by using the ATP-PPi exchange assay and His-CmaA. The y axis indicates the ATP-PPi exchange activity for various amino acid substrates compared to the activity for l-allo-isoleucine. The curved line indicates that the data for l-allo-isoleucine, at a relative activity of 100%, are off the scale relative to the data for the other substrates.
TABLE 1.
Kinetic parameters for CmaA-FLAG and His-CmaA as determined by the PPi exchange assay
| Substrate | CmaA-FLAG
|
His-CmaA
|
||||
|---|---|---|---|---|---|---|
| Km (mM) | kcat (min−1) | kcat/Km (min−1 mM−1) | Km (mM) | kcat (min−1) | kcat/Km (min−1 mM−1) | |
| l-allo-Isoleucine | 0.26 ± 0.02 | 17 ± 0.3 | 65.4 | 1.4 ± 0.2 | 40 ± 6.0 | 28.6 |
| l-Isoleucine | 1.0 ± 0.1 | 2.8 ± 0.1 | 2.8 | 0.12 ± 0.00 | 1.6 ± 0.1 | 13.3 |
| l-Leucine | 7.4 ± 1.1 | 16 ± 3 | 2.2 | NDa | ND | ND |
| l-Valine | 8.2 ± 1.3 | 12 ± 1 | 1.5 | ND | ND | ND |
| d-allo-Isoleucine | 0.84 ± 0.07 | 0.20 ± 0.06 | 0.24 | 4.9 ± 1.1 | 1.0 ± 0.1 | 0.20 |
| d-Isoleucine | 1.1 ± 0.2 | 1.1 ± 0.2 | 1.0 | 4.1 ± 2.1 | 0.80 ± 0.1 | 0.20 |
| dl-Coronamic acid | 1.5 ± 0.3 | 6.7 ± 0.4 | 4.5 | ND | ND | ND |
| dl-Norcoronamic acid | 11 ± 3 | 1.1 ± 0.2 | 0.10 | ND | ND | ND |
ND, not determined.
l-Isoleucine appears to be preferred over l-leucine and l-valine, as implied by the relatively small Km value and the relatively large kcat/Km value. Furthermore, CmaA is quite selective with respect to the absolute configuration of isoleucine; that is, the kcat and kcat/Km values of the d stereoisomer of isoleucine are roughly one-third those of the l stereoisomer, clearly indicating that there is a preference for the latter. However, after examination of the l-allo-isoleucine data, it became very apparent that this amino acid is by far the preferred substrate, as it yields the smallest Km, the greatest kcat, and the largest kcat/Km value of all the substrates tested. The preference for the l isomer of allo-isoleucine was also apparent, as d-allo-isoleucine resulted in a threefold increase in the Km, an 85-fold decrease in the kcat, and consequently a 272-fold decrease in the kcat/Km value. Interestingly, CMA, the ethylcyclopropyl amino acid component of COR which is thought to be the product of the cyclization of allo-isoleucine, was the second best substrate tested, as judged by the kcat/Km value. Norcoronamic acid, which is thought to be the product of cyclization of valine, was the worst substrate tested.
PPi release assay.
A continuous, spectrophotometric assay was also employed to determine the substrate specificity of CmaA (8). This assay measures the release of PPi by means of a coupled enzyme system in which PPi drives the reduction of NAD+ to NADH, a process that is conveniently monitored at 340 nm. Continuous release of PPi must be accompanied by the release of the aminoacyl-AMP derivative to regenerate the free A domain for catalysis of another reaction cycle. Thus, this assay is in effect a measure of the frequency of loss of the aminoacyl-AMP derivative from the A domain active site. Following adenylation, preferred amino acid substrates are expected to be held tightly by the enzyme, presumably to await transfer to the 4′-phosphopantetheine arm of the T domain. On the other hand, less preferred substrates have a tendency to leak from the adenylation site following the adenylation reaction. With this assay, therefore, highly preferred substrates should generate relatively small Km and kcat values (i.e., tight binding with little leakage), while the substrates that are less preferred should yield larger Km and kcat values.
Table 2 shows the kinetic values obtained in the PPi release assay when CmaA-FLAG was used in conjunction with various substrates. As observed with the PPi exchange assay (Table 1), the PPi release assay results illustrated the preference of CmaA for l-isoleucine over l-leucine and l-valine. Furthermore, the specificity of CmaA for l-isoleucine over d-isoleucine was also apparent when this assay was used. Most importantly, it is clear that l-allo-isoleucine was the most preferred substrate for CmaA of all the substrates tested, as it displayed the smallest Km and kcat values and the largest kcat/Km value (Table 2). These results are in good agreement with those of the PPi exchange assay.
TABLE 2.
Kinetic parameters for CmaA-FLAG as determined by the forward PPi assay
| Substrate | Km (mM) | kcat (min−1) | kcat/Km (min−1 mM−1) |
|---|---|---|---|
| l-allo-Isoleucine | 0.04 ± 0.01 | 0.09 ± 0.00 | 2.3 |
| l-Isoleucine | 0.50 ± 0.06 | 0.71 ± 0.03 | 1.4 |
| l-Leucine | 1.5 ± 0.1 | 1.8 ± 0.1 | 1.2 |
| l-Valine | 1.2 ± 0.1 | 1.4 ± 0.1 | 1.2 |
| d-allo-Isoleucine | 5.6 ± 1.0 | 0.51 ± 0.09 | 0.09 |
| d-Isoleucine | 5.3 ± 0.9 | 0.79 ± 0.09 | 0.15 |
| dl-Coronamic acid | 1.0 ± 0.2 | 1.3 ± 0.1 | 1.3 |
| dl-Norcoronamic acid | 4.5 ± 1.0 | 1.8 ± 0.4 | 0.40 |
Covalent loading of l-allo-isoleucine by CmaA.
With the preferred substrate of the adenylation reaction identified, the ability of CmaA to catalyze loading of its T domain with l-allo-isoleucine was examined. CmaA-FLAG was incubated with l-[G-3H]allo-isoleucine in the presence or absence of ATP. The proteins were subsequently denatured, electrophoresed, and analyzed by autoradiography. Figure 6 shows the autoradiograph obtained, which verified that there was ATP-dependent acylation of CmaA with l-allo-isoleucine. Identical results were obtained with His-CmaA (data not shown).
FIG. 6.
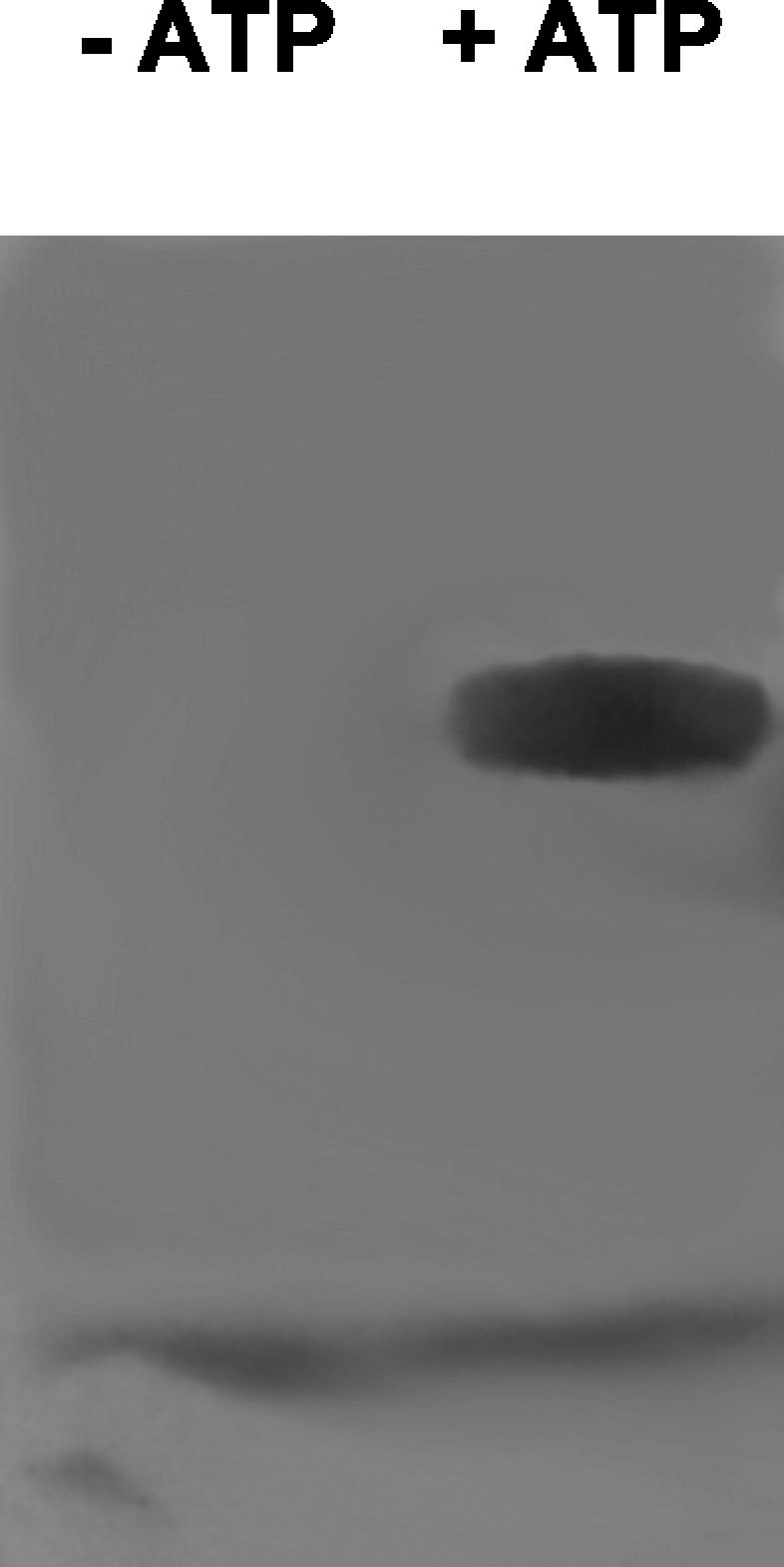
Covalent labeling of CmaA-FLAG by l-[G-3H]allo-isoleucine in the presence of ATP.
DISCUSSION
Biosynthesis of the plant phytotoxin COR by P. syringae appears to proceed by formation of CMA and CFA, which are then linked to produce COR (Fig. 1). A set of three genes, cmaA, cmaB, and cmaT, in the CMA region of the biosynthetic cluster (Fig. 3) have previously been implicated in CMA formation from the proteogenic amino acid l-isoleucine. To begin to decipher the mechanism of cyclopropane ring formation, we have undertaken biochemical characterization of the CmaA protein. This protein is predicted to be a 68-kDa, two-domain (A domain-T domain) protein that resembles the amino acid activation modules observed in nonribosomal peptide biosynthetic pathways.
Discrete A domain-T domain proteins in biosynthetic pathways often capture a proteogenic amino acid and modify it. The modified amino acid is then released to create a dedicated pool of nonproteinogenic monomers for natural product biosynthesis (7). For example, in the nikkomycin biosynthetic pathway, a discrete A domain-T domain enzyme specifically activates and loads histidine, after which a P450 enzyme hydroxylates the β-carbon position of the covalently sequestered histidine residue. Similar discrete A domain-T domain proteins are observed in numerous other natural product pathways, such as those for chloramphenicol and novobiocin (7). In this study we demonstrated that the logic of CMA biosynthesis is to activate l-allo-isoleucine and to link it to CmaA in the form of a thiolester. It is postulated that the covalently linked l-allo-isoleucine is cyclized to produce covalently bound CMA, which is subsequently hydrolyzed by CmaT to produce free CMA.
The cmaA gene from P. syringae pv. glycinea PG4180 and the cmaA gene from P. syringae pv. tomato DC3000 were cloned and expressed in a Pseudomonas host strain and in E. coli, respectively, and were purified by affinity chromatography. The protein was posttranslationally modified in the T domain with a 4′-phosphopantetheinyl moiety in vivo, either by the heterologously expressed Bacillus phosphopantetheinyl transferase Sfp in the E. coli expression system or by an endogenous Pseudomonas transferase. The kinetic data indicate that the CmaA that is produced in Pseudomonas is substantially more active than the enzyme that is produced in E. coli. The N-terminally tagged version produced in E. coli has a mutation in the protein sequence (Ser521Phe) which may contribute to the decrease in activity. However, a wild-type C-terminally His-tagged construct also exhibited substantially reduced activity compared to the activity of the Pseudomonas protein (data not shown). Therefore, we suspect that the differences in the kinetic data are due to the fact that E. coli is a poor expression vehicle for the COR biosynthetic proteins. CmaA protein folding may not occur properly in E. coli, or alternatively, the B. subtilis phosphopantetheinyl transferase may not modify CmaA efficiently. Incubation of purified protein from either background with l-[G-3H]allo-isoleucine and subsequent analysis by SDS gel electrophoresis followed by autoradiography revealed that the protein was covalently modified with this amino acid in an ATP-dependent manner (Fig. 6). Monitoring 32PPi exchange data, which measured the enzyme-dependent reversible formation of an aminoacyl-AMP derivative, indicated that l-allo-isoleucine is strongly favored as the substrate over all the other amino acids examined. The data suggest that free l-allo-isoleucine is biosynthesized by the phytopathogenic P. syringae strains prior to this step of CMA biosynthesis. How this unusual isoleucine diastereomer is synthesized remains unknown. The kinetic parameters for l-valine indicate that it is a rather poor substrate. This is somewhat surprising given the natural occurrence of norcoronamic acid, which is probably derived from l-valine by a process similar to CMA formation. However, norcoronatine is a minor constituent of P. syringae pv. glycinea fermentation, and so formation of norcoronamic acid may be a relatively inefficient process.
The A domains of nonribosomal peptide synthetases typically exhibit a consensus sequence that determines the amino acid substrate specificity. While the consensus sequence of l-isoleucine-activating domains is very similar to that of CmaA (Fig. 2), there are three changes in the consensus sequence that may be responsible for the preference for l-allo-isoleucine. Although other l-allo-isoleucine-containing natural products exist (23, 34), this is the first example of an A domain specific for the (2S,3R) stereochemistry of l-allo-isoleucine.
In this paper we describe the first characterization of the CMA biosynthetic pathway at the enzymatic level. The data reveal that there is a discrete A domain-T domain whose function is to tether l-allo-isoleucine, presumably for subsequent cyclization and hydrolysis by CmaT to produce CMA. Previous studies have shown that CmaT exhibits thioesterase activity with model substrates (28). In addition to the CmaA and CmaT genes, five other genes are present in the CMA biosynthetic region of P. syringae PG4180 (Fig. 3). The sequence of CmaB exhibits similarities to the sequences of α-ketoglutarate-dependent dioxygenases, BarB1/Bar2 encoded by the barbamide gene cluster (6), and SyrB2 encoded by the syringomycin gene cluster (15, 41). These similarities suggest that CmaB is a nonheme iron dioxygenase that may carry out hydroxylation or chlorination of the CmaA-bound l-allo-isoleucine at C-6 (Fig. 7). CmaC exhibits similarity to methylmalonyl coenzyme A mutases, which suggests that its role may be to deprotonate and cyclize CmaA-bound 6-hydroxy-allo-isoleucine or 6-chloro-allo-isoleucine to produce CmaA-bound CMA (Fig. 7). CmaD exhibits similarities to acyl carrier proteins, while CmaE shows similarities to proteins with an α/β hydrolase fold. The role played by these two proteins is unclear. Characterization of the later steps in the CMA biosynthetic pathway is under way.
FIG. 7.

Hypothetical biosynthetic pathway for conversion of l-allo-isoleucine into CMA.
Acknowledgments
We acknowledge support of this study by the National Institutes of Health (grant GM26564 to R.J.P. and grant GM20011 to C.T.W.), by the Robert A. Welch Foundation (grant C-0729 to R.J.P.), and by an Irving S. Sigal postdoctoral fellowship (to S.E.O).
Construction of vector pSU20-sfp by M. G. Thomas is also gratefully acknowledged.
REFERENCES
- 1.Bartolome, B., Y. Jubete, E. Martinez, and F. de la Cruz. 1991. Construction and properties of a family of pACYC184-derived cloning vectors compatible with pBR322 and its derivatives. Gene 102:75-78. [DOI] [PubMed] [Google Scholar]
- 2.Bender, C., H. Liyanage, D. Palmer, M. Ullrich, S. Young, and R. Mitchell. 1993. Characterization of the genes controlling biosynthesis of the polyketide phytotoxin coronatine including conjugation between coronafacic and coronamic acids. Gene 133:31-38. [DOI] [PubMed] [Google Scholar]
- 3.Bender, C. L., F. Alarcon-Chaidez, and D. C. Gross. 1999. Pseudomonas syringae phytotoxins: mode of action, regulation, and biosynthesis by peptide and polyketide synthetases. Microbiol. Mol. Biol. Rev. 63:266-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Budde, I. P., B. H. Rohde, C. L. Bender, and M. S. Ullrich. 1998. Growth phase and temperature influence promoter activity, transcript abundance, and protein stability during biosynthesis of the Pseudomonas syringae phytotoxin coronatine. J. Bacteriol. 180:1360-1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Challis, G. L., J. Ravel, and C. A. Townsend. 2000. Predictive, structure-based model of amino acid recognition by nonribosomal peptide synthetase adenylation domains. Chem. Biol. 7:211-224. [DOI] [PubMed] [Google Scholar]
- 6.Chang, Z., P. Flatt, W. Gerwick, V. Nguyen, C. Willis, and D. Sherman. 2002. The barbamide biosynthetic gene cluster: a novel marine cyanobacterial system of mixed polyketide synthase (PKS)-non-ribosomal peptide synthetase (NRPS) origin involving an unusual trichloroleucyl starter unit. Gene 296:235-247. [DOI] [PubMed] [Google Scholar]
- 7.Chen, H., M. G. Thomas, S. E. O'Connor, B. K. Hubbard, M. D. Burkart, and C. T. Walsh. 2001. Aminoacyl-S-enzyme intermediates in beta-hydroxylations and alpha, beta-desaturations of amino acids in peptide antibiotics. Biochemistry 40:11651-11659. [DOI] [PubMed] [Google Scholar]
- 8.Chen-Goodspeed, M., J. L. VanHooke, H. M. Holden, and F. M. Raushel. 1999. Kinetic mechanism of kanamycin nucleotidyltransferase from Staphylococcus aureus. Bioorg. Chem. 27:395-408. [Google Scholar]
- 9.Couch, R., H. Seidle, and R. J. Parry. 2002. Construction of expression vectors to produce affinity-tagged proteins in Pseudomonas. BioTechniques 32:1230-1236. [DOI] [PubMed] [Google Scholar]
- 10.Fersht, A. 1985. Enzyme structure and mechanism. Freeman, New York, N.Y.
- 11.Feys, B. J. F., C. E. Benedetti, C. N. Penfold, and J. G. Turner. 1994. Arabidopsis mutants selected for resistance to the phytotoxin coronatine are male sterile, insensitive to methyl jasmonate, and resistant to a bacterial pathogen. Plant Cell 6:751-759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garde, S., and C. L. Bender. 1991. DNA probes for detection of copper resistance genes in Xanthomonas campestris pv. vesicatoria. Appl. Environ. Microbiol. 57:2435-2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gnanamanickam, S. S., A. N. Starratt, and E. W. B. Ward. 1982. Coronatine production in vitro and in vivo and its relation to symptom development in bacterial blight of soybean. Can. J. Bot. 60:645-650. [Google Scholar]
- 14.Greulich, F., T. Yoshihara, and A. Ichihara. 1995. Coronatine, a bacterial phytotoxin, acts as a stereospecific analog of jasmonate type signals in tomato cells and potato tissues. J. Plant Physiol. 147:359-366. [Google Scholar]
- 15.Guenzi, E., G. Galli, I. Grgurina, D. C. Gross, and G. Grandi. 1998. Characterization of the syringomycin synthetase gene cluster. A link between prokaryotic and eukaryotic peptide synthetases. J. Biol. Chem. 273:32857-32863. [DOI] [PubMed] [Google Scholar]
- 16.King, E. O., M. K. Ward, and D. E. Raney. 1954. Two simple media for the demonstration of pyocyanin and fluorescein. J. Lab. Clin. Med. 44:301-307. [PubMed] [Google Scholar]
- 17.Koda, Y., K. Takahashi, Y. Kikuta, F. Greulich, H. Toshima, and A. Ichihara. 1996. Similarities of the biological activities of coronatine and coronafacic acid to those of jasmonic acid. Phytochemistry 41:93-96. [Google Scholar]
- 18.Liyanage, H., C. Penfold, J. Turner, and C. L. Bender. 1995. Sequence, expression, and transcriptional analysis of the coronafacate ligase-encoding gene required for coronatine biosynthesis by Pseudomonas syringae. Gene 153:17-23. [DOI] [PubMed] [Google Scholar]
- 19.Marahiel, M. A., T. Stachelhaus, and H. D. Mootz. 1997. Modular peptide synthetases involved in nonribosomal peptide synthesis. Chem. Rev. 97:2651-2674. [DOI] [PubMed] [Google Scholar]
- 20.Mitchell, R. E. 1985. Norcoronatine and N-coronafacoyl-l-valine, phytotoxic analogs of coronatine produced by a strain of Pseudomonas syringae pv. glycinea. Phytochemistry 24:1485-1487. [Google Scholar]
- 21.Mitchell, R. E., and H. Young. 1985. N-coronafacoyl-l-isoleucine and N-coronafacoyl-l-alloisoleucine, potential biosynthetic intermediates of the phytotoxin coronatine. Phytochemistry 24:2716-2717. [Google Scholar]
- 22.Mofid, M. R., M. A. Marahiel, R. Ficner, and K. Reuter. 1999. Crystallization and preliminary crystallographic studies of Sfp: a phosphopantetheinyl transferase of modular peptide synthetases. Acta Crystallogr. Sect. D Biol. Crystallogr. 55:1098-1100. [DOI] [PubMed] [Google Scholar]
- 23.Nakajima, M., M. Inukai, T. Haneishi, A. Terahara, M. Arai, T. Kinoshita, and C. Tamura. 1978. Globomycin, a new peptide antibiotic with spheroplast-forming activity. III. Structural determination of globomycin. J. Antibiot. 31:426-432. [DOI] [PubMed] [Google Scholar]
- 24.Nakano, M. M., N. Corbell, J. Besson, and P. Zuber. 1992. Isolation and characterization of sfp: a gene that functions in the production of the lipopeptide biosurfactant, surfactin, in Bacillus subtilis. Mol. Gen. Genet. 232:313-321. [DOI] [PubMed] [Google Scholar]
- 25.Parry, R. J., M.-T. Lin, A. E. Walker, and S. V. Mhaskar. 1991. The biosynthesis of coronatine: investigations of the biosynthesis of coronamic acid. J. Am. Chem. Soc. 113:1849-1850. [Google Scholar]
- 26.Parry, R. J., S. V. Mhaskar, M.-T. Lin, A. E. Walker, and R. Mafoti. 1994. Investigations of the biosynthesis of the phytotoxin coronatine. Can. J. Chem. 72:86-99. [Google Scholar]
- 27.Parry, R. J., S. Jiralerspong, S. V. Mhaskar, L. Alemany, and R. Willcott. 1996. Investigations of coronatine biosynthesis. Elucidation of the mode of incorporation of pyruvate into coronafacic acid. J. Am. Chem. Soc. 118:703-704. [Google Scholar]
- 28.Patel, J., J. C. Hoyt, and R. J. Parry. 1998. Investigations of coronatine biosynthesis. Overexpression and assay of CmaT, a thioesterase involved in coronamic acid biosynthesis. Tetrahedron 54:15927-15936. [Google Scholar]
- 29.Penaloza-Vazquez, A., and C. L. Bender. 1998. Characterization of CorR, a transcriptional activator which is required for biosynthesis of the phytotoxin coronatine. J. Bacteriol. 180:6252-6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Penfold, C. N., C. L. Bender, and J. G. Turner. 1996. Characterization of genes involved in biosynthesis of coronafacic acid, the polyketide component of the phytotoxin coronatine. Gene 183:167-173. [DOI] [PubMed] [Google Scholar]
- 31.Rangaswamy, V., and C. L. Bender. 2000. Phosphorylation of CorS and CorR, regulatory proteins that modulate production of the phytotoxin coronatine in Pseudomonas syringae. FEMS Microbiol. Lett. 193:13-18. [DOI] [PubMed] [Google Scholar]
- 32.Rangaswamy, V., S. Jiralerspong, R. Parry, and C. L. Bender. 1998. Biosynthesis of the Pseudomonas polyketide coronafacic acid requires monofunctional and multifunctional polyketide synthase proteins. Proc. Natl. Acad. Sci. 95:15469-15474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sambrook, J. E., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
- 34.Shoji, J. 1973. Configuration of the N-methylalloisoleucine contained in quinoxaline antibiotics. J. Antibiot. 26:302-303. [DOI] [PubMed] [Google Scholar]
- 35.Stachelhaus, T., H. D. Mootz, and M. A. Marahiel. 1999. The specificity-conferring code of adenylation domains in nonribosomal peptide synthetases. Chem. Biol. 6:493-505. [DOI] [PubMed] [Google Scholar]
- 36.Sundin, G. W., and C. L. Bender. 1993. Ecological and genetic analysis of copper and streptomycin resistance in Pseudomonas syringae pv. syringae. Appl. Environ. Microbiol. 59:1018-1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ullrich, M., A. C. Guenzi, R. E. Mitchell, and C. L. Bender. 1994. Cloning and expression of genes required for coronamic acid (2-ethyl-1-aminocyclopropane 1-carboxylic acid), an intermediate in the biosynthesis of the phytotoxin coronatine. Appl. Environ. Microbiol. 60:2890-2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ullrich, M., A. Penaloza-Vazquez, A.-M. Bailey, and C. L. Bender. 1995. A modified two-component regulatory system is involved in temperature-dependent biosynthesis of the Pseudomonas syringae phytotoxin coronatine. J. Bacteriol. 177:6160-6169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weiler, E. W., T. M. Kutchan, T. Gorba, W. Brodschelm, U. Niesel, and F. Bublitz. 1994. The Pseudomonas phytotoxin coronatine mimics octadecanoid signalling molecules of higher plants. FEBS Lett. 345:9-13. [DOI] [PubMed] [Google Scholar]
- 40.Young, S. A., S. K. Park, C. Rodgers, R. E. Mitchell, and C. L. Bender. 1992. Physical and functional characterization of the gene cluster encoding the polyketide phytotoxin coronatine in Pseudomonas syringae pv. glycinea. J. Bacteriol. 174:1837-1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang, J. H., N. B. Quigley, and D. C. Gross. 1995. Analysis of the syrB and syrC genes of Pseudomonas syringae pv. syringae indicates that syringomycin is synthesized by a thiotemplate mechanism. J. Bacteriol. 177:4009-4020. [DOI] [PMC free article] [PubMed] [Google Scholar]