

The crystal structure of SMU.595, a putative dihydroorotate dehydrogenase (DHOD) from S. mutans, is reported at 2.4 Å resolution.
Keywords: dihydroorotate dehydrogenases, Streptococcus mutans, pyrimidine biosynthesis
Abstract
Streptococcus mutans is one of the pathogenic species involved in dental caries, especially in the initiation and development stages. Here, the crystal structure of SMU.595, a putative dihydroorotate dehydrogenase (DHOD) from S. mutans, is reported at 2.4 Å resolution. DHOD is a flavin mononucleotide-containing enzyme which catalyzes the oxidation of l-dihydroorotate to orotate, which is the fourth step and the only redox reaction in the de novo biosynthesis of pyrimidine nucleotides. The reductive lysine-methylation procedure was applied in order to improve the diffraction qualities of the crystals. Analysis of the S. mutans DHOD crystal structure shows that this enzyme is a class 1A DHOD and also suggests potential sites that could be exploited for the design of highly specific inhibitors using the structure-based chemotherapeutic design technique.
1. Introduction
Dihydroorotate dehydrogenase (DHOD), also known as dihydroorotate oxidase, catalyzes the stereospecific oxidation of (S)-dihydroorotate to orotate, which is the fourth step in de novo pyrimidine biosynthesis and is the only redox reaction in this pathway. Sequence alignments have shown that all DHODs contain a polypeptide chain encoded by a pyrD gene (Andersen et al., 1994 ▶). This polypeptide forms the catalytic core structure, folding into an (α/β) TIM barrel. DHODs can be divided into two main classes. The class 1 DHODs, also called cytosolic enzymes, are primarily found in Gram-positive bacteria. The class 2 DHODs are membrane-associated enzymes that are primarily found in eukaryotic mitochondria and Gram-negative bacteria. The class 1 DHODs can be further divided into subclasses 1A and 1B, which differ in quaternary structure and in their use of electron acceptors. Class 1A enzymes are PyrD homodimers with an FMN located inside the barrel of each monomer and use fumarate as their natural electron acceptor. Class 1B enzymes, in contrast, are heterotetramers composed of a homodimer resembling the class 1A enzymes and two additional PyrK subunits which contain FAD and a 2Fe–2S cluster (Andersen et al., 1996 ▶). These additional groups allow the enzyme to use NAD+ as its natural electron acceptor (Jensen & Bjornberg, 1998 ▶). The class 2 membrane-associated enzymes are monomers that use respiratory quinones as the physiological electron acceptor.
The crystal structure of Lactococcus lactis DHOD, a class 1A DHOD, is available and reveals a homodimer of two PyrD subunits. Each subunit forms a TIM-barrel fold with a bound FMN cofactor located near the top of the barrel. Above the isoalloxazine ring, a small cavity has been defined as the binding site for the substrate (S)-dihydroorotate (DHO; Rowland et al., 1997 ▶). The structures of the class 2 enzymes have an FMN-containing TIM-barrel domain as found in the class 1 PyrD subunit and an additional N-terminal α-helix that folds into a separate domain which contains the binding sites for the respiratory quinones and two inhibitors (Hansen et al., 2004 ▶; Liu et al., 2000 ▶).
Streptococcus mutans is a facultatively anaerobic Gram-positive bacterium that is commonly found in the human oral cavity and is a significant contributor to dental caries. SMU.595 (gi:24379073; GeneID 1028049; EC 1.3.3.1; 311 amino-acid residues; 34.3 kDa) is a putative DHOD from S. mutans and is one of the selected targets in the S. mutans structural genomics project that is in progress at Peking University (Su et al., 2006 ▶). The amino-acid sequence alignment of SMU.595 with other class 1A DHODs implies that this protein is a member of class 1A (Fig. 1 ▶).
Figure 1.

Multiple alignment of amino-acid sequences of DHODs. The alignment was produced for S. mutans SMU.595, L. lactis DHOD (LlDHOD), T. brucei DHOD (TbDHOD) and Homo sapiens DHOD (HuDHOD). Strictly conserved residues are boxed in red and similar residues are represented by red letters. The alignment was performed using the programs ClustalX (Larkin et al., 2007 ▶) and ESPript v.2.2 (Gouet et al., 1999 ▶).
Here, we present structural evidence that SMU.595 is a class 1A DHOD. The protein was overexpressed in Escherichia coli and was purified by affinity chromatography. The reductive lysine-methylation procedure was adopted to improve the diffraction quality of the crystal. The structure was solved to 2.4 Å resolution and demonstrates a typical structure of a class 1A DHOD.
2. Materials and methods
2.1. Cloning, expression and purification
The smu.595 gene was amplified from genomic S. mutans DNA by polymerase chain reaction (PCR). Using conventional cloning methods, the PCR-amplified fragment was digested with BamHI and XhoI (NEB) and cloned into the expression vector pET-28a (Novagen). The cloned sequence corresponds to residues 1–311 plus an N-terminal His6 tag followed by 28 residues of the vector (MGSSHHHHHHSSGLVPRGSHMASMTGGQQMGRGS). The recombinant plasmid was transformed into E. coli BL21 (DE3) (Invitrogen) competent cells for expression. Overexpression of SMU.595 was induced with 1 mM isopropyl β-d-1-thiogalactopyranoside when the cell density reached an OD600 of 0.6–0.8. After further incubation for 4 h at 310 K, the cells were harvested by centrifugation and suspended in buffer A (50 mM HEPES pH 7.5, 500 mM NaCl). The resuspended cells were lysed by sonication on ice and cell debris was removed by centrifugation (46 400g, 40 min, 277 K). The supernatant was loaded onto an Ni2+-chelating affinity column (GE Healthcare, USA) pre-equilibrated with buffer A. Impurities were washed out with buffer A containing 100 mM imidazole and the target protein was eluted with a higher concentration of imidazole (50 mM HEPES pH 7.5, 500 mM NaCl, 500 mM imidazole). Fractions containing the target protein were pooled and concentrated by ultrafiltration with a Millipore centrifugal ultrafiltration device (Amicon Ultra, 30 kDa cutoff). The purity of the target protein was examined by SDS–PAGE during each step.
2.2. Lysine methylation
The protein was diluted to less than 1 mg ml−1 in buffer consisting of 50 mM HEPES pH 7.5, 250 mM NaCl. 20 µl freshly prepared 1 M dimethylamine–borane complex (ABC; Fluka product No. 15584) and 40 µl 1 M formaldehyde (made from 37% stock; Fluka product No. 33220) were added per millilitre of protein solution and the reaction was gently mixed and incubated at 277 K for 2 h. A further 20 µl ABC and 40 µl formaldehyde were added and incubation was continued for 2 h. Following a final addition of 10 µl ABC, the reaction mixture was incubated overnight at 277 K. The soluble methylated protein was loaded onto a gel-filtration column (Superdex 200 16/60 on an ÄKTApurifier 100 system; GE Healthcare) pre-equilibrated in 20 mM Tris–HCl pH 7.5, 200 mM NaCl. The pooled peak fractions were concentrated and exchanged into buffer consisting of 20 mM Tris–HCl pH 7.5, 200 mM NaCl, 3 mM dithiothreitol, 5% glycerol.
2.3. Protein crystallization and data collection
After the gel filtration, SMU.595 was concentrated to ∼26 mg ml−1 for crystallization. The protein concentration was checked using a Bio-Rad protein-assay kit (Bio-Rad Laboratories, USA) based on the method of Bradford (1976 ▶). Initial crystallization screening was carried out at 293 K by the sitting-drop vapour-diffusion method using commercially available crystallization screens from Hampton Research (USA). The crystals appeared within 4 h in the condition 0.1 M MES pH 6.5, 12% PEG 20 000. After optimization, crystals suitable for diffraction experiments were obtained using 0.1 M MES pH 6.0, 12% PEG 20 000 and 0.03%(v/v) dichloromethane with a protein concentration of 20 mg ml−1.
For data collection, a crystal was flash-frozen in a nitrogen stream at 100 K after soaking in reservoir solution supplemented with 20% glycerol for several seconds. Data were collected on beamline 3W1A at Beijing Synchrotron Radiation Facility (BSRF). Autoindexing, data reduction and scaling were performed using the HKL-2000 program suite (Otwinowski & Minor, 1997 ▶). The crystal belonged to space group C2221, with unit-cell parameters a = 103.1, b = 158.3, c = 198.3 Å. Data-collection statistics are summarized in Table 1 ▶.
Table 1. Data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| Data collection | |
| Wavelength (Å) | 1.0 |
| Space group | C2221 |
| Unit-cell parameters (Å) | a = 103.1, b = 158.3, c = 198.3 |
| Resolution (Å) | 2.4 (2.44–2.40) |
| No. of unique reflections | 60852 (2865) |
| Completeness (%) | 98 (93.5) |
| Multiplicity | 10 (9.1) |
| Mean I/σ(I) | 26 (3.6) |
| Molecules in asymmetric unit | 4 |
| Rmerge† (%) | 11.2 (42.3) |
| Structure refinement | |
| Resolution range (Å) | 50–2.4 |
| No. of atoms | |
| Protein | 1239 |
| Water | 493 |
| FMN | 4 |
| Glycerol | 4 |
| Rwork‡/Rfree§ (%) | 20.2/24.8 |
| Average B factors (Å2) | |
| Main chain (monomer A/B/C/D) | 27.5/42.1/28.8/33.0 |
| Side chain (monomer A/B/C/D) | 29.5/43.5/31.2/35.6 |
| Flavin atoms | 25.6 |
| Waters | 32.7 |
| Ramachandran plot (%) | |
| Most favoured | 97.6 |
| Allowed | 2.4 |
| Disallowed | 0.0 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.008 |
| Bond angles (°) | 1.171 |
R
merge = 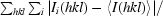
 , where Ii(hkl) is the intensity of reflection hkl and
, where Ii(hkl) is the intensity of reflection hkl and 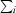 is the sum over all i measurements of reflection hkl.
is the sum over all i measurements of reflection hkl.
The R factor R
work = 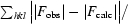
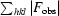 , where F
obs and F
calc are the observed and calculated structure factors, respectively.
, where F
obs and F
calc are the observed and calculated structure factors, respectively.
R free is the R factor calculated over a subset of the data that were excluded from refinement.
2.4. Structure determination and refinement
Initial phases were obtained by molecular replacement (MR) with the program Phaser (McCoy et al., 2007 ▶) from the CCP4 package (Collaborative Computational Project, Number 4, 1994 ▶). The crystal structure of DHOD from L. lactis (LIDHOD; PDB code 1dor; Rowland et al., 1997 ▶), which shares 63% sequence identity with SMU.595, was used as the search model. It was modified by the program CHAINSAW (Stein, 2008 ▶) in order to change its sequence to the target sequence before MR. The structure provided by Phaser was refined using phenix.refine v.1.6.4 (McCoy et al., 2007 ▶). Coot (Emsley & Cowtan, 2004 ▶) was used to improve the model and add ligands (FMN and glycerol). The quality of the final structure was assessed by PROCHECK (Laskowski et al., 1993 ▶). The R factor and R free of the final model were 20.15% and 24.76%, respectively. Refinement statistics and model parameters are given in Table 1 ▶.
3. Results and discussion
3.1. Overall structure
The SMU.595 structure was solved and refined to 2.4 Å resolution in space group C2221. Consistent with previously determined structures of the class 1A DHOD family, the overall SMU.595 monomer folds as a α/β barrel consisting of a core of central eight parallel β-strands surrounded by a ring of eight α-helices. Some secondary-structural elements (including αA–αC and βA–βE) and loops form a subdomain as insertions towards the top of the barrel. The FMN group is located between the top of the barrel and the subdomain (Fig. 2 ▶ a). The asymmetric unit of the crystal structure contains four monomers that are arranged as two distinct homodimers that are nearly perpendicular to each other, i.e. the tetramer has the shape of the letter ‘L’ (Fig. 2 ▶ b). The monomers of each dimer are related by a noncrystallographic twofold axis. In the dimer structure, the β6–α6 loop protrudes from one subunit towards α6 and α7 of the other subunit to form the dimer interface (Fig. 2 ▶ c). The dimer overlays well with the LlDHOD dimer and the Trypanosoma brucei DHOD dimer (TbDHOD; PDB code 2b4g; Arakaki et al., 2008 ▶; Fig. 2 ▶ d).
Figure 2.

Structure of SMU.595. (a) The overall crystal structure of SMU.595 is shown as a ribbon diagram with secondary-structure elements labelled in cyan (helices), violet (strands) and pink (loops). The FMN cofactor is represented as green sticks. (b) The crystal structure of the SMU.595 tetramer is shown as a ribbon diagram. The FMN cofactor is represented as orange sticks. (c) The dimer structure viewed from above the twofold axis. The β6–α6 loop and helices α6 and α7, which participate in dimer-interface interactions, are shown in pink and deep blue, respectively. The FMN cofactor is represented as orange sticks. (d) Left, structural overlay of the homodimers of SMU.595 (wheat yellow), TbDHOD (light blue) and LlDHOD (light pink). Right, the secondary structures (including the β2–α2, β4–αA and β6–α6 loops) surrounding the FMN cofactor. FMNs are shown in green, blue and pink. The amino-acid residues that are well conserved in the three sequences and that participate in interactions with FMN cofactors are shown as orange, blue and pink sticks, respectively. The methylated lysines are shown in yellow.
3.2. The effect of lysine methylation on crystal structure
We adopted the protocol developed in the Oxford Protein Production Facility (OPPF) and the Division of Structural Biology at Oxford University (Walter et al., 2006 ▶). Our analytical gel-filtration chromatography results suggested that the native enzyme exists as a dimer in solution. Following lysine methylation, a change in the oligomeric state was observed: there was a shift from a dimer to a monomer (results not shown). Although crystals of unmodified SMU.595 were easy to grow in the primary crystallization screen, extensive efforts to optimize the crystal quality were fruitless. Using the synchrotron beamline at BSRF, the rod-shaped and needle-shaped crystals only diffracted to 7.8 Å resolution and showed high mosaicity. Conversely, crystals of methylated SMU.595 grew with a much better shape (Fig. 3 ▶) and showed improved diffraction to 2.4 Å resolution. After solution of the methylated structure, we observed that seven dimethylated lysines (Lys45, Lys114, Lys137, Lys184, Lys210, Lys251 and Lys289) out of the 20 lysine residues in the monomer were visible according to the electron-density map (Fig. 4 ▶). Structural alignment of TbDHOD and SMU.595 shows that the conformation of the crystal structure and the active site of the FMN group are not influenced by reductive lysine methylation (Fig. 2 ▶ d). The FMN forms hydrogen bonds to residues Ala21, Lys45, Thr46, Lys165, Ile193 and Thr247 and hydrophobic interactions with residues Ala20, Gly22, Met71, Tyr60 and Asn69 (Fig. 5 ▶ a). In addition, the active sites of the conserved residues responsible for interaction with the product orotate, which are Asn69, Met71, Gly72, Leu73, Asn128, Asn133, Asn194 and Ser195, remain unchanged (Fig. 5 ▶ b). The only difference exists in the region 126–143, which is strongly conserved in all class 1A DHODs and can be considered to be a pyrimidine-binding motif. In the crystal structure of SMU.595 the loop 132–138 is in an open conformation, while in TbDHOD it is in a relatively closed conformation like a lid over the orotate. In the crystal structures of human DHOD, which belongs to the class 2 DHODs, the corresponding loop is poorly ordered. Therefore, the flexible loop might act as a lid that allows dihydroorotate to enter and orotate to exit (Liu et al., 2000 ▶; Arakaki et al., 2008 ▶). For these reasons, our SMU.595 crystal structure is accurate and credible.
Figure 3.

Crystals of S. mutans DHOD as grown after lysine methylation.
Figure 4.

Dimethylated lysines (orange) are shown in stick representation. The homodimer of SMU.595 is shown as ribbon models in cyan and green.
Figure 5.

Structural overlay of residues interacting with FMN and orotate. The residues, FMN and orotate are shown as stick models. SMU.595 is shown in pale green and TbDHOD is shown in light pink. SMU.595 residues are labelled in parentheseses. The hydrogen bonds between FMN (or orotate) and side chains of residues are shown as dotted lines. (a) The FMNs of SMU.595 and TbDHOD are shown in green and pink, respectively. (b) The orotate of TbDHOD is shown in orange. Residues 123–146 are shown as a ribbon model. Figs. 2, 3, 4 and 5 were generated using PyMOL (DeLano, 2002 ▶).
The methylation process is essential for crystal growth. According to thermodynamics, the entropic cost of burying surface residues at crystal-contact regions may seriously impede the crystallization process. Statistically, lysines are predominantly located on the surface, with 68% exposed, 26% partly exposed and only 6% buried (Baud & Karlin, 1999 ▶), making them the most solvent-exposed residues in proteins. In this case, the dimethylated lysines did not affect the conformation of the homodimer and no evidence of drastic changes in the intermolecular forces (i.e. hydrogen bonds, salt bridges, van der Waals contacts and hydrophobic effects) could be observed in the structure. However, methylation does decrease the solubility and change the oligomeric state in solution and lead to the formation of diffraction-quality crystals. One explanation for the change might be that the protein has a high concentration of surface lysine residues, the side chains of which are likely to destabilize the crystal lattice through entropic effects. It is well known that methyl groups are themselves hydrophobic and can alter the structure of water in the vicinity. The dimethylated lysines probably have an effect on the nature of the crystal contacts. In principle, the nature of the crystal contacts is the primary determinant of the physical qualities of the crystal. Thus, the hydrophobic nature of the methylated lysines might favour protein–protein interactions and thus form the novel arrangements of modified protein that exhibit superior diffraction.
4. Summary and conclusions
In summary, the crystal structure of SMU.595 indicates that it belongs to the class 1A DHOD family and presents evidence that will help in our understanding of its molecular mechanism and in further study of its substrate-binding and inhibitor-binding specificities, which could provide some promising clues for the further design of chemotherapeutics based on the SMU.595 structure.
Supplementary Material
PDB reference: SMU.595, 3oix
Acknowledgments
This work was supported by the grants from the National Natural Science Foundation of China (10979005) and the National Basic Research Program of China (2009CB918600).
References
- Andersen, P. S., Jansen, P. J. G. & Hammer, K. (1994). J. Bacteriol. 176, 3975–3982. [DOI] [PMC free article] [PubMed]
- Andersen, P. S., Jansen, P. J. G. & Hammer, K. (1996). J. Bacteriol. 178, 5005–5012. [DOI] [PMC free article] [PubMed]
- Arakaki, T. L., Buckner, F. S., Gillespie, J. R., Malmquist, N. A., Phillips, M. A., Kalyuzhniy, O., Luft, J. R., DeTitta, G. T., Verlinde, C. L. M. J., Van Voorhis, W. C., Holl, W. G. J. & Merritt, E. A. (2008). Mol. Microbiol. 68, 37–50. [DOI] [PMC free article] [PubMed]
- Baud, F. & Karlin, S. (1999). Proc. Natl Acad. Sci. USA, 96, 12494–12949. [DOI] [PMC free article] [PubMed]
- Bradford, M. M. (1976). Anal. Biochem. 72, 248–254. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- DeLano, W. L. (2002). PyMOL http://www.pymol.org. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Gouet, P., Courcelle, E., Stuart, D. I. & Métoz, F. (1999). Bioinformatics, 15, 305-308. [DOI] [PubMed]
- Hansen, M., Le Nours, J., Johansson, E., Antal, T., Ullrich, A., Löffler, M. & Larsen, S. (2004). Protein Sci. 13, 1031–1042. [DOI] [PMC free article] [PubMed]
- Jensen, K. F. & Bjornberg, O. (1998). Paths Pyrimidines, 6, 20–28.
- Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., Valentin, F., Wallace, I. M., Wilm, A., Lopez, R., Thompson, J. D., Gibson, T. J. & Higgins, D. G. (2007). Bioinformatics, 23, 2947–2948. [DOI] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst. 26, 283–291.
- Liu, S., Neidhardt, E. A., Grossman, T. H., Ocain, T. & Clardy, J. (2000). Structure Fold. Des. 8, 25–33. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Rowland, P., Nielsen, F. S., Jensen, K. F. & Larsen, S. (1997). Structure, 5, 239–252. [DOI] [PubMed]
- Stein, N. (2008). J. Appl. Cryst. 41, 641–643.
- Su, X.-D., Liang, Y., Li, L., Nan, J., Bröstromer, E., Liu, P., Dong, Y. & Xian, D. (2006). Acta Cryst. D62, 843–851. [DOI] [PubMed]
- Walter, T. S., Meier, C., Assenberg, R., Au, K. F., Ren, J.-S., Verma, A., Nettleship, J. E., Owens, R. J., Stuart, D. I. & Grimes, J. M. (2006). Structure, 14, 1617–1622. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: SMU.595, 3oix