

Activity-based protein profiling (ABPP) research is directed towards the development of tools and techniques that report on enzyme activity in complex biological samples.[1–4] With the aid of activity-based probes (ABPs)—small molecules designed to react specifically, covalently, and irreversibly with the active site residues of an enzyme or enzyme family—enzymatic activity levels are detected, rather than the protein expression levels that are measured by means of conventional proteomics techniques. A typical ABP consists of three parts: 1) a “warhead”, the reactive group that binds covalently and irreversibly to the enzyme active site, 2) a recognition element targeting the ABP to a certain enzyme (family), and 3) an affinity tag or a fluorophore for visualization and/or enrichment purposes. In most ABPs that report on enzyme activity, the reporter group is directly attached to the probe, with obvious advantages with respect to experimental design. Incorporation of, for instance, a biotin or large fluorophore in an ABP, however, might have a detrimental effect either on bioavailability (cell permeability) or on enzyme reactivity of the probe, or on both. With the aim of alleviating these problems, the two-step labeling approach is an important alternative in ABPP. We and Cravatt and co-workers simultaneously reported that this approach is also versatile in the profiling of enzyme families: namely the proteasome and serine hydrolases, respectively.[5, 6] In two-step ABPP approaches a small biocompatible reactive group, normally an azide or an acetylene, is introduced into an ABP. After covalent modification of a target protein (family), a reporter group is introduced in a chemoselective manner, by means either of Staudinger–Bertozzi ligation[6–8] or of Huisgen [2+3] cycloaddition (the “click reaction”, of which both copper(I)-catalyzed[5, 9–13] and copper-free[14,15] versions exist). Key to the success of such two-step ABPP experiments are the selectivity (in terms of cross-reactivity towards endogenous functional groups in a biological sample) and efficiency (in terms of chemical yields with which the azide- or acetylene-modified proteins are converted) of the chemoselective ligation step by which the reporter group is attached to the modified proteins. There are several reports on the selectivity of both Staudinger–Bertozzi and click ligations.[11,14] Here we describe a compatible set of one-step and two-step proteasome ABPs 4 and 6 (Scheme 1) and demonstrate that with these the efficiency of the Staudinger–Bertozzi ligation in the two-step ABPP of the proteasome catalytic activities is estimated to proceed in a quantitative fashion.
Scheme 1.

Reagents and conditions: a) N-hydroxysuccinimide, EDC, DCM, 2 h, 68 %. b) DBU, DMF, 5 min. c) HOBt, 1 min. d) 2, DiPEA, 30 min, 86 %. e) 5, 10 mol % CuSO4, 20 mol % sodium ascorbate, tBuOH/H2O 1:1, RT, 15 h, quant.
The design of probes 4 and 6 is based on the new bifunctional azido-BODIPY acid derivative 1, which can be incorporated into ABPs and subsequently functionalized either before or after enzyme labeling by both Staudinger–Bertozzi and click ligation. We have recently demonstrated that the BODIPY-TMR-modified proteasome inhibitor 8 (MV151) labels all proteasome catalytic sites both in cell lysates and in living cells.[16] The capability to introduce a biotin moiety into 4 at will at any time in the profiling experiment provides flexibility in designing the optimal ABP (one-step or two-step), depending on the nature of the ABPP experiment.
The title compound, azido-BODIPY acid 1, was synthesized by adaptation of the literature procedures for the synthesis of BODIPY-TMR[16,17] (Supporting Information) and was subsequently converted into the corresponding succinimidyl ester 2 (Scheme 1). Removal of the Fmoc protective group in the hexapeptide vinyl sulfone 3,[16] followed by condensation with azido-BODIPY-OSu 2, gave ABP 4. Copper(I)-catalyzed Huisgen [2+3] cycloaddition[9, 10] with biotin-propargylamide (5) gave rise to the fluorescent and affinity-tagged ABP 6.
Having synthesized probes 4 and 6, we assessed their ability to label the proteolytically active proteasome subunits both in cell lysates (Figure 1) and in living cells (Figure 2). EL-4 cell lysates containing both the constitutive proteasome and the immunoproteasome[18] were treated with increasing concentrations of 4 or 6 for 1 h at 37°C. The lysates treated with 4 were then exposed to biotin-phosphane 7 for 1 h at 37°C. All samples were precipitated, and their protein contents were resolved by SDS-PAGE. Direct in-gel read-out of the wet gel slabs showed uniform labeling of the proteasome catalytic subunits (β1, β2, β5, β1i, β2i, β5i) by both ABPs in a concentration-dependent manner. The observed patterns are similar to those demonstrated previously (see the labeling pattern of 8, Figure 1A lane 10 for a representative example).[16] Preincubation with epoxomicin[19,20] (Figure 1A, lane 9, Figure 1C, lane 8) abolished all labeling, which further confirms the activity-based mechanism of ABPs 4 and 6. ABP 4 appears to be slightly more reactive than its biotinylated counterpart 6 (compare Figure 1A, lanes 3–5 and Figure 1C, lanes 3–5). Quantitative Staudinger–Bertozzi ligation on proteasome subunits modified by ABP 4 is evidenced by the gel shift of those samples exposed to 100 μM biotin-phosphane 7 (Figure 1A; compare lanes 3–7 and 8). The efficiency of the ligation is also apparent when the streptavidin blots we prepared from the same gels are compared (Figures 1B and D). Again, the two patterns are highly similar, and the intensities of the signals are similar for those experiments in which we applied 10 μM concentrations of either 4 or 6 (Figures 1B and D, lanes 7).
Figure 1.
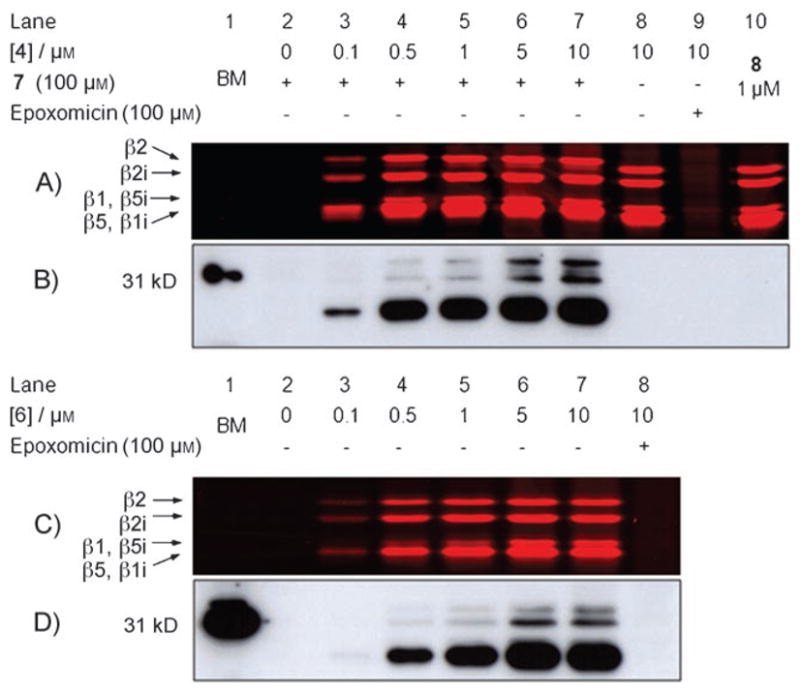
Fluorescence readout (A and C) and streptavidin blot (B and D) of labeled proteasomes in cell lysate. A) and B) EL-4 cell lysates (25 μg total protein) were treated with 4 for 1 h at 37 °C, followed by Staudinger ligation (100 μM biotin-phosphane 7) and SDS-PAGE. C) and D) EL-4 cell lysates (25 μg total protein) were treated with 6 for 1 h at 37 °C, followed by SDS-PAGE. BM = biotinylated marker.
Figure 2.
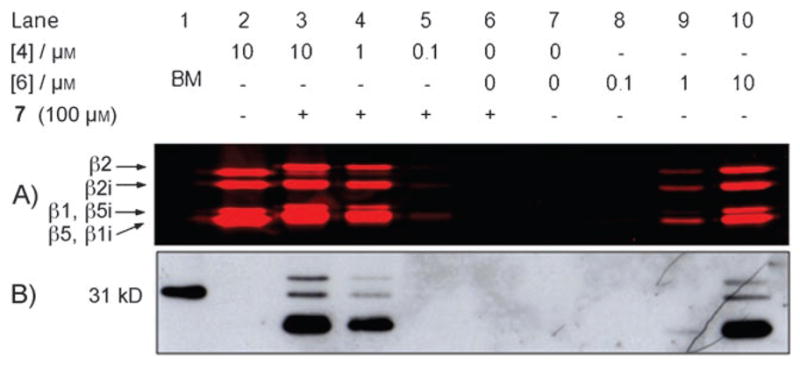
A) Fluorescence readout, and B) streptavidin blot of labeled proteasomes in living cells. Living EL4 cells were exposed to the indicated probes for 2 h at 37 °C, before being harvested and lysed. Lanes 3–6: 25 μg total protein was treated with biotin-phosphane 7 (100 μM) for 1 h at 37 °C. Lanes 7–10: 25 μg total protein was loaded on SDS-PAGE. BM = biotinylated marker.
The proteasome labeling potential of ABPs 4 and 6 in living cells was established by incubating EL-4 cells with either of the two probes at various concentrations for 2 h at 37°C. The exposed cells were harvested, washed, and lysed, and the lysates were processed as before (Figure 2). The outcomes of these experiments are highly reminiscent of those involving the ABPP labeling of lysates depicted in Figure 1. However, the main, and important, difference is found in the divergent labeling efficiency now observed for the two probes. In contrast with the proteasome profiling experiments on lysates, in which both probes appeared about equally efficient, we estimate that the two-step ABP 4 is at least five times more efficient in targeting the proteasome catalytic activities in living cells. As both probes are equally efficient in labeling proteasomes in lysates, this difference must be based on the relative cell permeabilities of the two probes.
In conclusion, we have demonstrated the versatility of the bifunctional fluorophore azido-BODIPY acid 1 as a new tool in ABPP experiments. We have established that the Staudinger–Bertozzi ligation proceeds in quantitative yield under the conditions applied here. This result essentially means that two-step ABPP may proceed with an efficiency equal to that of contemporary one-step ABPP approaches with respect to protein tagging. The efficiency thus depends on the reactivity of the ABP towards the target protein (family), and not on the chemoselective ligation employed in the second step. The advantage of two-step ABPP is evident from the results presented here demonstrating that ABP 4 is better than biotinylated analogue 6 at labeling proteasomes in living cells. We expect that BODIPY derivative 1 will be useful to the chemical biology community outside the proteasome field for several reasons. Firstly, the system presented here should be of assistance in optimizing Staudinger–Bertozzi ligation conditions, in reaction time and in the amount of phosphane used with respect to the azido modified biomolecule, for instance. Further, azido-BODIPY acid 1 can be readily transposed to different ABPP experimental settings. These include not only those directed towards the profiling of different enzyme families (entailing the incorporation of 1 into other ABPs), but also those directed towards the development or employment of other bio-orthogonal ligation strategies. An obvious extension of the work reported here is evaluation of the efficiency of the Huisgen cycloaddition reaction, but modification of the azide in 1 to encompass reaction partners for new bio-orthogonal ligations are envisaged as well. We are currently pursuing research in these directions.
Experimental Section
General
All reagents were commercial grade and were used as received unless indicated otherwise. Toluene (Tol, purum), ethyl acetate (EA, puriss.), diethyl ether, and light petroleum ether (PE, puriss.) were obtained from Riedel–de Ha+n and distilled prior to use. Dichloroethane (DCE), dichloromethane (DCM), dimethyl formamide (DMF), and dioxane (Biosolve) were stored over molecular sieves (4 Å). Methanol and N-methylpyrrolidone (NMP) were obtained from Biosolve. Tetrahydrofuran (THF, Biosolve) was distilled from LiAlH4 prior to use. Reactions were monitored by TLC analysis on DC-Alufolien (Merck, Kieselgel 60, F254), with detection variously by UV absorption (254 nm), spraying with H2SO4 in ethanol (20 %), and subsequent charring at ~150°C, by spraying with a solution of(NH4)6Mo7O24·4 H2O (25 gL−1) and (NH4)4Ce(SO4)4·2H2O (10 g L−1) in sulfuric acid (10 %), followed by charring at ~150°C, or by spraying with an aqueous solution of KMnO4 (7%) and KOH (2 %). Column chromatography was performed on Screening Devices B.V. silica gel (40–63 μm, 60 Å). LC/MS analysis was performed on a LCQ Advantage Max (Thermo Finnigan) fitted with a Gemini C18 column (Phenomenex). The applied buffers were: A) H2O, B) MeCN, and C) aq. TFA (1.0 %). HRMS were recorded on a LTQ Orbitrap (Thermo Finnigan). 1H and 13C APT-NMR spectra were recorded on Jeol JNM-FX-200 (200/50), Bruker DPX 300 (300/75 MHz), or Bruker AV400 (400/100 MHz) instruments fitted with a pulsed field gradient accessory or on a Bruker DMX 600 (600/150 MHz) instrument with a cryoprobe. Chemical shifts are given in ppm relative to tetramethylsilane as internal standard. Coupling constants are given in Hz. All 13C-APT spectra presented are proton-decoupled. UV spectra were recorded on a Perkin–Elmer Lambda 800 UV/Vis spectrometer.
Synthesis
3-{7-[4-(3-Azidopropoxy)phenyl]-4,4-difluoro-1,3-dimethyl-4-bora-3a,4a-diaza-s-indacene-2-yl}propionic acid N-succimidyl ester (2)
Azido-BODIPY acid 1 (30 mg, 64 μmol) was coevaporated thrice with toluene, before being dissolved in DCM (1 mL). After the addition of N-hydroxysuccinimide (29 mg, 0.25 mmol. 4 equiv) and EDC (48 mg, 0.25 mmol, 4 equiv), the reaction mixture was stirred for 2 h. Next, the reaction mixture was diluted with EtOAc, washed with aq. HCl (0.5 M), dried over MgSO4, and concentrated. Purification by column chromatography (0→4 % EtOAc in Tol) furnished title compound 2 (24 mg, 43 μmol, 68 %). 1H NMR (200 MHz, CDCl3): δ = 7.88 (d, J = 9.1 Hz, 2 H), 7.12 (s, 1 H), 6.97 (d, J = 8.8 Hz, 2 H), 6.96 (m, 1H), 6.56 (d, J =4.4 Hz, 1H), 4.11 (t, J =5.8 Hz, 2 H), 3.54 (t, J =6.6 Hz, 2 H), 2.79 (m, 8H), 2.56 (s, 3 H), 2.14 (s, 3H), 2.08 ppm (dt, J =6.0 Hz, 2 H); 13C NMR (50.1 MHz, CDCl3): δ = 168.98, 167.55, 159.45, 158.30, 155.91, 139.71, 135.10, 133.95, 130.65, 128.37, 128.22, 125.43, 123.12, 118.45, 114.09, 64.35, 48.07, 30.69, 28.60, 25.39, 18.96, 12.95, 9.47 ppm; HRMS: calcd for C27H27BF2N6O5H: 565.21768; found: 565.21783; for C27H27BFN6O5: 545.21145; found: 545.21130.
N3-BODIPY-Ahx3L3VS (4)
DBU (3.3 μL, 22 μmol, 1 equiv) was added to a solution of Fmoc-Ahx3L3VS (3,[16] 21.2 mg, 22 μmol) in DMF. After 5 min. of stirring, HOBt (13.4 mg, 0.1 mmol, 4.5 equiv) was added. Compound 2 (12.4 mg, 22 μmol, 1 equiv) and DiPEA (22 μL, 0.13 mmol, 6 equiv) were added to this mixture, which was then stirred for 30 min before being concentrated in vacuo. Purification by column chromatography (0.1 % TEA in DCM→3 % MeOH, 0.1 % TEA in DCM) afforded N3-Bodipy-Ahx3L3VS (4, 22.8 mg, 19 μmol, 86%). 1H NMR (400 MHz, CDCl3/MeOD): δ = 7.86 (d, J = 8.85 Hz, 2 H), 7.75–7.60 (m, 3H), 7.51–7.44 (m, 2H), 7.43–7.36 (m, 1 H), 7.28–7.22 (m, 1 H), 7.20 (s, 1H), 7.03–6.95 (m, 3H), 6.86–6.77 (m, 1H), 6.56 (m, 2 H), 4.73–4.60 (m, 1 H), 4.38–4.26 (m, 2 H), 4.13 (t, J =5.89 Hz, 2 H), 3.55 (t, J =6.62 Hz, 2H), 3.21–3.09 (m, 6 H), 2.98 (s, 3H), 2.74 (t, J = 7.43 Hz, 2H), 2.54 (s, 3 H), 2.31 (t, J =7.34 Hz, 2H), 2.27–2.20 (m, 5 H), 2.16 (t, J =7.51 Hz, 2 H), 2.13–2.05 (m, 4 H), 1.73–1.18 (m, 27 H), 1.03–0.84 ppm (m, 18H); 13C NMR (100 MHz, CDCl3/MeOD): δ = 174.52, 174.45, 174.08, 173.20, 172.75, 172.72, 172.63, 159.31, 159.11, 154.85, 147.32, 139.97, 134.64, 134.09, 130.27, 128.72, 127.62, 125.36, 122.67, 117.85, 113.82, 64.23, 51.89, 51.85, 47.58, 46.09, 42.06, 41.95, 40.06, 39.87, 39.85, 38.80, 38.69, 35.66, 35.53, 35.38, 35.33, 28.42, 28.31, 25.96, 25.91, 25.80, 24.96, 24.90, 24.80, 24.41, 24.38, 24.33, 22.33, 22.29, 22.26, 21.12, 21.08, 21.01, 19.91, 8.85, 8.10 ppm; UV/Vis: λabs = 541.94 nm, λem = 570.00 nm, ε = 62488 Lmol−1cm−1; HRMS: calcd for C61H94BF2N11O9SH: 1206.70906; found: 1206.71092; for C61H94BF2N11O9SNa: 1228.69100; found: 1228.69269; for C61H94BF2N11O9SK: 1244.66494; found: 1244.66770.
Biotin-BODIPY-Ahx3L3VS (6)
N3-BODIPY-Ahx3L3VS (4, 5.6 mg, 4.6 μmol) was dissolved in tBuOH (0.25 mL), after which aqueous solutions of CuSO4 (3.7 mM, 125 μL, 10 mol%) and sodium ascorbate (7.4 mM, 125 μL, 20 mol %) were added. The reaction mixture was stirred for 12 h, concentrated, and purified by size-exclusion chromatography (Sephadex LH-20, eluent: MeOH) to give the title compound as a brown/red solid (6.9 mg, 4.6 μmol, quant.). 1H NMR (600 MHz, CDCl3/MeOD): δ = 7.88–7.85 (m, 1 H), 7.82 (d, J = 8.74 Hz, 2H), 7.55 (s, 1H), 7.08 (d, J =3.97 Hz, 1H), 6.95 (d, J =8.77 Hz, 2 H), 6.66 (dd, J1 = 15.20 Hz, J2 = 5.02 Hz, 1H), 6.62 (d, J = 3.97 Hz, 1 H), 6.54 (d, J =15.23 Hz, 1H), 4.57–4.48 (m, 3 H), 4.34–4.27 (m, 3 H), 4.27–4.19 (m, 2H), 4.13–4.08 (m, 1 H), 4.01 (t, J =5.89 Hz, 2 H), 3.10–3.04 (m, 1H), 3.03–2.95 (m, 6 H), 2.91 (s, 3H), 2.79 (dd, J1 = 12.53 Hz, J2 = 5.00 Hz, 1 H), 2.62 (t, J = 7.43 Hz, 2H), 2.58 (d, J = 12.54 Hz, 1 H), 2.43 (s, 3H), 2.31–2.25 (m, 2H), 2.23–2.17 (m, 5H), 2.16–2.06 (m, 4 H), 2.02 (t, J =7.42 Hz, 2H), 1.99 (t, J =7.47 Hz, 2H), 1.65–1.07 (m, 33H), 0.91–0.74 ppm (m, 18H); 13C NMR (150 MHz, CDCl3/MeOD): δ =173.79, 173.23, 173.03, 172.32, 172.19, 160.34, 159.72, 154.49, 147.35, 146.45, 145.65, 141.19, 135.36, 134.72, 131.72, 131.37, 131.02, 130.99, 130.96, 130.93, 130.07, 129.52, 128.61, 125.91, 124.40, 124.36, 123.39, 123.33, 118.35, 114.77, 114.71, 65.09, 61.78, 60.00, 56.07, 52.08, 51.97, 47.12, 42.50, 42.46, 40.87, 40.79, 40.30, 35.97, 35.93, 35.72, 35.57, 34.65, 30.14, 29.42, 29.33, 28.81, 28.59, 26.68, 26.64, 25.79, 25.69, 25.66, 25.62, 24.89, 24.86, 24.73, 23.17, 23.14, 21.83, 21.76, 21.52, 13.44, 13.12, 9.38 ppm; UV/Vis: λabs = 551.94 nm, λem = 574.05 nm, ε=59325 Lmol−1cm−1; HRMS: calcd for C74H113BF2N14O11S2: 1487.82885; found: 1487.83093.
Two-step labeling of fluorescently labeled proteasomes in living cells
EL4 cells were cultured on DMEM supplemented with fetal calf serum (FCS, 10%), penicillin (10 units mL−1), and streptomycin (10 μg mL−1) in a CO2 (5 %) humidified incubator at 37 °C. Some 2·106 cells were seeded in 6 cm Petri dishes and allowed to grow overnight in medium (1 mL). The cells were exposed to 0, 0.1, 1, 10 μM probe (1 μL 100× solution in DMSO) for 2 h, before being washed with PBS (2×) and harvested. After flash freezing (N2 (l)) the cells were lysed in digitonin lysis buffer [Tris pH 7.5 (50 mM), sucrose (250 mM), MgCl2 (5 mM), dithiothreitol (DTT; 1 mM), digitonin (0.025%), 50 μL] for 5 min. on ice and centrifuged at 16100 rcf (relative centrifugal force) for 20 min at 4 °C. The supernatant containing the cytosolic fraction was collected, and the protein content was determined by Bradford assay. Some 25 μg of total protein were incubated with biotin-phosphine 7 (100 μM) in lysis buffer (20 μL) containing DTT (5 mM) for 1 h at 37 °C. The reaction was terminated by a chloroform/methanol precipitation of the proteins.[21] The pellet was solubilized by boiling for 5 min in 1× Laemli’s sample buffer containing β-mercaptoethanol. The proteins were resolved by SDS-PAGE (12.5 %). Labeled proteasome subunits were visualized by in-gel fluorescence scanning on a Typhoon variable mode imager (Amersham Biosciences) followed by Western blotting. The blots were blocked with BSA (1 %) in TBS-Tween 20 (0.1 % Tween 20) over 30 min at RT, hybridized for 30 min with streptavidin/HRP (1:10 000) in blocking buffer, washed, and visualized with the aid of an ECL + kit (Amersham Biosciences).
Supplementary Material
Acknowledgments
This work was supported by the Netherlands Organization for Scientific Research (NWO) and the Netherlands Proteomics Centre.
Footnotes
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author.
References
- 1.Evans MJ, Cravatt BF. Chem Rev. 2006;106:3279–3301. doi: 10.1021/cr050288g. [DOI] [PubMed] [Google Scholar]
- 2.van Swieten PF, Leeuwenburgh MA, Kessler BM, Overkleeft HS. Org Biomol Chem. 2005;3:20–27. doi: 10.1039/b412558d. [DOI] [PubMed] [Google Scholar]
- 3.Sadaghiani AM, Verhelst SH, Bogyo M. Curr Opin Chem Biol. 2007;11:20–28. doi: 10.1016/j.cbpa.2006.11.030. [DOI] [PubMed] [Google Scholar]
- 4.Prescher JA, Bertozzi CR. Nat Chem Biol. 2005;1:13–21. doi: 10.1038/nchembio0605-13. [DOI] [PubMed] [Google Scholar]
- 5.Speers AE, Adam GC, Cravatt BF. J Am Chem Soc. 2003;125:4686–4687. doi: 10.1021/ja034490h. [DOI] [PubMed] [Google Scholar]
- 6.Ovaa H, van Swieten PF, Kessler BM, Leeuwenburgh MA, Fiebiger E, van den Nieuwendijk AMCH, Galardy PJ, van der Marel GA, Ploegh HL, Overkleeft HS. Angew Chem. 2003;115:3754–3757. doi: 10.1002/anie.200351314. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2003;42:3626–3629. doi: 10.1002/anie.200351314. [DOI] [PubMed] [Google Scholar]
- 7.Saxon E, Bertozzi CR. Science. 2000;287:2007–2010. doi: 10.1126/science.287.5460.2007. [DOI] [PubMed] [Google Scholar]
- 8.Hang HC, Loureiro J, Spooner E, van der Velden AW, Kim YM, Pollington AM, Maehr R, Starnbach MN, Ploegh HL. ACS Chem Biol. 2006;1:713–723. doi: 10.1021/cb600431a. [DOI] [PubMed] [Google Scholar]
- 9.Tornøe CW, Christensen C, Meldal M. J Org Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 10.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew Chem. 2002;114:2708–2711. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 11.Speers AE, Vravatt BF. Chem Biol. 2004;11:535–546. doi: 10.1016/j.chembiol.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 12.Wang Q, Chan TR, Hilgraf R, Fokin VV, Sharpless KB, Finn MG. J Am Chem Soc. 2003;125:3192–3193. doi: 10.1021/ja021381e. [DOI] [PubMed] [Google Scholar]
- 13.Link AJ, Tirrell DA. J Am Chem Soc. 2003;125:11164–11165. doi: 10.1021/ja036765z. [DOI] [PubMed] [Google Scholar]
- 14.Agard NJ, Baskin JM, Prescher JA, Lo A, Bertozzi CR. ACS Chem Biol. 2006;1:644–648. doi: 10.1021/cb6003228. and references therein. [DOI] [PubMed] [Google Scholar]
- 15.Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, Lo A, Codelli JA, Bertozzi CR. Proc Natl Acad Sci USA. 2007;104:16793–16797. doi: 10.1073/pnas.0707090104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Verdoes M. Chem Biol. 2006;13:1217–1226. doi: 10.1016/j.chembiol.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 17.Haugland, et al. 4774339. United States Patent. 1988; Kang, et al. 5274113. United States Patent. 1993; Kang, et al. 5451663. United States Patent. 1995
- 18.Baumeister W, Walz J, Zuhl F, Seemuller E. Cell. 1998;92:367–380. doi: 10.1016/s0092-8674(00)80929-0. [DOI] [PubMed] [Google Scholar]
- 19.Hanada M, Sugawara K, Kaneta K, Toda S, Nishiyama Y, Tomita K, Yamamoto H, Konishi M, Oki T. J Antibiot. 1992;45:1746–1752. doi: 10.7164/antibiotics.45.1746. [DOI] [PubMed] [Google Scholar]
- 20.Sin N, Kim KB, Elofsson M, Meng L, Auth H, Kwok BH, Crews CM. Bioorg Med Chem Lett. 1999;9:2283–2288. doi: 10.1016/s0960-894x(99)00376-5. [DOI] [PubMed] [Google Scholar]
- 21.Wessel D, Flügge UI. Anal Biochem. 1984;138:141–143. doi: 10.1016/0003-2697(84)90782-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.