

Crystallization conditions are described for the cis- and trans-imide bond-containing SH2 domain of IL-2-inducible T-cell kinase.
Keywords: proline cis–trans isomerization, prolyl imide bond, IL-2-inducible T-cell kinase, SH2 domain
Abstract
Proline is a unique amino acid owing to the relatively small energy difference between the cis and trans conformations of its peptide bond. The X–Pro imide bond readily undergoes cis–trans isomerization in the context of short peptides as well as some proteins. However, the direct detection of cis–trans proline isomerization in folded proteins is technically challenging. NMR spectroscopy is well suited to the direct detection of proline isomerization in folded proteins. It is less clear how well X-ray crystallography can reveal this conformational exchange event in folded proteins. Conformational heterogeneity owing to cis–trans proline isomerization in the Src homology 2 (SH2) domain of the IL-2-inducible T-cell kinase (ITK) has been extensively characterized by NMR. Using the ITK SH2 domain as a test system, an attempt was made to determine whether proline isomerization could be detected in a crystal structure of the ITK SH2 domain. As a first step towards this goal, the purification, crystallization and preliminary characterization of the ITK SH2 domain are described.
1. Introduction
Planar peptide bonds within folded proteins show an overwhelming preference for the trans conformation (Stewart et al., 1990 ▶). One notable exception to this conformational preference is the X–proline imide bond (where X is any amino acid), which can adopt either the cis or trans conformation owing to the small energy difference (2.09 kJ mol−1) between the cis and trans conformers (Maigret et al., 1970 ▶). A survey of protein structures shows that ∼5.2% of X–proline imide bonds occur in the cis conformation, compared with ∼0.03% of nonproline peptide bonds (Weiss et al., 1998 ▶). Additionally, the activation-energy barrier for cis–trans isomerization about an X–proline imide bond is lower than that for a nonproline peptide bond (54.43 versus 83.74 kJ mol−1, respectively; Schulz & Schirmer, 1979 ▶; Jorgensen & Gao, 1988 ▶; Schnur et al., 1989 ▶). Thus, a proline-containing polypeptide has the ability to exist in either the cis or trans conformation about the X–proline imide bond or in some cases exist in an equilibrium that consists of both the cis and trans conformers (Andreotti, 2003 ▶).
It has long been appreciated that proline isomerization plays a role in controlling the rate of protein folding (Wedemeyer et al., 2002 ▶; Schmid et al., 1993 ▶). More recently, this conformational exchange event has been shown to modulate a variety of processes, including ion-channel gating (Lummis et al., 2005 ▶), histone lysine methylation (Nelson et al., 2006 ▶), phage infectivity (Eckert et al., 2005 ▶), ligand-binding specificity (Breheny et al., 2003 ▶; Santiveri et al., 2004 ▶), enzyme function (Grochulski et al., 1994 ▶; OuYang et al., 2008 ▶), amyloid plaque formation (Pastorino et al., 2006 ▶; Eakin et al., 2006 ▶) and cell signaling (Brazin et al., 2002 ▶; Zhou et al., 2000 ▶; Wulf et al., 2005 ▶; Yaffe et al., 1997 ▶; Fischer & Aumüller, 2003 ▶).
While the biological significance of proline isomerization is becoming evident, the detection of proline isomerization in proteins is still technically challenging. Proline isomerization evades detection by most biochemical techniques. In fact, in most low-resolution crystal structures of proteins the X–proline imide-bond conformer is presumed to adopt the trans conformation. It is therefore possible that proline isomerization is underestimated among the protein structures available in the Protein Data Bank. The cis- or trans-proline imide-bond conformers of several proteins have been crystallized separately under different crystallization conditions (Cotton et al., 1979 ▶; Hynes & Fox, 1991 ▶; Loll & Lattman, 1989 ▶; Szebenyi & Moffat, 1986 ▶). However, the simultaneous detection of both proline imide-bond conformers of a protein in a given crystal structure has been rare (Svensson et al., 1992 ▶). Moreover, since isomerizing proline residues are frequently present in flexible loops, the poor electron density corresponding to these regions potentially obscures the underlying conformational heterogeneity (Feng et al., 1997 ▶; Mallis et al., 2002 ▶; Grochulski et al., 1994 ▶; Golmohammadi et al., 1993 ▶).
Conformational heterogeneity owing to an isomerizing proline (Pro287) within the Src homology 2 (SH2) domain of the IL-2-inducible T-cell kinase (ITK) has been extensively characterized by NMR (Mallis et al., 2002 ▶; Breheny et al., 2003 ▶; Pletneva et al., 2006 ▶; Severin et al., 2009 ▶). Both the cis- and trans-imide bond-containing SH2-domain structures have been solved from a single NMR data set (Mallis et al., 2002 ▶). Moreover, cis–trans isomerization about the ITK SH2 Asn286–Pro287 imide bond has been shown to direct the ligand-binding preference of the ITK SH2 domain, which may have functional implications in T-cell signaling (Breheny et al., 2003 ▶). The trans-imide bond-containing conformer of the ITK SH2 domain has higher affinity for a classical phosphotyrosine-containing ligand (Pletneva et al., 2006 ▶). The cis-imide bond-containing conformer of the ITK SH2 domain mediates a nonclassical interaction with the ITK SH3 domain (Severin et al., 2009 ▶). Given that the ITK SH2 domain adopts two interconverting conformers in solution, we decided to investigate the extent to which this conformational heterogeneity can be observed in a structure of the ITK SH2 domain derived from X-ray crystallographic approaches. The description of the initial crystallization conditions of the ITK SH2 domain provided here is the first step towards addressing this question.
2. Materials and methods
2.1. Cloning and expression
The construct for the ITK SH2 domain (ITK; gene ID 16428) has been described previously (Brazin et al., 2000 ▶). Briefly, the mouse ITK SH2 domain (residues 231–338) was cloned into the pGEX-2T vector (GE Healthcare) with an N-terminal (vector-derived) GST tag and a thrombin cleavage site between the GST tag and the ITK SH2 domain. The construct was verified by sequencing at the Iowa State DNA Sequencing and Synthesis Facility prior to transformation into Escherichia coli strain BL21 (DE3) cells (Novagen). For protein expression, cells were grown in LB medium containing 100 µg ml−1 ampicillin at 310 K until the optical density of the culture at 600 nm reached 0.7. The temperature of the culture was then lowered to 293 K and it was induced with 1 mM β-d-1-thiogalactopyranoside (IPTG) for 24 h. The cells were harvested and resuspended in lysis buffer (50 mM HEPES pH 7.4, 75 mM NaCl, 2 mM DTT, 0.02% NaN3) with 0.5 mg ml−1 lysozyme and stored at 193 K. To produce selenomethionyl (SeMet) labeled ITK SH2 domain, cells were grown in modified M9 medium containing l-selenomethionine (Calbiochem) as described previously (Van Duyne et al., 1993 ▶). The incorporation of l-selenomethionine was confirmed by MALDI–TOF mass-spectrometric analysis of the tryptic digests of the labeled protein at the Iowa State Protein Facility.
2.2. Protein purification
The ITK SH2 domain was purified as described previously with several modifications (Brazin et al., 2000 ▶). The cells were lysed upon thawing at room temperature by the addition of protease inhibitor (1 mM PMSF) and DNase I (Sigma; 50 µl of 10 mg ml−1 stock). The cell lysate was clarified by centrifugation and the resulting supernatant was loaded onto two 5 ml glutathione–agarose columns (Sigma). Each column was washed with 200 ml lysis buffer (50 mM HEPES pH 7.4, 75 mM NaCl, 2 mM DTT, 0.02% NaN3). The GST-fusion protein was eluted in lysis buffer containing 10 mM glutathione. The protein was concentrated and buffer-exchanged in order to remove the glutathione and was then cleaved overnight with thrombin (Calbiochem) at room temperature. After cleavage, the protein was passed over two 5 ml glutathione–agarose columns to remove GST, concentrated and loaded onto a gel-filtration column (a C26/100 column packed with Sephacryl S-100 HR resin; GE Healthcare) equilibrated with the same buffer (50 mM HEPES pH 7.4, 75 mM NaCl, 2 mM DTT, 0.02% NaN3). Fractions containing protein were analyzed for purity by SDS–PAGE; the pure fractions were pooled and the salt concentration of the buffer was increased from 75 to 150 mM NaCl. The pooled protein was then passed over a 10 ml benzamidine column (GE Healthcare) equilibrated with the same buffer (50 mM HEPES pH 7.4, 150 mM NaCl, 2 mM DTT, 0.02% NaN3). The purified ITK SH2 domain was concentrated to 13.5 mg ml−1 in the same buffer (50 mM HEPES pH 7.4, 150 mM NaCl, 2 mM DTT, 0.02% NaN3) and filter-sterilized prior to setting up crystal trays. The final purified ITK SH2 domain has two additional vector-derived N-terminal residues (G and S) and eight vector-derived C-terminal residues (GSPGIHRD).
2.3. Crystallization
The ideal protein concentration for crystallization screening of the ITK SH2 domain was determined to be 1.35 mg ml−1 using the PCT Pre-Crystallization test (Hampton Research). Initial screening was performed at 1.35 mg ml−1 ITK SH2 domain using Crystal Screen and Crystal Screen 2 (Hampton Research). Crystals were screened at room temperature and 277 K by the hanging-drop method in vapor-diffusion VDX plates (Hampton Research). 2 µl protein solution was mixed with an equal volume of well solution and the mixture was equilibrated against 500 µl well solution. Microcrystals were observed after 2 d at room temperature in condition No. 40 of Crystal Screen [20% 2-propanol, 0.1 M sodium citrate pH 5.6, 20%(w/v) PEG 4000]. After several rounds of buffer optimization and additional screening (Additive Screen and Detergent Screen from Hampton Research), single crystals that diffracted to 2.4 Å resolution were obtained. The final crystals were grown by mixing 3.5 µl of a 1:1 ratio of 1.35 mg ml−1 ITK SH2 domain and reservoir solution [0.1 M sodium citrate pH 5.3, 10% 2-propanol, 20%(w/v) PEG 4000 and 2 mM DTT] with 0.5 µl (12.5 mM final concentration) of a 100 mM stock solution of glycyl-glycyl-glycine (Sigma) as an additive in a final drop volume of 4 µl. All drops were equilibrated against 500 µl reservoir solution. The plate-like crystals grew to approximately 50–100 µm in two weeks and were flash-cooled directly (without additional cryoprotectants) in liquid nitrogen.
2.4. X-ray diffraction
Crystals were screened on a Rigaku R-AXIS IV++ rotating-anode/image-plate system using Cu Kα radiation and an Osmic confocal optics system at Iowa State University. Diffraction data for native and SeMet-labeled ITK SH2 domain were collected from single crystals at a crystal-to-detector distance of 140 mm on beamline 4.2.2 of the Advanced Light Source, Lawrence Berkeley Laboratory. The crystal was rotated through 180° with a 1° oscillation range per frame. For the SeMet crystals, complete anomalous data sets were obtained at wavelengths corresponding to the peak absorbance (0.9790 Å), the inflection point (0.9793 Å) and a remote wavelength (0.9951 Å) from the absorption edge of selenium. The program d*TREK (Pflugrath, 1999 ▶) was used to index, integrate, scale and merge the intensities, which were then converted to structure factors using the TRUNCATE program from CCP4 (Collaborative Computational Project, Number 4, 1994 ▶; French & Wilson, 1978 ▶).
3. Results and discussion
The ITK SH2 domain was purified to greater than 98% homogeneity (as assessed by Coomassie Blue staining of an SDS–PAGE gel) for crystallization screening (Fig. 1 ▶ a). Incorporation of SeMet in the SeMet-labeled ITK SH2 domain was confirmed by MALDI–TOF MS analysis of tryptic digests of labeled and unlabeled ITK SH2 domains. Incorporation of SeMet leads to an increase in mass of the ITK SH2-domain peptide fragment EGAFMVR from 809 to 857 Da (Figs. 1 ▶ b and 1 ▶ c, respectively). The ideal protein concentration for crystallization screening was determined to be 1.35 mg ml−1 using the PCT Pre-Crystallization test (Hampton Research). Indeed, crystallization screens (Crystal Screens and Crystal Screen 2, Hampton Research) set up using a higher ITK SH2 concentration of 13.5 mg ml−1 resulted in immediate precipitation in 90% of the conditions tested. The crystallization concentration requirement (1.35 mg ml−1) of the ITK SH2 domain could be a reflection of increased stability of the ITK SH2 domain at lower protein concentrations: while a 1.35 mg ml−1 solution of the ITK SH2 domain was stable at 277 K for several weeks, a 13.5 mg ml−1 solution of the ITK SH2 domain showed visible precipitate formation within a week at 277 K. Crystallization screens were set up at both room temperature and 277 K. Microcrystals were observed in Crystal Screen condition No. 40 [20% 2-propanol, 0.1 M sodium citrate pH 5.6, 20%(w/v) PEG 4000] in 2 d at room temperature. Microcrystals were also observed after one month at 277 K under the same buffer conditions. Larger plate-like crystals that diffracted were obtained after optimization of the buffer conditions and additional (additive) screening (Fig. 2 ▶). The final crystallization conditions were 0.1 M sodium citrate pH 5.3, 10% 2-propanol, 20%(w/v) PEG 4000, 2 mM DTT with 12.5 mM glycyl-glycyl-glycine as an additive (see §2). SeMet-incorporated ITK SH2 domain crystals were also obtained using the same crystallization conditions. The native and SeMet-labeled crystals diffracted to approximately 2.4 Å resolution (Fig. 3 ▶). The space group was determined to be I222, with unit-cell parameters a = 53.6, b = 57.4, c = 83.2 Å and a = 53.5, b = 57.4, c = 81.3 Å for the native and SeMet-labeled crystals, respectively. A summary of the data-collection statistics is provided in Table 1 ▶. The calculated Matthews coefficient (V M) of 2.35 Å3 Da−1, with a solvent content of 47.7%, suggests the presence of one molecule per asymmetric unit. Efforts to solve the structure using the multiwavelength anomalous diffraction data are currently under way.
Figure 1.

Characterization of the ITK SH2 domain. (a) Chromatogram of the Sephacryl S-100 HR gel-filtration column along with SDS–PAGE analysis of the column fractions (inset). (b, c) MALDI–TOF MS analysis of the tryptic digests of native ITK SH2 domain (b) and SeMet-labelled ITK SH2 domain (c). Incorporation of SeMet leads to an increase in the mass of the ITK SH2-domain peptide fragment EGAFMVR from 809 to 857 Da.
Figure 2.

A representative crystal of the ITK SH2 domain. The crystals are approximately 50–100 µm in size.
Figure 3.

X-ray diffraction image collected from a native crystal of the ITK SH2 domain.
Table 1. Crystal parameters and data-collection statistics.
Values in parentheses are for the outermost resolution shell.
| SeMet | ||||
|---|---|---|---|---|
| Native | Peak | Inflection | Remote | |
| No. of crystals | 1 | 1 | ||
| Beamline | 4.2.2, Advanced Light Source, Lawrence Berkeley Laboratory | |||
| Wavelength (Å) | 1.2398 | 0.9790 | 0.9793 | 0.9951 |
| Detector | NOIR-1 MBC system | |||
| Crystal-to-detector distance (mm) | 140 | 200 | ||
| Rotation range per image (°) | 1 | |||
| Total rotation range (°) | 180 | |||
| Exposure time per image (s) | 12 | 15 | ||
| Resolution (Å) | 47.22–2.35 (2.43–2.35) | 46.50–2.40 (2.49–2.40) | 46.87–2.70 (2.80–2.70) | 47.00–2.80 (2.90–2.80) |
| Space group | I222 | I222 | ||
| Unit-cell parameters (Å) | a = 53.6, b = 57.4, c = 83.2 | a = 53.5, b = 57.4, c = 81.3 | ||
| Mosaicity (°) | 2.00 | 2.07 | 2.09 | 2.05 |
| Total reflections | 29274 | 31798 | 25015 | 22686 |
| Unique reflections | 5487 | 4923 | 3636 | 3308 |
| Average multiplicity | 5.34 (4.42) | 6.46 (4.61) | 6.88 (7.12) | 6.86 (7.07) |
| Average I/σ(I) | 10.3 (4.1) | 8.4 (2.0) | 7.9 (2.1) | 9.1 (2.1) |
| Completeness (%) | 97.8 (93.1) | 97.9 (83.5) | 99.9 (100) | 99.9 (100) |
| Rmerge† (%) | 9.4 (34.7) | 9.5 (51.1) | 10.8 (56.0) | 9.5 (52.9) |
| Rmeas‡ (%) | 10.42 (39.45) | 10.33 (57.75) | 11.68 (60.40) | 10.28 (61.61) |
| Ranom§ (%) | 5.68 | 4.76 | 3.55 | |
| Overall B factor from Wilson plot (Å2) | 51.80 | 23.26 | ||
R
merge = 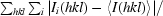
 , where Ii(hkl) is the scaled intensity of the ith reflection with indices hkl and 〈I(hkl)〉 is the mean intensity for this set of reflections.
, where Ii(hkl) is the scaled intensity of the ith reflection with indices hkl and 〈I(hkl)〉 is the mean intensity for this set of reflections.
R
meas = 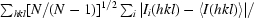 , where N is the data multiplicity.
, where N is the data multiplicity.
R
anom = 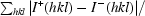
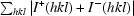 , where I
+(hkl) and I
−(hkl) are the averages of I
+(hkl) and I
−(hkl), respectively.
, where I
+(hkl) and I
−(hkl) are the averages of I
+(hkl) and I
−(hkl), respectively.
Acknowledgments
This work was supported by grants from the National Institutes of Health to AHA (National Institute of Allergy and Infectious Diseases, AI043957 and AI075150) and to RBH (NS010546). The use of the beamline at the Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences of the US Department of Energy under Contract No. DE-AC02-05CH11231.
References
- Andreotti, A. H. (2003). Biochemistry, 42, 9515–9524. [DOI] [PubMed]
- Brazin, K. N., Fulton, D. B. & Andreotti, A. H. (2000). J. Mol. Biol. 302, 607–623. [DOI] [PubMed]
- Brazin, K. N., Mallis, R. J., Fulton, D. B. & Andreotti, A. H. (2002). Proc. Natl Acad. Sci. USA, 99, 1899–1904. [DOI] [PMC free article] [PubMed]
- Breheny, P. J., Laederach, A., Fulton, D. B. & Andreotti, A. H. (2003). J. Am. Chem. Soc. 125, 15706–15707. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Cotton, F. A., Hazen, E. E. Jr & Legg, M. J. (1979). Proc. Natl Acad. Sci. USA, 76, 2551–2555. [DOI] [PMC free article] [PubMed]
- Eakin, C. M., Berman, A. J. & Miranker, A. D. (2006). Nature Struct. Mol. Biol. 13, 202–208. [DOI] [PubMed]
- Eckert, B., Martin, A., Balbach, J. & Schmid, F. X. (2005). Nature Struct. Mol. Biol. 12, 619–623. [DOI] [PubMed]
- Feng, Y., Hood, W. F., Forgey, R. W., Abegg, A. L., Caparon, M. H., Thiele, B. R., Leimgruber, R. M. & McWherter, C. A. (1997). Protein Sci. 6, 1777–1782. [DOI] [PMC free article] [PubMed]
- Fischer, G. & Aumüller, T. (2003). Rev. Physiol. Biochem. Pharmacol. 148, 105–150. [DOI] [PubMed]
- French, S. & Wilson, K. (1978). Acta Cryst. A34, 517–525.
- Golmohammadi, R., Valegård, K., Fridborg, K. & Liljas, L. (1993). J. Mol. Biol. 234, 620–639. [DOI] [PubMed]
- Grochulski, P., Li, Y., Schrag, J. D. & Cygler, M. (1994). Protein Sci. 3, 82–91. [DOI] [PMC free article] [PubMed]
- Hynes, T. R. & Fox, R. O. (1991). Proteins, 10, 92–105. [DOI] [PubMed]
- Jorgensen, W. L. & Gao, J. (1988). J. Am. Chem. Soc. 110, 4212–4216.
- Loll, P. A. & Lattman, E. E. (1989). Proteins, 5, 183–201. [DOI] [PubMed]
- Lummis, S. C., Beene, D. L., Lee, L. W., Lester, H. A., Broadhurst, R. W. & Dougherty, D. A. (2005). Nature (London), 438, 248–252. [DOI] [PubMed]
- Maigret, B., Perahia, D. & Pullman, B. (1970). J. Theor. Biol. 29, 275–291. [DOI] [PubMed]
- Mallis, R. J., Brazin, K. N., Fulton, D. B. & Andreotti, A. H. (2002). Nature Struct. Biol. 9, 900–905. [DOI] [PubMed]
- Nelson, C. J., Santos-Rosa, H. & Kouzarides, T. (2006). Cell, 126, 905–916. [DOI] [PubMed]
- OuYang, B., Pochapsky, S. S., Dang, M. & Pochapsky, T. C. (2008). Structure, 16, 916–923. [DOI] [PMC free article] [PubMed]
- Pastorino, L., Sun, A., Lu, P.-J., Zhou, X. Z., Balastik, M., Finn, G., Wulf, G., Lim, J., Li, S.-H., Li, X., Xia, W., Nicholson, L. K. & Lu, K. P. (2006). Nature (London), 440, 528–534. [DOI] [PubMed]
- Pflugrath, J. W. (1999). Acta Cryst. D55, 1718–1725. [DOI] [PubMed]
- Pletneva, E. V., Sundd, M., Fulton, D. B. & Andreotti, A. H. (2006). J. Mol. Biol. 357, 550–561. [DOI] [PubMed]
- Santiveri, C. M., Pérez-Cañadillas, J. M., Vadivelu, M. K., Allen, M. D., Rutherford, T. J., Watkins, N. A. & Bycroft, M. (2004). J. Biol. Chem. 279, 34963–34970. [DOI] [PubMed]
- Schmid, F. X., Mayr, L. M., Mücke, M. & Schönbrunner, E. R. (1993). Adv. Protein Chem. 44, 25–66. [DOI] [PubMed]
- Schnur, D. M., Yuh, Y. H. & Dalton, D. R. (1989). J. Org. Chem. 54, 3779–3785.
- Schulz, G. E. & Schirmer, R. H. (1979). Principles of Protein Structure, p. 25. New York: Springer.
- Severin, A., Joseph, R. E., Boyken, S., Fulton, D. B. & Andreotti, A. H. (2009). J. Mol. Biol. 387, 726–743. [DOI] [PMC free article] [PubMed]
- Stewart, D. E., Sarkar, A. & Wampler, J. E. (1990). J. Mol. Biol. 214, 253–260. [DOI] [PubMed]
- Svensson, L. A., Thulin, E. & Forsén, S. (1992). J. Mol. Biol. 223, 601–606. [DOI] [PubMed]
- Szebenyi, D. M. & Moffat, K. (1986). J. Biol. Chem. 261, 8761–8777. [PubMed]
- Van Duyne, G. D., Standaert, R. F., Karplus, P. A., Schreiber, S. L. & Clardy, J. (1993). J. Mol. Biol. 229, 105–124. [DOI] [PubMed]
- Wedemeyer, W. J., Welker, E. & Scheraga, H. A. (2002). Biochemistry, 41, 14637–14644. [DOI] [PubMed]
- Weiss, M. S., Jabs, A. & Hilgenfeld, R. (1998). Nature Struct. Biol. 5, 676. [DOI] [PubMed]
- Wulf, G., Finn, G., Suizu, F. & Lu, K. P. (2005). Nature Cell Biol. 7, 435–441. [DOI] [PubMed]
- Yaffe, M. B., Schutkowski, M., Shen, M., Zhou, X. Z., Stukenberg, P. T., Rahfeld, J.-U., Xu, J., Kuang, J., Kirschner, M. W., Fischer, G., Cantley, L. C. & Lu, K. P. (1997). Science, 278, 1957–1960. [DOI] [PubMed]
- Zhou, X. Z., Kops, O., Werner, A., Lu, P.-J., Shen, M., Stoller, G., Küllertz, G., Stark, M., Fischer, G. & Lu, K. P. (2000). Mol. Cell, 6, 873–883. [DOI] [PubMed]