

The thermostable direct haemolysin from G. hollisae has been purified and crystallized in two crystal forms using the vapour-diffusion method.
Keywords: thermostable direct haemolysin, Grimontia hollisae
Abstract
Vibrio hollisae, a halophilic species recently reclassified as Grimontia hollisae, is a causative agent of gastroenteritis and septicaemia. One important pathogenic Vibrio factor, thermostable direct haemolysin (TDH), has been purified and crystallized in two crystal forms using the vapour-diffusion method. The crystals belonged to an orthorhombic space group, with unit-cell parameters a = 104.8, b = 112.4, c = 61.3 Å and a = 122.9, b = 123.3, c = 89.8 Å. The crystals contained either four or eight molecules per asymmetric unit, with predicted solvent contents of 49.4 and 46.3% and Matthews coefficients (V M) of 2.4 and 2.3 Å3 Da−1, respectively. These crystals were suitable for structure determination, which would yield structural details related to the cytotoxicity and oligomeric structure of this pore-forming toxin.
1. Introduction
The halophilic Gram-negative bacterium Grimontia hollisae (formerly named Vibrio hollisae) can cause wound infections, gastroenteritis, hypovolemic shock, bacteraemia and septicaemia in humans related to the consumption of contaminated marine foods or to contact with environmental reservoirs (Hickman et al., 1982 ▶; Morris et al., 1982 ▶; Lowry et al., 1986 ▶; Thompson et al., 2003 ▶; Hinestrosa et al., 2007 ▶). In addition to the well studied V. cholerae O1 and O139, one major virulence factor of Vibrio species that is epidemiologically associated with disease is thermostable direct haemolysin (TDH). TDH exhibits a variety of biological activities, including the haemolysis of various species of erythrocytes, cytotoxicity, lethal toxicity to small experimental animals and cardiotoxicity (Sakurai et al., 1976 ▶; Goshima et al., 1977 ▶; Miyamoto et al., 1969 ▶; Honda & Iida, 1993 ▶; Tang et al., 1995 ▶, 1997 ▶; Raimondi et al., 2000 ▶; Naim et al., 2001 ▶; Lang et al., 2004 ▶).
Almost all clinical pathogenic V. parahaemolyticus strains and all strains of G. hollisae produce TDH proteins. The tdh genes from both V. parahaemolyticus and G. hollisae contain an open reading frame (ORF) of 570 nucleotides and code for a 189-amino-acid protein that contains a 24-amino-acid signal peptide in the N-terminal region and a 165-residue mature peptide at the C-terminus. Comparative amino-acid sequence analysis between G. hollisae TDH (Gh-rTDH) and V. parahaemolyticus TDH (Vp-TDH) revealed a difference of 20 amino acids over the whole protein (Yoh et al., 1989 ▶). In addition, the produced Gh-rTDH is antigenetically and genetically related to, but different from, Vp-TDH. For example, the haemolytic activity of Gh-TDH is heat-labile at temperatures above 333 K, whereas Vp-TDH exhibits an Arrhenius effect of inactivation on heating to 333 K but reactivation on additional heating to above 353 K (Yoh et al., 1986 ▶; Fukui et al., 2005 ▶). Many physiochemical explanations, including an increase in hydrogen bonding, better hydrophobic internal packing, improved electrostatic interactions, salt-bridge optimization, enhanced secondary-structure stability, an increase in aromatic interactions and a strengthening of inter-subunit associations, could contribute to the enhanced thermostability (Vogt et al., 1997 ▶).
Vp-TDH has been proposed to be a pore-forming toxin which causes colloidal osmotic lysis of erythrocytes (Honda et al., 1992 ▶). However, the detailed mechanism of TDH haemolysis has remained unclear. Biophysical characterization of Vp-TDH using small-angle X-ray scattering and ultracentrifugation techniques suggested that Vp-TDH exists as a tetramer in solution (Hamada et al., 2007 ▶). The C4 symmetry of the Vp-TDH tetramer determined by transmission electron microscopy was consistent with our finding for Gh-rTDH (data not shown). Recently, the structure of Vp-TDH has been determined (Parker & Feil, 2005 ▶; Yanagihara et al., 2010 ▶). The Gh-rTDH and Vp-TDH enzymes share 87.8% amino-acid sequence identity, but have rather different thermostabilities. A comparison of the two enzymes is important in order to understand the structural features that are essential for protein thermostability and to investigate whether these differences can provide further information on the catalytic mechanism of haemolytic activity, cytotoxicity and membrane recognition. Here, we describe the crystallization and preliminary X-ray diffraction studies of the Gh-rTDH protein from G. hollisae.
2. Materials and methods
2.1. Molecular cloning, protein expression and purification
The recombinant pTOPO-Gh-tdh expression vector was obtained by direct insertion of the Taq polymerase-amplified PCR product of the full-length Gh-tdh gene from G. hollisae (ATCC 33564) into topoisomerase-activated pCR 2.1-TOPO vector (Invitrogen, Carlsbad, California, USA) according to the manufacturer’s protocol. The recombinant plasmid was sequenced using an ABI PRISM 3100 genetic analyzer to ensure its fidelity in the subsequent protein-expression and purification experiments. An aliquot of 10 ml overnight culture of Escherichia coli BL21 (DE3) pLysS transformed with the expression vector was inoculated at a 1:500 ratio into 1.0 l LB medium (10 g l−1 tryptone, 5 g l−1 yeast extract, 10 g l−1 NaCl) containing kanamycin (50 µg ml−1) and the cell culture was shaken at 250 rev min−1 at 310 K for 16 h without IPTG induction. The cells were harvested by centrifugation, resuspended in 40 ml lysis buffer (20 mM Tris–HCl pH 7.0) and lysed by sonication: 2 s on, 1 s off at 35% amplitude for six cycles at 277 K. The lysate was clarified by centrifugation at 12 000g for 30 min and subsequently filtered through an 0.22 µm cellulose membrane. The solution was applied onto a Phenyl-Sepharose 6 Fast Flow column (GE Healthcare) pre-equilibrated with 20 mM Tris–HCl pH 7.0 and the fraction with TDH activity passed directly through the column. The partially purified protein solution with solid NaCl added to a final concentration of 0.2 M was passed through the same Phenyl-Sepharose 6 Fast Flow column washed with 20 and 50% ethylene glycol, respectively, and pre-equilibrated in buffer consisting of 20 mM Tris–HCl pH 7.0 and 0.2 M NaCl. After three isocratic washing steps with two column volumes of the same buffer containing 200, 100 and 50 mM NaCl, the active protein was eluted with 20 mM Tris–HCl pH 7.0. The peak fractions were pooled, concentrated to 2, 4, 8 and 10 mg ml−1 and stored at 193 K. The homogeneity of the protein solution was determined by SDS–PAGE analysis of serially diluted protein samples to check for contaminants and degradation. For determination of the haemolytic activity of TDH, 100 µl purified protein [10%(v/v) in phosphate-buffered saline (PBS)] was mixed with 100 µl rabbit erythrocytes [4%(v/v) in PBS] and incubated at 310 K for 1 h. The solution was centrifuged at 1000g for 1 min and assayed for change in absorbance at 540 nm.
2.2. Crystallization and data collection
The hanging-drop vapour-diffusion method was used to screen initial crystallization conditions using Crystal Screen and Crystal Screen 2 (Hampton Research) and Cryo II sparse-matrix (Emerald BioSystems) crystallization kits. The crystallization droplets consisted of 1 µl protein solution with a concentration in the range 2–10 mg ml−1 mixed with 1 µl reservoir solution and were equilibrated against 150 µl reservoir solution in CrystalEX 96-well plates (Corning) at 293 K.
Flaky plate-shaped crystals appeared within 2–5 d in several crystallization droplets using various conditions containing 2-propanol, 1,6-hexanediol, ethylene glycol or 1,2-propanediol as a key precipitant. Further manual refinement of these conditions in hanging drops (4 µl) yielded slightly thicker plate crystals (0.3 × 0.2 × 0.05 mm) with an optimized composition of the reservoir solution consisting of 100 mM Tris–HCl pH 8.5, 200 mM MgCl2, 25% 1,2-propanediol and 2 or 10% glycerol. Two crystal forms were obtained in the same drop consisting of 2 µl protein solution mixed with 2 µl reservoir solution and equilibrated against 400 µl reservoir solution at 293 K. These protein crystals were initially screened for diffraction quality on Biological Crystallography Facility beamlines BL13B1 and BL13C1 of the National Synchrotron Radiation Research Center (NSRRC, Taiwan). A single crystal harvested directly from the crystallization drop using a 0.1–0.5 mm rayon-fibre cryoloop was flash-cooled in a cold nitrogen-gas stream at 100 K. Single-wavelength diffraction data were collected on an ADSC Quantum 315 CCD detector using a crystal-to-detector distance of 250 mm and 153 images were collected with a 0.5° incremental oscillation width. The diffraction data were indexed, integrated and scaled using the HKL-2000 program suite and analysis of the pseudo-precession images was performed using the program HKLVIEW (Collaborative Computational Project, Number 4, 1994 ▶; Otwinowski & Minor, 1997 ▶). The statistics of data collection are summarized in Table 1 ▶.
Table 1. Data-collection and processing statistics for the TDH crystals.
Values in parentheses are for the highest resolution shell.
| Form I | Form II | |
|---|---|---|
| Possible space group | P21212 | |
| Unit-cell parameters (Å) | a = 104.8, b = 112.4, c = 61.3 | a = 122.9, b = 123.3, c = 89.8 |
| Resolution range (Å) | 30.0–2.7 (2.8–2.7) | 30.0–2.6 (2.7–2.6) |
| Total reflections | 126357 | 250747 |
| Unique reflections | 20575 (1999) | 42546 (4092) |
| Mosaicity (°) | 0.509 | 0.702 |
| Multiplicity | 6.1 (6.1) | 5.9 (5.4) |
| Completeness (%) | 100 (100) | 100 (97) |
| Mean I/σ(I) | 20.0 (3.9) | 14.9 (2.7) |
| Rmerge† | 0.095 (0.487) | 0.109 (0.484) |
R
merge = 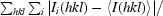
 , where Ii(hkl) is the observed intensity of reflection hkl and 〈I(hkl)〉 is the average intensity of symmetry-related reflections.
, where Ii(hkl) is the observed intensity of reflection hkl and 〈I(hkl)〉 is the average intensity of symmetry-related reflections.
3. Results and discussion
The Gh-tdh gene was amplified from G. hollisae ATCC 33564 genomic DNA and subcloned into the plasmid pCR 2.1-TOPO to generate the pTOPO-Gh-tdh recombinant plasmid. The recombinant pTOPO-Gh-tdh gene was subjected to DNA sequencing, sequence analysis and protein overexpression. A comparison of the deduced amino-acid sequences of Gh-rTDH and Vp-TDH (shown in Fig. 1 ▶) revealed several residues that are putatively involved in changing side-chain polarity and/or hydrogen bonding. However, no apparent amino-acid residues involved in either subunit contact or thermostability could be deduced from sequence comparison between these two genes. Gh-rTDH was overproduced in E. coli and purified to homogeneity. Electrophoresis of the purified Gh-rTDH revealed a single band at approximately 22 kDa according to SDS–PAGE analysis. Nevertheless, nondenaturing PAGE showed a single band of approximately 90 kDa, indicating that the Gh-rTDH protein exists as a monomer under denatured conditions and associates as a homotetramer in solution (Fig. 2 ▶).
Figure 1.

Amino-acid sequence alignment of G. hollisae TDH and V. parahaemolyticus TDH2.
Figure 2.

Purification of the Gh-rTDH protein. (a) The crude protein expressed from BL21 (DE3) pLysS strain (lane 1) was passed through Phenyl Sepharose 6 Fast Flow twice (lanes 2 and 3) to obtain a homogenous protein with a molecular mass of ∼22 kDa as shown by SDS–PAGE. (b) Native PAGE of Gh-rTDH, showing a molecular mass of ∼90 kDa; lane 1, crude protein; lane 2, purified protein; lane M, molecular-weight standards (kDa).
Initial attempts using hanging-drop crystallization screening were able to suggest several potential precipitants such as 2-propanol, 1,6-hexanediol, ethylene glycol and 1,2-propanediol. After manually fine-tuning the crystallization conditions to reduce the clustering and to increase the thickness of the plate crystals, the crystallization conditions were refined to protein concentrations of 2 and 10 mg ml−1 with corresponding glycerol concentrations of 10 and 2%, respectively (Fig. 3 ▶). Nevertheless, it was necessary to screen for diffraction-quality protein crystals on a synchrotron beamline because crystals of different space groups may be present in the same crystallization drop; most screened crystals showed reflections with diffuse spots or a poor resolution limit beyond 3 Å. After screening crystals with good morphology produced by optimizing the crystallization condition, diffraction to 2.7 Å resolution was obtained on NSRRC beamline BL13B1 (Fig. 4 ▶). Data processing using HKL-2000 indicated that the protein had crystallized in two polymorphic forms that were suitable for structure determination (Table 1 ▶). The form I and II crystals both belonged to the orthorhombic system and had unit-cell parameters a = 104.8, b = 112.4, c = 61.3 and a = 122.9, b = 123.3, c = 89.8 Å, respectively. Cell-content analyses indicated either four or eight subunits in the asymmetric unit, with Matthews volumes of 2.4 and 2.3 Å3 Da−1 and solvent contents of 49.4 and 46.3%, respectively. Based on the systematic absences of the axial reflections (all even for h00, 0k0, 00l), the space group was identified as P21212. The self-rotation function obtained from the form I crystal showed no additional twofold symmetry apart from crystallographic symmetry (Fig. 5 ▶). However, the native Patterson map calculation revealed a large non-origin peak at (u, v, w) = (0.5, 0.378, 0.5), indicating that noncrystallographic twofold axes, if present, must be parallel to twofold crystallographic axes in the c direction at the position (x, y) = (0.25, 0.19) (Fig. 6 ▶). Since the biochemical data suggested that the protein was in a tetrameric form, we speculate that each dimer of the tetrameric protein is probably related to the neighbouring dimer by a noncrystallographic twofold-symmetry operation. At present, we are focusing on structure determination of the form I crystal, as recent preliminary diffraction data screening of SeMet-substituted Gh-rTDH crystals showed similar unit-cell parameters. The molecular details of this quaternary pore-forming toxin will be resolved by structure determination using the multiple-wavelength anomalous diffraction (MAD) method.
Figure 3.

Two different single-crystal forms of thermostable direct haemolysin from G. hollisae (Gh-rTDH) with dimensions of around 0.3 × 0.2 × 0.05 mm observed by cross-polarized light microscopy.
Figure 4.

X-ray diffraction image of a Gh-rTDH crystal: a typical 1° oscillation photograph. The inner ring marked in red indicates the 2.6 Å resolution shell.
Figure 5.

Self-rotation function, κ = 180° section. This was calculated using all data from the form I crystal in the range 30–2.7 Å with a radius of integration in the Patterson function of 30 Å.
Figure 6.

Native Patterson map calculated from 30 to 2.7 Å resolution for the form I crystal. The plot shows fractional coordinates u and v ranging from 0.0 to 0.5 and w = 0.5 at the Harker section. An intense pseudo-origin peak appears at (u, v) = (0.5, 0.378). The figure was prepared using FFT from the CCP4 suite.
Acknowledgments
We thank the National Chiao Tung University, the MOE ATU program and the National Science Council (NSC-98-2627-M-009-007 and NSC-98-2627-M-009-008) for financial support of this research. We also thank the support staff at NSRRC beamlines BL13B1 and BL13C1 for their help and technical assistance during X-ray data collection and processing.
References
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Fukui, T., Shiraki, K., Hamada, D., Hara, K., Miyata, T., Fujiwara, S., Mayanagi, K., Yanagihara, K., Iida, T., Fukusaki, E., Imanaka, T., Honda, T. & Yanagihara, I. (2005). Biochemistry, 44, 9825–9832. [DOI] [PubMed]
- Goshima, K., Honda, T., Hirata, M., Kikuchi, K., Takeda, Y. & Miwatani, T. (1977). J. Mol. Cell. Cardiol. 9, 191–213. [DOI] [PubMed]
- Hamada, D., Higurashi, T., Mayanagi, K., Miyata, T., Fukui, T., Iida, T., Honda, T. & Yanagihara, I. (2007). J. Mol. Biol. 365, 187–195. [DOI] [PubMed]
- Hickman, F. W., Farmer, J. J. III, Hollis, D. G., Fanning, G. R., Steigerwalt, A. G., Weaver, R. E. & Brenner, D. J. (1982). J. Clin. Microbiol. 15, 395–401. [DOI] [PMC free article] [PubMed]
- Hinestrosa, F., Madeira, R. G. & Bourbeau, P. P. (2007). J. Clin. Microbiol. 45, 3462–3463. [DOI] [PMC free article] [PubMed]
- Honda, T. & Iida, T. (1993). Rev. Med. Microbiol. 4, 106–113.
- Honda, T., Ni, Y., Miwatani, T., Adachi, T. & Kim, J. (1992). Can. J. Microbiol. 38, 1175–1180. [DOI] [PubMed]
- Lang, P. A., Kaiser, S., Myssina, S., Birka, C., Weinstock, C. & Northoff, H. (2004). Cell. Microbiol. 6, 391–400. [DOI] [PubMed]
- Lowry, P. W., McFarland, L. M. & Threefoot, H. K. (1986). J. Infect. Dis. 154, 730–731. [DOI] [PubMed]
- Miyamoto, Y., Kato, T., Obara, Y., Akiyama, S., Takizawa, K. & Yamai, S. (1969). J. Bacteriol. 100, 1147–1149. [DOI] [PMC free article] [PubMed]
- Morris, J. G. Jr, Wilson, R., Hollis, D. G., Weaver, R. E., Miller, H. G., Tacket, C. O., Hickman, F. W. & Blake, P. A. (1982). Lancet, 319, 1294–1296. [DOI] [PubMed]
- Naim, R., Yanagihara, I., Iida, T. & Honda, T. (2001). FEMS Microbiol. Lett. 195, 237–244. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Parker, M. W. & Feil, S. C. (2005). Prog. Biophys. Mol. Biol. 88, 91–142. [DOI] [PubMed]
- Raimondi, F., Kao, J. P. Y., Fiorentini, C., Fabbri, A., Donelli, G. & Gasparini, N. (2000). Infect. Immun. 68, 3180–3185. [DOI] [PMC free article] [PubMed]
- Sakurai, J., Honda, T., Junguji, Y., Arita, M. & Miwatani, T. (1976). Infect. Immun. 13, 876–883. [DOI] [PMC free article] [PubMed]
- Tang, G., Iida, T., Inoue, H., Yutsudo, M., Yamamoto, K. & Honda, T. (1997). Biochim. Biophys. Acta, 1360, 277–282. [DOI] [PubMed]
- Tang, G.-Q., Iida, T., Yamamoto, K. & Honda, T. (1995). FEMS Microbiol. Lett. 134, 233–238. [DOI] [PubMed]
- Thompson, F. L., Hoste, B., Vandemeulebroecke, K. & Swings, J. (2003). Int. J. Syst. Evol. Microbiol. 53, 1615–1617. [DOI] [PubMed]
- Yanagihara, I., Nakahira, K., Yamane, T., Kaieda, S., Mayanagi, K., Hamada, D., Fukui, T., Ohnishi, K., Kajiyama, S., Shimizu, T., Sato, M., Ikegami, T., Ikeguchi, M., Honda, T. & Hashimoto, H. (2010). J. Biol. Chem. 285, 16267–16274. [DOI] [PMC free article] [PubMed]
- Vogt, G., Woell, S. & Argos, P. (1997). J. Mol. Biol. 269, 631–643. [DOI] [PubMed]
- Yoh, M., Honda, T. & Miwatani, T. (1986). Can. J. Microbiol. 32, 632–636. [DOI] [PubMed]
- Yoh, M., Honda, T., Miwatani, T., Tsunasawa, S. & Sakiyama, F. (1989). J. Bacteriol. 171, 6859–6861. [DOI] [PMC free article] [PubMed]