

A disulfide cross-linked complex between bovine poly(A) polymerase and a chemically modified RNA was crystallized. X-ray diffraction data were collected to 2.25 Å resolution from crystals that belonged to space group P2.
Keywords: mRNA 3′ processing, protein–RNA complexes, poly(A) polymerase, disulfide cross-linking, oligo(A) RNA
Abstract
Poly(A) polymerase (PAP) synthesizes the polyadenine tail at the 3′-end of messenger RNA. A disulfide cross-linking strategy was implemented to obtain a complex between bovine PAP (bPAP) and a 15-mer oligo(A). All seven endogenous cysteines were mutated to eliminate nonspecific cross-linked complexes. A cysteine residue was introduced at several different positions and A152C was found to achieve maximum specific cross-linking efficiency. The resulting bPAP construct was active and, when mixed with a chemically modified RNA, yielded crystals of a bPAP–RNA complex. The crystals, which belonged to space group P2 and harbored two protein–RNA complexes per asymmetric unit, diffracted X-rays to 2.25 Å resolution.
1. Introduction
Poly(A) polymerase (PAP) is an essential protein factor in the maturation of eukaryotic mRNA (Mandel et al., 2008 ▶). After endonucleolytic cleavage of the 3′-untranslated region of mRNA, PAP adds a polyadenine tail to the 3′-end of mRNA (Mandel et al., 2008 ▶). Similar to type I CCA transferase (Xiong & Steitz, 2004 ▶), an enzyme which also belongs to the DNA polymerase β superfamily, PAP synthesizes the poly(A) tail in the 5′-to-3′ direction in a template-independent manner. Kinetics experiments (Balbo et al., 2005 ▶), together with the crystal structures of PAP bound to an ATP analog (Balbo et al., 2007 ▶; Bard et al., 2000 ▶; Martin et al., 2000 ▶, 2004 ▶) and of yeast PAP–5-mer oligo(A) complexes (Balbo & Bohm, 2007 ▶), suggested an induced-fit mechanism for the specific recognition of adenine through open/closed states similar to those described in DNA polymerases (Doublié et al., 1998 ▶, 1999 ▶). The translocation mechanism, on the other hand, has yet to be elucidated. Intriguingly, a mechanism in which the RNA loops or wraps around the polymerase during translocation has been described for a noncanonical PAP, VP55, from vaccinia virus (Li et al., 2009 ▶). A crystal structure of bPAP in complex with a long RNA primer would provide a framework to investigate the translocation mechanism in canonical PAPs. Multiple attempts at cocrystallizing PAP with RNA oligomers of different lengths failed to produce well diffracting crystals. Soaking RNA into preformed crystals was equally unsuccessful. A disulfide cross-linking approach has been applied in the successful crystallization of several protein–DNA complexes (Qi et al., 2009 ▶; Corn & Berger, 2007 ▶; Ho et al., 2006 ▶; Banerjee & Verdine, 2006 ▶; Banerjee et al., 2006 ▶; Buck & Wells, 2005 ▶; Fromme et al., 2004 ▶; Sarafianos et al., 2003 ▶; He & Verdine, 2002 ▶; Huang et al., 1998 ▶). The basic strategy is to use a modified thiolated DNA base with engineered surface cysteines to form a stable Cys–thio-DNA covalent link (Verdine & Norman, 2003 ▶). Here, we present for the first time a strategy to obtain a disulfide cross-linked complex between a protein and an RNA oligomer. In the following, we describe the preparation, crystallization and X-ray diffraction analysis of a complex of a PAP variant and a modified 15-mer oligo(A).
2. Materials and methods
2.1. Preparation, expression and purification of bPAP513 and mutants
The wild-type C-terminal truncation construct bPAP513 (residues 1–513) was cloned into pGM10 vector with a His6 tag at the N-terminus (Martin & Keller, 1996 ▶). bPAP513 retains a catalytic activity similar to that of the full-length enzyme, which comprises 739 residues; the C-terminal region (residues 514–739) was omitted because it is disordered (Martin et al., 2000 ▶). DNA primers for mutagenesis were purchased from IDT (Illinois, USA). bPAP513 mutants were made using a QuikChange II XL mutagenesis kit (Stratagene) as described previously (Yang et al., 2010 ▶) and were confirmed by DNA sequencing. The bPAP513 variants were transformed into Rosetta (DE3) pLysS cells (Novagen) and expressed using autoinduction (Studier, 2005 ▶). bPAP513 variants were purified to homogeneity using Ni–NTA resin (Qiagen), HiTrap heparin, HiTrap Blue and HiTrap Q columns on an ÄKTApurifier system (all from GE Healthcare) according to a previously described protocol (Martin & Keller, 1996 ▶). Proteins were dialyzed into buffer consisting of 20 mM Tris–HCl pH 8.0, 100 mM KCl and 10%(v/v) glycerol to eliminate any dithiothreitol (DTT) used in previous purification steps. Protein variants were concentrated to 5 mg ml−1 using an Amicon YM-10 concentrator (Millipore), flash-frozen in liquid N2 and stored at 193 K.
2.2. Synthesis, modification and purification of oligo(A) RNA
One 15-mer oligomer, 5′-(A*)12-X-A*-A-3′, was ordered from TriLink BioTechnologies (San Diego, California, USA). The other 15-mer, 5′-(A*)13-X-A-3′, and two 14-mer oligomers, 5′-(A*)11-X-A*-A-3′ and 5′-(A*)12-X-A-3′, were synthesized by established methods using an ABI392 DNA/RNA synthesizer (Applied Biosystems; Pinard et al., 1999 ▶), where A* represents 2′-O-methyl A and X represents O 6-phenyl-dI. Phosphoramidites were from Glen Research Inc. (Sterling, Virginia, USA). The oligomers were deprotected as described previously (Wincott et al., 1995 ▶). After deprotection, the lyophilized oligomers were subsequently dissolved in 1 ml 1 M cystamine pH 12 and incubated at 333 K for 18 h. The solution was cooled to room temperature and neutralized with glacial acetic acid. The solution was passed through a HiTrap Q column on an ÄKTApurifier system using a NaCl gradient containing 10 mM NaOH. 10 mM HCl was subsequently added to individual fractions. The purity of the fractions was checked by γ-32P isotope labeling and visualization on a 20% denaturing polyacrylamide gel. Fractions containing pure modified RNA were pooled, dialyzed against H2O, lyophilized and subsequently dissolved in H2O to a final concentration of 1 mM and stored at 193 K.
2.3. Cross-linking and purification of the bPAP513–oligo(A) cross-linked complex
The protein–RNA complex was formed by mixing a bPAP513 variant with the modified 15-mer oligo(A) in a 1:1.2 molar ratio. The cross-linking reaction was carried out in a buffer consisting of 20 mM Tris–HCl pH 8.0, 30 mM KCl, 10%(v/v) glycerol and 0.2 mM MnCl2 for 16 h at 293 K. The cross-linked complex was purified using a MonoQ column (GE Healthcare) and a KCl gradient containing 20 mM Tris–HCl pH 8.0 and 10%(v/v) glycerol. The purified complex was then concentrated to ∼3 mg ml−1 using an Amicon YM-10 concentrator (Millipore), flash-frozen in liquid N2 and stored at 193 K.
2.4. Crystallization, data collection and processing
96-well crystallization plates (Corning) were set up with a Gilson 925 PC Workstation by mixing 0.6 µl of the protein–RNA complex at two concentrations (1.5 and 3 mg ml−1) with 0.6 µl crystallization solution from the following commercial crystallization screen kits: Crystal Screen HT, Index HT, PEG/Ion HT, SaltRx (Hampton Research), Wizard I and II (Emerald BioSystems) and ProPlex (Molecular Dimensions). However, no crystals were obtained after ∼12 months, at which point the plates had completely dried out. Crystallization experiments were then set up manually in 24-well VDX plates (Hampton Research) using the alternative-reservoir hanging-drop vapor-diffusion method (Newman, 2005 ▶; Dunlop & Hazes, 2005 ▶). The purified protein–RNA complex was mixed with the crystallization solution in a 1:1 volume ratio. Instead of the crystallization solution, 0.5 ml 2 M NaCl was deposited into the reservoirs. Crystal needle clusters were initially identified using both PEG/Ion screen and Crystal Screen (Hampton Research), with the exception of condition No. 36 of the PEG/Ion screen, which yielded individual needles. Further optimization was performed by several rounds of hanging-drop experiments in which the volume of NaCl in the reservoir, the precipitant concentration, the pH and divalent-ion additives were varied. Crystals appeared in one week and grew to up to 0.5 mm within three weeks at 293 K. Crystals were cryoprotected during the course of crystal growth by equilibration against a 2 M NaCl reservoir solution so that no further procedure was required prior to cryocooling. Diffraction data were collected from a single crystal at 100 K at a wavelength of 1.5418 Å on a MAR345 detector (MAR Research) mounted on a Rigaku RU-200 rotating-anode generator equipped with Xenocs focusing mirrors. The diffraction data were indexed using XDS (Kabsch, 2010 ▶) and scaled with XSCALE. Diffraction statistics are shown in Table 1 ▶.
Table 1. X-ray data-collection and processing statistics.
Values in parentheses are for the highest resolution shell.
| Wavelength (Å) | 1.5418 |
| Space group | P2 |
| Unit-cell parameters (Å, °) | a = 98.44, b = 56.04, c = 132.54, α = γ = 90.00, β = 102.99 |
| Resolution (Å) | 20–2.25 (2.3–2.25) |
| Rmerge (%)† | 8.2 (37.3) |
| Total reflections | 294620 |
| Unique reflections | 66007 |
| 〈I/σ(I)〉 | 14.18 (4.61) |
| Completeness (%) | 97.8 (94.8) |
| Multiplicity | 4.5 (3.5) |
R
merge = 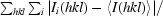
 , where Ii(hkl) is the observed intensity and 〈I(hkl)〉 is the average intensity for multiple measurements.
, where Ii(hkl) is the observed intensity and 〈I(hkl)〉 is the average intensity for multiple measurements.
3. Results and discussion
3.1. Strategy to obtain a disulfide cross-linked protein–RNA complex
Successful crystallizations of disulfide cross-linked protein–DNA complexes have been reported in several instances (Ho et al., 2006 ▶; Huang et al., 1998 ▶; Qi et al., 2009 ▶; Verdine & Norman, 2003 ▶; Corn & Berger, 2007 ▶). For this work, we adapted the cross-linking protocol to obtain a disulfide cross-linked PAP–RNA complex. Structural comparisons between bovine poly(A) polymerase (bPAP; Martin et al., 2000 ▶, 2004 ▶) and the yeast poly(A) polymerase–RNA complex (Balbo & Bohm, 2007 ▶) and between bPAP and the class I CCA-transferase–RNA complex (Xiong & Steitz, 2004 ▶) indicated that residues Ala152, Phe153 and Val154 of bPAP would be attractive candidates for introducing a cysteine mutation, since these residues lie outside the active site yet are predicted to be in close proximity to an RNA base. Of these three residues, the A152C variant yielded the highest cross-linking efficiency (Fig. 1 ▶). We previously determined using gel-shift assays that the RNA should have a minimal length of 14 nucleotides in order to form a complex with bovine PAP. Moreover, RNA oligomers longer than 21-mer were shown to supershift, indicating the possibility that more than one polymerase was bound. For this study, we therefore focused on 14-mer and 15-mer RNA oligomers. A significant hurdle in making RNA containing a single tethered thiol is that the chemical modification requires high pH (pH 12.0) and high temperature (333 K), which is a lethal combination for RNA. 2′-O-Methyl RNA was therefore used instead of the naturally occurring 2′-OH to prevent hydrolysis. The base at the 3′-end of the oligomer was not modified. Two 14-mer and two 15-mer RNAs with a modified base located at either the second or third position from the 3′-end were designed. Biochemical assays indicated that bPAP513 binds all four 2′-OMe-modified RNA oligomers, all of which act as substrates for polyadenylation under reducing conditions (data not shown). The A152C variant cross-linked with a 15-mer RNA containing a modified base at the antepenultimate position [5′-(A*)12-X-A*-A-3′) with the highest efficiency (∼70%) and was therefore selected for use in crystallization trials.
Figure 1.

Analysis of cross-linking reactions using various bPAP variants. A 15% SDS–PAGE gel is shown. The molecular weight of the His6-bPAP513 variants is ∼59 kDa and that of the 15-mer poly(A) RNA is ∼6 kDa. Lane M, protein molecular-weight markers (kDa); lane 1, 5S2V; lane 2, wild-type bPAP513; lanes 3 and 6, 5S2V A152C; lane 4, V154C; lane 5, A152C; lane 7, dissolved crystals with 5 mM DTT; lane 8, dissolved crystals without reducing reagent. In lanes 3 and 6 the lower band corresponds to the protein only and the upper band corresponds to the cross-linked protein–RNA complex.
3.2. Purification of a homogenous protein–RNA complex
A sharp peak corresponding to the bPAP513 A152C–RNA complex was observed on a chromatogram corresponding to the MonoQ purification step, but an SDS–PAGE gel revealed the presence of several bands. This result suggested that endogenous cysteine residues were able to establish nonspecific cross-links with the modified RNA, which was indeed the case, as shown in Fig. 1 ▶. Owing to the heterogeneous nature of the bPAP513 A152C–RNA complex, crystallization trials did not produce any crystals. To overcome this problem, all seven endogenous cysteines of bPAP513 were mutated. Five cysteines located at the surface (Cys36, Cys160, Cys197, Cys257 and Cys293) were mutated to serines, whereas the other two (Cys118 and Cys204), which were surrounded by hydrophobic residues, were mutated to valines (this construct will be referred to as 5S2V in the following). 5S2V is able to bind RNA at a level similar to that of wild-type PAP (data not shown), but no longer forms nonspecific cross-links with the modified RNA (Fig. 1 ▶). A construct in which all seven cysteines were mutated to serines nearly abolished RNA binding (data not shown). A homogenous cross-linked complex was obtaining by introducing the A152C mutation into the 5S2V construct (Fig. 1 ▶).
3.3. Crystallization and cryoprotection of the 5S2V A152C–RNA complex
Crystallization experiments were initially set up using conventional vapor-diffusion methods in which either sitting drops or hanging drops were equilibrated with crystallization solutions. However, no crystals were obtained from any of the commercially available crystallization screen kits tested, even after ∼12 months. Intrigued by the previously reported alternative-reservoir method (Dunlop & Hazes, 2005 ▶; Newman, 2005 ▶), we opted to use 2 M NaCl as the reservoir solution; crystals were subsequently observed using PEG/Ion screen condition Nos. 20, 25, 28, 32, 33, 36, 40 and 41; hence, 2 M NaCl was used as the reservoir solution for the subsequent optimization work. After several rounds of optimization based on condition No. 36, large single crystals (0.4 × 0.08 × 0.08 mm; Fig. 2 ▶) were obtained for the 5S2V A152C–RNA complex from a hanging drop made up of 1 µl protein–RNA complex (3 mg ml−1) and 1 µl crystallization solution consisting of 120 mM sodium tartrate, 14 mM MnCl2, 9.5%(w/v) PEG 8000 and 100 mM HEPES pH 7.0 equilibrated against 500 µl 2 M NaCl reservoir at 293 K. Crystals appeared in one week and grew to full size in 2–3 weeks. The protein–RNA complex remained intact in the crystal, which was verified by loading dissolved crystals onto an SDS–PAGE gel (Fig. 1 ▶). We found that after equilibrating against 2 M NaCl over the period of crystal growth the crystals were already cryoprotected and no additional cryoprotectant solution was needed.
Figure 2.

Crystal of the bPAP513 5S2V A152C–15-mer modified oligo(A) complex. The dimensions of this crystal are about 0.4 × 0.08 × 0.08 mm
3.4. Initial X-ray diffraction analysis
A complete data set was collected from a crystal which diffracted to 2.25 Å resolution. The data were indexed in the monoclinic P2 space group with good statistics (Table 1 ▶). No systematically absent reflections were observed, suggesting that the space group is P2, with unit-cell parameters a = 98.44, b = 56.04, c = 132.54 Å, β = 102.99°. The calculated Matthews coefficient (V M) of 2.97 Å3 Da−1, with a corresponding solvent content of 58.6%, suggests the presence of two protein–RNA complexes per asymmetric unit (Matthews, 1968 ▶). We plan to solve the complex structure by molecular replacement using the previously published high-resolution bPAP513 structure (Martin et al., 2004 ▶) as a search model. If molecular replacement were to fail, we will engineer the selenomethionyl variant of the enzyme (Doublié, 2007 ▶) and screen for heavy-atom derivatives (Dauter & Dauter, 2007 ▶).
Acknowledgments
We are grateful to K. Zahn and Drs P. Aller, M. Rould and K. Imamura for discussions and assistance with data collection. We thank G. Martin and Dr W. Keller for the bovine PAP plasmid and for discussions and M. Fay and Dr J. Burke for help with RNA preparation. This work was supported by NIH award GM62239 (to SD).
References
- Balbo, P. B. & Bohm, A. (2007). Structure, 15, 1117–1131. [DOI] [PMC free article] [PubMed]
- Balbo, P. B., Meinke, G. & Bohm, A. (2005). Biochemistry, 44, 7777–7786. [DOI] [PubMed]
- Balbo, P. B., Toth, J. & Bohm, A. (2007). J. Mol. Biol. 366, 1401–1415. [DOI] [PMC free article] [PubMed]
- Banerjee, A., Santos, W. L. & Verdine, G. L. (2006). Science, 311, 1153–1157. [DOI] [PubMed]
- Banerjee, A. & Verdine, G. L. (2006). Proc. Natl Acad. Sci. USA, 103, 15020–15025. [DOI] [PMC free article] [PubMed]
- Bard, J., Zhelkovsky, A. M., Helmling, S., Earnest, T. N., Moore, C. L. & Bohm, A. (2000). Science, 289, 1346–1349. [DOI] [PubMed]
- Buck, E. & Wells, J. A. (2005). Proc. Natl Acad. Sci. USA, 102, 2719–2724. [DOI] [PMC free article] [PubMed]
- Corn, J. E. & Berger, J. M. (2007). Structure, 15, 773–780. [DOI] [PMC free article] [PubMed]
- Dauter, M. & Dauter, Z. (2007). Methods Mol. Biol. 364, 149–158. [DOI] [PubMed]
- Doublié, S. (2007). Methods Mol. Biol. 363, 91–108. [DOI] [PubMed]
- Doublié, S., Sawaya, M. R. & Ellenberger, T. (1999). Structure, 7, R31–R35. [DOI] [PubMed]
- Doublié, S., Tabor, S., Long, A. M., Richardson, C. C. & Ellenberger, T. (1998). Nature (London), 391, 251–258. [DOI] [PubMed]
- Dunlop, K. V. & Hazes, B. (2005). Acta Cryst. D61, 1041–1048. [DOI] [PubMed]
- Fromme, J. C., Banerjee, A., Huang, S. J. & Verdine, G. L. (2004). Nature (London), 427, 652–656. [DOI] [PubMed]
- He, C. & Verdine, G. L. (2002). Chem. Biol. 9, 1297–1303. [DOI] [PubMed]
- Ho, W. C., Fitzgerald, M. X. & Marmorstein, R. (2006). J. Biol. Chem. 281, 20494–20502. [DOI] [PubMed]
- Huang, H., Chopra, R., Verdine, G. L. & Harrison, S. C. (1998). Science, 282, 1669–1675. [DOI] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Li, C., Li, H., Zhou, S., Sun, E., Yoshizawa, J., Poulos, T. L. & Gershon, P. D. (2009). Structure, 17, 680–689. [DOI] [PubMed]
- Mandel, C. R., Bai, Y. & Tong, L. (2008). Cell. Mol. Life Sci. 65, 1099–1122. [DOI] [PMC free article] [PubMed]
- Martin, G. & Keller, W. (1996). EMBO J. 15, 2593–2603. [PMC free article] [PubMed]
- Martin, G., Keller, W. & Doublié, S. (2000). EMBO J. 19, 4193–4203. [DOI] [PMC free article] [PubMed]
- Martin, G., Möglich, A., Keller, W. & Doublié, S. (2004). J. Mol. Biol. 341, 911–925. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Newman, J. (2005). Acta Cryst. D61, 490–493. [DOI] [PubMed]
- Pinard, R., Heckman, J. E. & Burke, J. M. (1999). J. Mol. Biol. 287, 239–251. [DOI] [PubMed]
- Qi, Y., Spong, M. C., Nam, K., Banerjee, A., Jiralerspong, S., Karplus, M. & Verdine, G. L. (2009). Nature (London), 462, 762–766. [DOI] [PMC free article] [PubMed]
- Sarafianos, S. G., Clark, A. D. Jr, Tuske, S., Squire, C. J., Das, K., Sheng, D., Ilankumaran, P., Ramesha, A. R., Kroth, H., Sayer, J. M., Jerina, D. M., Boyer, P. L., Hughes, S. H. & Arnold, E. (2003). J. Biol. Chem. 278, 16280–16288. [DOI] [PubMed]
- Studier, F. W. (2005). Protein Expr. Purif. 41, 207–234. [DOI] [PubMed]
- Verdine, G. L. & Norman, D. P. (2003). Annu. Rev. Biochem. 72, 337–366. [DOI] [PubMed]
- Wincott, F., DiRenzo, A., Shaffer, C., Grimm, S., Tracz, D., Workman, C., Sweedler, D., Gonzalez, C., Scaringe, S. & Usman, N. (1995). Nucleic Acids Res. 23, 2677–2684. [DOI] [PMC free article] [PubMed]
- Xiong, Y. & Steitz, T. A. (2004). Nature (London), 430, 640–645. [DOI] [PubMed]
- Yang, Q., Gilmartin, G. M. & Doublié, S. (2010). Proc. Natl Acad. Sci. USA, 107, 10062–10067. [DOI] [PMC free article] [PubMed]