

Abstract
Background
Familial Alzheimer’s disease (AD) due to PSEN1 mutations provides an opportunity to examine AD biomarkers in persons in whom the diagnosis is certain.
Methods
We describe a 55 year-old woman with clinically probable AD and a novel PSEN1 mutation who underwent genetic, clinical, biochemical and magnetic resonance and nuclear imaging assessments. We also describe neuropathological findings in her similarly affected brother.
Results
Neuropsychological testing confirmed deficits in memory, visuospatial and language function. CSF t-tau and p-tau181 were markedly elevated and Aβ42 levels reduced. FDG-PET revealed hypometabolism in the left parietotemporal cortex. FDDNP-PET showed increased binding of tracer in medial temporal and parietal lobes and in the head of the caudate and anterior putamen bilaterally. Neuropathological examination of her brother showed the typical findings of AD and the striatum demonstrated amyloid pathology and marked neurofibrillary pathology beyond that typically seen in late-onset AD. A novel S212Y substitution in PSEN1 was present in the index patient and her affected brother but not in an older unaffected sister. An in-vitro assay in which the S212Y mutation was introduced in cell culture confirmed that it was associated with increased production of Aβ42.
Conclusions
We describe biochemical, imaging, and neuropathological changes in a pedigree with a novel PSEN1 mutation. This allows us to validate the pathogenicity of this mutation and the indices used to assess AD.
Keywords: Alzheimer’s disease, PSEN1, S212Y, amyloid imaging, FDDNP, cerebrospinal fluid, FDG, Positron emission tomography, tau, beta-amyloid, striatum
Introduction
Three genes have been identified, alteration of which cause an autosomal dominant form of Alzheimer’s disease (AD) of young onset (familial AD or FAD). Alterations in the PSEN1 gene are the most common with 174 alterations in PSEN1 being associated with FAD (www.molgen.ua.ac.be/ADMutations). Many cause amino acid changes along the transmembrane portion of the protein[4]. This may cause a conformational change that modifies the way the Presenilin-1 protein (PS1) cleaves amyloid precursor protein (APP)[1]. Though evidence of pathogenicity is convincing for most mutations, the relationship of some to clinical disease is uncertain[6]. We describe two affected members of a family with young-onset AD in whom a novel PSEN1 mutation segregates with the disease.
Our ability to diagnose AD during life is imperfect. Because of this and the need for indices to serve as outcome measures for clinical trials, the relationships of biochemical (e.g. Aβ42 and tau levels in cerebrospinal fluid, or CSF) and neuroimaging (e.g. hippocampal volumetry, positron emission tomography using flurodeoxyglucose and amyloid and tau ligands) measures to dementia diagnosis and stage are being explored. In this report we describe biomarker results in an index patient with a novel PSEN1 mutation and the neuropathological findings in her affected brother.
Methods and Materials
CSF Analysis
CSF was collected at various times of the day by aspiration using a 24 gauge Sprotte needle. CSF was initially placed in polystyrene tubes and within 2 hours was centrifuged, aliquotted into 0.5 cc siliconized polypropylene Eppendorf tubes and frozen to -80°C until being analyzed for Aβ42 using Luminex reagents and X-MAP technology. CSF samples were also sent overnight on dry ice to Athena Diagnostics who measured total tau (t-tau) and p-tau181 using ELISA methodology as provided by Innnogenetics, N.V. (Innotest™ hTAU Ag, and Innotest™ Phospho-tau).
In-vitro study of APP metabolism
To examine the effect on APP processing of the putative mutation causing FAD in this family, the S212Y mutation was introduced by site-directed mutagenesis into a cDNA construct containing wild-type PS1 using a Quick Change II site-directed mutagenesis kit (Stratagene)[5]. Human embryonic kidney 293 (HEK293) cells were transiently transfected with mutant or wild-type PSEN1 vectors or control vector (GFP) and human APP cDNA with the Swedish mutation for 48 hours. The culture medium was replaced with fresh medium and incubated for 16 hrs. Secreted Aβ40 and Aβ42 levels were measured in the culture media by ELISA with specific antibodies recognizing Aβ1-40 and Aβ1-42 according to the manufacturer’s protocol (Invitrogen).
Neuropathological Analysis
Standard gross and microscopic neuropathologic evaluation was performed. Histologic stains included hematoxylin and eosin, Luxol Fast Blue, Bielschowsky, and Gallyas. Immunohistologic evaluation used the following antibodies: Aβ peptide (6E10, Signet Labs, 1:400), alpha-synuclein (LB509, 1:1000 and syn 303, 1:500, gifts J.Q. Trojanowski), tau (AT8, Endogen, 1:250), TDP43 (Protein Tech, 1:2000), and ubiquitin (Dako, 1:150).
Results
Case Reports
Index patient
The index patient is a Caucasian female assessed at age 55. At the request of her adult children, she presented to the Genetics Department for testing for the etiology of early-onset AD. Healthy except for migraine headaches and chronic anxiety, she had the onset of memory problems at age 38. Her problems slowly progressed such that she quit working as a nurse at age 43 and was diagnosed with dementia at age 44. When seen at age 55 she was reported to have additional difficulties with language and no longer drove an automobile nor cooked. Her Clinical Dementia Rating (CDR) scale total score was 1.0 indicating mild dementia. Mini-Mental Status Examination (MMSE) score was 24/30. Psychiatric evaluation revealed a life-long history of anxiety that had increased with the development of dementia such that at the time of her evaluation she qualified for an Axis I diagnosis of generalized anxiety disorder with depressive features. Her physical examination was normal except for brisk reflexes in the upper extremities without any notable parkinsonism, gait abnormalities or cerebellar signs. Neuropsychological testing revealed poor performance (<2nd percentile) in verbal and nonverbal memory on the Wechsler Memory Scale-III Logical Memory, California Verbal Learning Test - II, and the Rey-Osterrieth Complex Figure Test. She scored in the low average range (13th percentile) on the Spatial Localization test and in the impaired range (3rd percentile) on the Working Memory tests of the English version of the Spanish-English Neuropsychological Assessment Scale (SENAS)[8]. Language performance was average on the SENAS Object Naming (32nd percentile) and Phonemic Fluency (42nd percentile) tests, but she was only able to name 12 animals in a minute’s time (3rd percentile). Visuospatial performance on Block Design was average (25-27th percentile).
Her father had died at age 70 with a progressive dementia syndrome with onset around age 56. Details of his condition or of his prior family history are not known. She had two brothers and one sister. One brother had a similar illness with onset around age 41 and died at age 55 while another brother had been affected by the disease since age 44 and died at age 55 and underwent autopsy (see below). A 66 year-old sister was thought to be unaffected.
In comparison to 5 controls (mean age 38 years) who underwent lumbar punctures, t-tau level was 18.8 SDs higher (696 pg/ml) and p-tau181 levels were 9 SDs higher (113 pg/ml) in the index case. Aβ42 levels in CSF were undetectable using an assay that had a mean level of 344 pg/ml in controls and was sensitive down to 6 pg/ml[12], corresponding to a decrement of approximately 4 SD’s.
T1-weighted 3-D magnetic resonance imaging was normal except for atrophy of the left cerebral hemisphere relative to the right on visual inspection. When hippocampal volumes were measured and compared to controls from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) cohort they were found to be non-significantly larger, indicating a lack of significant hippocampal atrophy. FDG-PET revealed diffuse left cortical hypometabolism, particularly evident in the parietal cortex (Figure 1), where FDG metabolism fell 2.6 SD’s below the mean of normal controls. The subject also underwent PET imaging with the radioactively labeled ligand 2-(1-{6-[(2-[F-18]-fluoroethyl)(methyl)amino]-2-napththyl}-ethylidene)malononitrile (FDDNP). FDDNP binds to amyloid plaques and tau-containing paired helical filaments in the brains of AD patients and has been investigated as a diagnostic tool[14]. Using the Logan graphic method normalized to cerebellum, Distribution Volume Ratios (DVRs) were calculated for the medial and lateral temporal, frontal, parietal, posterior cingulate regions and for the striatum. DVRs were elevated by at least 2.8 SD’s in all regions assessed and elevations were more evident in the left lateral temporal lobe compared to the right and superiorly in the right parietal lobe compared to the left. Global DVR was 5 SD’s higher than that of 30 non-demented controls. FDDNP binding was also substantially elevated in the heads of the caudate nuclei and anterior putamina bilaterally (Figure 1) with DVRs 5 SD’s higher than in controls and 1.5 SDs above that in AD patients with a mean age of 72 years.
Figure 1.
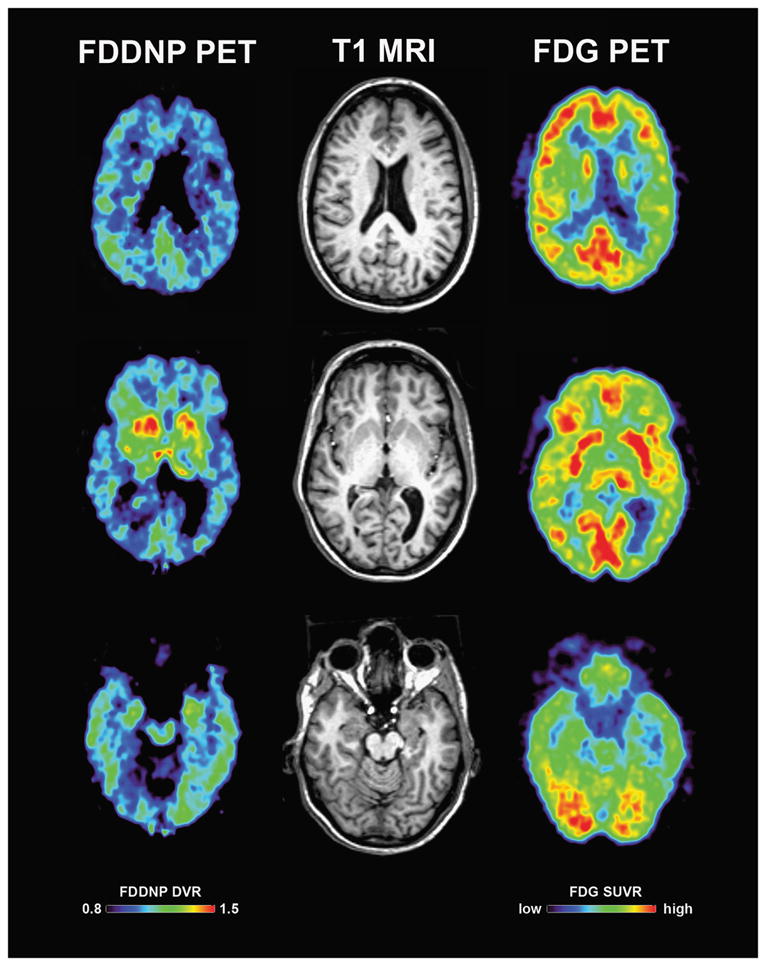
Representative FDDNP-PET (left), T1-weighted MRI (middle) and FDG-PET (right) images in the index patient. Note the left lateral parietotemporal area with relatively decreased metabolism on FDG, and the more inferior lateral temporal area with increased FDDNP signal. Also seen is increased FDDNP signal in the caudate head and anterior putamina bilaterally.
Index patient’s affected brother
The patient’s brother presented to the University of Washington at age 49 years with a four-year history of cognitive and behavioral changes. He was initially noted to have problems “multi-tasking” at work and then developed increasing forgetfulness and problems with navigation while driving. He also had symptoms of depression and sertraline was begun with improvement in mood though without benefits in cognition. MMSE score was 19/30, CDR score was 1.0. On the NYU paragraph recall test he had an immediate recall score of 2 and delayed recall of 0. A bilateral postural tremor was present, rapid alternating movements were slowed bilaterally, mild apraxia was present, and gait was normal. A diagnosis of probable AD was made. Subsequent evaluations at years 2, 3, and 4 revealed MMSE scores of 12, 7, and untestable, respectively. At visit 4 he had developed significant aphasia, and possible hallucinations and his CDR score was 3.0. He died at age 55.
Neuropathology Results
The brain of the index patient’s brother weighed 1,030 gm, with moderate frontal and mild parietal and temporal atrophy. Bielschowsky staining revealed neurofibrillary tangle pathology consistent with a Braak stage VI and a neuritic plaque score of “frequent”. Striatal tau pathology was severe in comparison to sporadic Braak VI Alzheimer’s disease (Figure 2). In contrast, striatal Aβ deposition was no greater than observed in sporadic Alzheimer’s disease. The cerebellum and occipital cortex demonstrated severe amyloid angiopathy. Alpha-synuclein pathology was limited to the amygdala. TDP-43 staining was negative.
Figure 2.
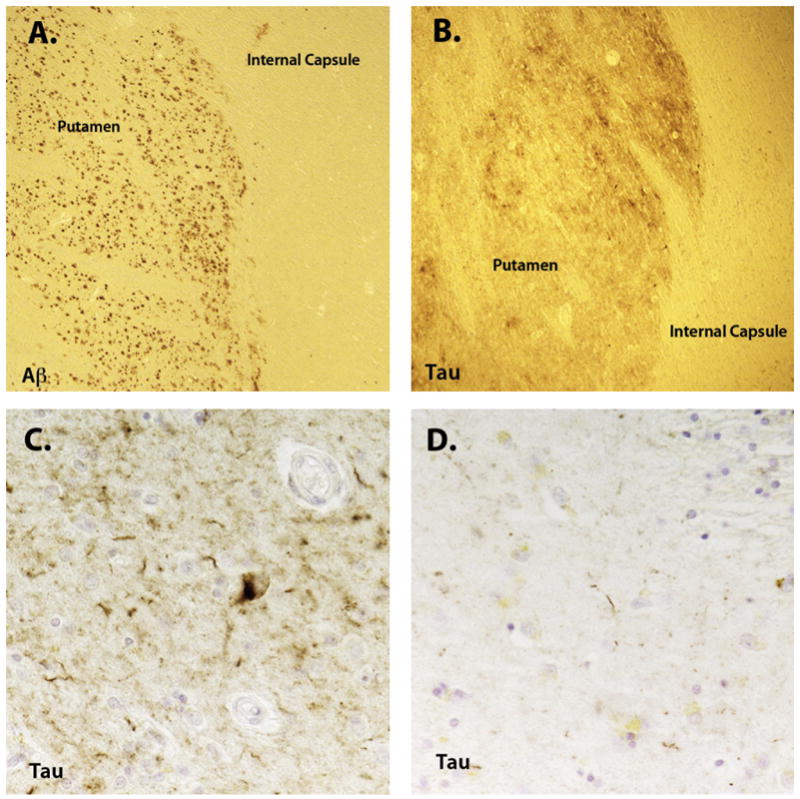
Striatal Aβ (A, 20X) and tau pathology (B, 40X and D, 400X) in the case with an S212Y PSEN1 mutation. In comparison to a sporadic Alzheimer’s disease (AD) case (C, 400X, Braak stage VI), the tau pathology was substantially more severe. No difference was observed in the severity of Aβ deposition between the familial AD case and sporadic AD cases.
Genetic results
Genetic testing of the index patient and her affected brother through Athena Diagnostics revealed a novel cytosine to adenine point mutation at nucleotide position 635. This causes a serine to tyrosine substitution at codon position 212 (S212Y) in the fourth transmembrane spanning region of the protein. This amino acid is conserved in orangutans and macaques but not in the dog, rat or mouse PSEN1 homologues. There have been no previously reported pathogenic PSEN1 mutations at this codon though three mutations each at nearby codons 209 and 213 have been reported in association with FAD. The index patient’s Apolipoprotein E genotype was 3/3. Genetic testing of the 66 year-old unaffected sister was negative for the PSEN1 mutation. Occurrence of the S212Y sequence variant was checked in the preliminary data release of the 1000 Genomes Project (http://www.1000genomes.org/, V4 of the Pilot3 Release, dated 2009-11-20, aimed at resequencing targeted gene regions, including PSEN1). No variants were reported at this position among 694 subjects of multiple ethnicities.
Biochemical Results
The assay analyzing the effect of the S212Y mutation on APP metabolism confirmed an effect in elevating the levels of Aβ40, Aβ42 and the Aβ42/Aβ40 ratio relative to that produced by wild-type PS1. Levels of Aβ42 produced by cells in which the S212Y mutation was introduced were 2.4x those produced by cells with WT PS1 (p = 4.5 × 10-8, Figure 3).
Figure 3.
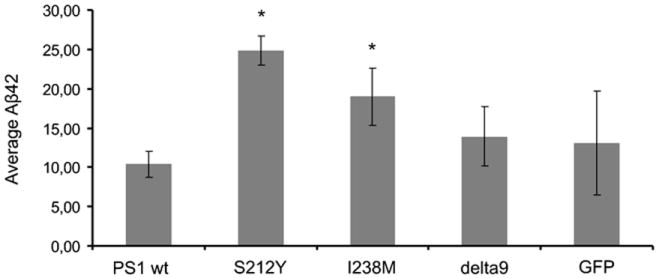
Average production of Aβ42 in a cellular model in which wild type PS1 (PS1 WT), the S212Y substitution, I238M substitution (another potential novel mutation), the delta 9 deletion, and control (GFP) constructs have been introduced. *p ≤ 0.001
Discussion
We describe a novel mutation in the PSEN1 gene associated with early-onset autosomal dominant AD. Neuropathological findings of an affected sibling confirmed the diagnosis of AD. Findings were typical of severe AD of late onset other than disproportionate tau pathology in the striatum. The resulting amino acid substitution, similar to many other previously described PSEN1 mutations, occurs in the transmembrane portion of the protein and may exert its effects by altering the way PS1 is positioned in the membrane and changing the way it cleaves APP. An in-vitro assay in which the S212Y mutation was introduced into a cellular construct verified that it caused an increase in the amount of Aβ42 produced. As increased absolute or relative production of Aβ42 is thought to be the mechanism by which PSEN1 mutations cause FAD, this is confirmatory evidence of the pathogenicity of this mutation.
The presence of this S212Y substitution in an affected and absence in an unaffected sibling and a database of 694 controls provide further evidence for its pathogenicity. Though most reported PSEN1 mutations associated with AD are pathogenic, a minority of such alterations have been shown to be polymorphisms[6]. We feel that genetic counseling regarding this possibility and confirmation of its presence in other affected family members is important when a patient is found to have a genetic alteration not previously reported as the implications for family members of an essentially fully-penetrant mutation are considerable.
The clinical presentation in this family was typical for AD except for the possibly protracted course in the index patient and the motor features evident in her brother. The index patient had the onset of cognitive symptoms at age 38 though at age 55 was still in a relatively mild stage of dementia. It is possible that she had subtle cognitive deficits as early as age 38 though increased concern may have contributed to her cognitive complaints at that time. The knowledge of her family history and “at-risk” status may have led to hightened anxiety regarding her cognitive function. However, we have previously observed higher levels of depressive symptoms in female carriers of PSEN1 mutations[11]. Though this finding has yet to be confirmed, it suggests the alternative explanation that her chronic anxiety disorder might have been the earliest manifestation of the disease.
Though the age of symptom onset in some kindreds with PSEN1 mutations is consistent among family members, it is more variable in others. Though we have found a consistent age of symptom onset around age 40 in persons from diverse families with the A431E PSEN1 [9], the age of onset in the Columbian kindreds featuring the E280A mutation appears to be more variable with a range from 34 - 62 years[7]. When the index patient was examined at age 55, the age of death from the disease of her two brothers, she was still in a relatively mild stage of the illness and her father reportedly did not have symptoms until age 56 and lived to be 70 years of age. From studies in the Columbian kindreds, it has been estimated that concurrently carrying the APOE ε4 allele reduces the age of onset of familial AD by approximately 2-4 years[10]. In a study of families carrying the common N141I substitution in PSEN2, also representing a founder effect, it was estimated that APOE, other undefined genetic loci, and undefined environmental influences accounted for 2, 6.5, and 8.5% of the variance, respectively, in the age of disease onset[18]. APOE genotype was not available in the brother whose age of onset was earlier than the index patient who was of the 3/3 genotype. The variable age of disease onset in this family and in others harboring FAD mutations therefore remains to be fully explained.
CSF t-tau levels were 19 SD’s and p-tau181 levels 9 SD’s above controls from our studies. T-tau levels in AD patients with a mean MMSE score of 24/30 (as in our subject) in the ADNI cohort were only 2 SD’s and p-tau181 levels 1 S.D. above controls[13]. In our studies of FAD, t-tau and p-tau181 tend to be elevated early in the presymptomatic stage of disease [12] and the results in our index patient are the highest we have observed so far. The finding of extremely high levels of CSF tau in persons with early-onset FAD is consistent with prior observations that suggest such high levels reflect more aggressive disease[17]. As studies in late-onset AD have found that diminished CSF Aβ appears to precede tau elevations[3], this may represent a distinction between the behavior of these markers in late-onset and familial AD.
CSF Aβ42 levels were undetectable and therefore at least 4 SD’s below control levels in our index patient. In the ADNI study, similarly impaired AD patients had CSF Aβ42 levels approximately 1 SD below controls[13]. In our on-going studies of persons with or at-risk for FAD, only one other subject out of 16 had undetectable CSF Aβ42 levels[12]. It is possible that the transient placement of CSF in polystyrene tubes artifactually lowered Aβ42 levels as it has been shown that Aβ adheres to such surfaces[15]. Though this might affect absolute quantification of Aβ, all samples in our study were handled in the same fashion. The fact that the other subject with undetectable levels was in a more advanced stage of dementia (CDR score of 2, MMSE score of 5/30) validates the utility of reduced CSF Aβ42 levels as a marker for established disease.
FDG-PET demonstrated a disproportionate degree of hypometabolism in the left posterior perisylvian area. The pattern and degree of FDDNP binding on PET imaging were generally consistent with that previously observed in sporadic AD of later onset. However, FDG-PET did not demonstrate hypometabolism in the basal ganglia where increased FDDNP signal was seen as has previously been reported in symptomatic subjects carrying FAD mutations imaged with Pittsburgh Compound B[16]. This dissociation and the lack of significant parkinsonism in our subject indicate that the basal ganglia may be able to sustain significant amyloid and tau pathology without functional consequences.
The use of the cerebellum as a reference region for the FDDNP analysis is potentially problematic as patients with FAD have been found to have higher degrees of amyloid pathology there relative to AD of late onset. Indeed, the brother of the index subject had significant amyloid angiopathy in the cerebellum. The effect of this pathology, if detected by FDDNP, would be to lower the amount of relative FDDNP seen elsewhere in the brain. It is therefore possible that FDDNP binding was actually greater than was seen. In the future, normalization of the FDDNP signal using whole brain white matter as the reference region might be considered[19]. The increased striatal signal seen with PIB in FAD is thought to reflect early amyloid deposition there and considering the striatal tau pathology seen in our subject’s affected brother, the FDDNP signal seen in the striatum in our index patient may reflect its binding to tau. However, the possibility of less specific binding of either FDDNP or PIB[2] to unidentified ligands present in the striatum cannot be ruled out.
We describe the clinical, cognitive, biochemical, neuroimaging, and neuropathological characteristics associated with a novel S212Y substitution in the PS1 protein. Defining these changes in persons in whom the diagnosis of AD is genetically confirmed assists in evaluating their utility as markers of AD pathology and progression. The demonstration of this mutation in an affected sibling and its absence in controls as well as the in-vitro demonstration of an effect on increasing Aβ42 provides confirmatory evidence for its pathogenicity. We recommend genetic consultation for families to facilitate confirmatory testing and to ensure that appropriate family members are tested in situations where previously undescribed genetic alterations are found.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Berezovska O, Lleo A, Herl LD, Frosch MP, Stern EA, Bacskai BJ, Hyman BT. Familial Alzheimer’s disease presenilin 1 mutations cause alterations in the conformation of presenilin and interactions with amyloid precursor protein. J Neurosci. 2005;25:3009–3017. doi: 10.1523/JNEUROSCI.0364-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cole GB, Keum G, Liu J, Small GW, Satyamurthy N, Kepe V, Barrio JR. Specific estrogen sulfotransferase (SULT1E1) substrates and molecular imaging probe candidates. Proc Natl Acad Sci U S A. 107:6222–6227. doi: 10.1073/pnas.0914904107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fagan AM, Head D, Shah AR, Marcus D, Mintun M, Morris JC, Holtzman DM. Decreased cerebrospinal fluid Abeta(42) correlates with brain atrophy in cognitively normal elderly. Ann Neurol. 2009;65:176–183. doi: 10.1002/ana.21559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hardy J, Crook R. Presenilin mutations line up along transmembrane alpha-helices. Neurosci Lett. 2001;306:203–205. doi: 10.1016/s0304-3940(01)01910-3. [DOI] [PubMed] [Google Scholar]
- 5.Hecimovic S, Wang J, Dolios G, Martinez M, Wang R, Goate AM. Mutations in APP have independent effects on Abeta and CTFgamma generation. Neurobiol Dis. 2004;17:205–218. doi: 10.1016/j.nbd.2004.04.018. [DOI] [PubMed] [Google Scholar]
- 6.Lee P, Medina L, Ringman JM. The Thr354Ile substitution in PSEN1∷ disease-causing mutation or polymorphism? Neurology. 2006;66:1955–1956. doi: 10.1212/01.wnl.0000219762.28324.a6. [DOI] [PubMed] [Google Scholar]
- 7.Lopera F, Ardilla A, Martinez A, Madrigal L, Arango-Viana JC, Lemere CA, Arango-Lasprilla JC, Hincapie L, Arcos-Burgos M, Ossa JE, Behrens IM, Norton J, Lendon C, Goate AM, Ruiz-Linares A, Rosselli M, Kosik KS. Clinical features of early-onset Alzheimer disease in a large kindred with an E280A presenilin-1 mutation. JAMA. 1997;277:793–799. [PubMed] [Google Scholar]
- 8.Mungas D, Reed BR, Crane PK, Haan MN, Gonzalez H. Spanish and English Neuropsychological Assessment Scales (SENAS): further development and psychometric characteristics. Psychol Assess. 2004;16:347–359. doi: 10.1037/1040-3590.16.4.347. [DOI] [PubMed] [Google Scholar]
- 9.Murrell J, Ghetti B, Cochran E, Macias-Islas MA, Medina L, Varpetian A, Cummings JL, Mendez MF, Kawas C, Chui H, Ringman JM. The A431E mutation in PSEN1 causing Familial Alzheimer’s Disease originating in Jalisco State, Mexico: an additional fifteen families. Neurogenetics. 2006;7:277–279. doi: 10.1007/s10048-006-0053-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pastor P, Roe CM, Villegas A, Bedoya G, Chakraverty S, Garcia G, Tirado V, Norton J, Rios S, Martinez M, Kosik KS, Lopera F, Goate AM. Apolipoprotein Eepsilon4 modifies Alzheimer’s disease onset in an E280A PS1 kindred. Ann Neurol. 2003;54:163–169. doi: 10.1002/ana.10636. [DOI] [PubMed] [Google Scholar]
- 11.Ringman JM, Diaz-Olavarrieta C, Rodriguez Y, Chavez M, Paz F, Murrell J, Macias MA, Hill M, Kawas C. Female preclinical presenilin-1 mutation carriers unaware of their genetic status have higher levels of depression than their non-mutation carrying kin. J Neurol Neurosurg Psychiatry. 2004;75:500–502. doi: 10.1136/jnnp.2002.005025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ringman JM, Younkin SG, Pratico D, Seltzer W, Cole GM, Geschwind DH, Rodriguez-Agudelo Y, Schaffer B, Fein J, Sokolow S, Rosario ER, Gylys KH, Varpetian A, Medina LD, Cummings JL. Biochemical markers in persons with preclinical familial Alzheimer disease. Neurology. 2008;71:85–92. doi: 10.1212/01.wnl.0000303973.71803.81. [DOI] [PubMed] [Google Scholar]
- 13.Shaw LM, Vanderstichele H, Knapik-Czajka M, Clark CM, Aisen PS, Petersen RC, Blennow K, Soares H, Simon A, Lewczuk P, Dean R, Siemers E, Potter W, Lee VM, Trojanowski JQ. Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann Neurol. 2009;65:403–413. doi: 10.1002/ana.21610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Small GW, Kepe V, Ercoli LM, Siddarth P, Bookheimer SY, Miller KJ, Lavretsky H, Burggren AC, Cole GM, Vinters HV, Thompson PM, Huang SC, Satyamurthy N, Phelps ME, Barrio JR. PET of brain amyloid and tau in mild cognitive impairment. N Engl J Med. 2006;355:2652–2663. doi: 10.1056/NEJMoa054625. [DOI] [PubMed] [Google Scholar]
- 15.Vanderstichele H, Van Kerschaver E, Hesse C, Davidsson P, Buyse MA, Andreasen N, Minthon L, Wallin A, Blennow K, Vanmechelen E. Standardization of measurement of beta-amyloid(1-42) in cerebrospinal fluid and plasma. Amyloid. 2000;7:245–258. doi: 10.3109/13506120009146438. [DOI] [PubMed] [Google Scholar]
- 16.Villemagne VL, Ataka S, Mizuno T, Brooks WS, Wada Y, Kondo M, Jones G, Watanabe Y, Mulligan R, Nakagawa M, Miki T, Shimada H, O’Keefe GJ, Masters CL, Mori H, Rowe CC. High striatal amyloid beta-peptide deposition across different autosomal Alzheimer disease mutation types. Arch Neurol. 2009;66:1537–1544. doi: 10.1001/archneurol.2009.285. [DOI] [PubMed] [Google Scholar]
- 17.Wallin AK, Blennow K, Zetterberg H, Londos E, Minthon L, Hansson O. CSF biomarkers predict a more malignant outcome in Alzheimer disease. Neurology. 74:1531–1537. doi: 10.1212/WNL.0b013e3181dd4dd8. [DOI] [PubMed] [Google Scholar]
- 18.Wijsman EM, Daw EW, Yu X, Steinbart EJ, Nochlin D, Bird TD, Schellenberg GD. APOE and other loci affect age-at-onset in Alzheimer’s disease families with PS2 mutation. Am J Med Genet B Neuropsychiatr Genet. 2005;132B:14–20. doi: 10.1002/ajmg.b.30087. [DOI] [PubMed] [Google Scholar]
- 19.Wong KP, Wardak M, Shao W, Dahlbom M, Kepe V, Liu J, Satyamurthy N, Small GW, Barrio JR, Huang SC. Quantitative analysis of [18F]FDDNP PET using subcortical white matter as reference region. Eur J Nucl Med Mol Imaging. 37:575–588. doi: 10.1007/s00259-009-1293-8. [DOI] [PMC free article] [PubMed] [Google Scholar]