

Abstract
We report an online non-enzymatic method for site-specific digestion of proteins to yield peptides that are well suited for collision induced dissociation (CID) tandem mass spectrometry (MS/MS). The method combines online microwave heating acid hydrolysis at aspartic acid and online electrochemical oxidation at tryptophan and tyrosine. The combined microwave/electrochemical (microwave/echem) digestion is reproducible and produces peptides with an average sequence length of 10 amino acids. This peptide length is similar to the average peptide length of 9 amino acids obtained by digestion of proteins with the enzyme trypsin. As a result, the peptides produced by this novel non-enzymatic digestion method, when analyzed by ESI-MS, produce protonated molecules with mostly +1 and +2 charge states. The combination of these two non-enzymatic methods overcomes shortcomings with each individual method in that: i) peptides generated by the microwave-hydrolysis method have an average amino acid length of 16 amino acids, and ii) the inability of the electrochemical-cleavage method to reproducibly digest proteins with molecular masses above 4 kDa. Preliminary results are presented on the application and utility of this rapid online digestion (total of 6 min digestion time) on a series of standard peptides and proteins as well as an E. coli protein extract.
Introduction
Trypsin is the enzyme of choice for bottom-up proteomic studies1 and cleaves proteins specifically at the basic residues arginine (R) and lysine (K). Due to the frequency of these amino acids in protein sequences and their side group high basicities, the resulting peptides are well suited for Collision Induced Dissociation (CID) fragmentation in tandem MS (MS/MS) measurements. This compatibility is a result of their size (average sequence length of 9 amino acids) and their inherent 2+ charge when ionized via electrospray ionization (ESI). For 2+ charged peptides, the presence of a basic residue amino acid at the C-terminus of the peptide implies that one proton remains fixed at this site, while the other proton remains free to move along the backbone of the peptide2–4. Because of this mobile proton, the resulting heterogeneous precursor ion population provides different charge-site-directed fragmentation pathways along the peptide backbone, yielding a more complete series of ions representative of the sequence of the precursor peptide.
The use of site-specific non-enzymatic methods for protein digestion has potential advantages over enzyme-based digestion methods in terms of speed, simplicity, robustness and ease of automation. Several non-enzymatic approaches are available for bottom-up proteomic protocols including acid hydrolysis at aspartic acid (Asp, D)5, cyanogen bromide (CNBr) cleavage at the N-terminus of methionine (Met, M)6, hydroxylamine cleaving at asparagine (Asn, N) and glycine (Gly, G) bonds7, iodosobenzoic acid cleaving at tyrosine (Tyr, Y) and tryptophan (Trp, W)8, dimethyl sulfoxide-hydrochloric acid and cyanogens bromide (DMSO/HCl, CNBr) cleavage at tryptophan residues9, and 2-nitro-5-thiocyanobenzoic acid cleaving at the N-terminus of cysteine (Cys, C)10. Simultaneous cleavage at W and Y residues can also be induced via electrochemical oxidation (echem WY-cleavage) at a platinum11–13 and carbon14 anode, with yields ranging from 20 to 50%. This electrochemical non-enzymatic approach is particularly attractive as it has the potential to perform protein digestions in a reagentless fashion. However, these non-enzymatic approaches tend to yield large peptides that are not well suited for CID (vide infra) because of the low frequency at which these amino acids appear in protein sequences, usually between 1–10 % of the sequence. This is significant since CID is the most widely used fragmentation mode in MS/MS of peptides. As a result, these non-enzymatic digestion techniques have been traditionally used, often in conjunction with other enzymatic digestion methods, to study post-translational modifications in a particular region of a protein sequence15 and in the identification of membrane proteins16–18. In these cases, the inclusion of non-enzymatic digestion steps ensures a larger protein sequence coverage, overcomes solubility issues or produces short peptides for efficient in-gel extraction.
Two non-enzymatic techniques, acid hydrolysis at D and the electrochemical oxidation at W and Y (echem WY-cleavage) are of particular interest since they provide advantages in terms of speed of analysis, reagentless operation and online compatibility with downstream ESI-MS detection. For example, acid hydrolysis at D can utilize volatile organic acids like formic or acetic acid to lower the solution pH, while the echem WY-cleavage is reagentless. These attributes are well suited for the development of automated proteomic-based techniques that can be applied for high throughput analysis, rapid bacteria detection and/or disease diagnosis.
In the original acid hydrolysis at D method5, the protein sample is dissolved in aqueous formic acid (pH 2), sealed in a glass ampoule and heated at 130 °C for 2 hours. The resulting peptides are the result from site specific cleavages at the N- and C-termini of aspartic acid induced via a side chain carbonyl oxygen (β-carboxyl group) attacking either the C- or N-peptide bond carbonyl carbon forming either a cyclic anhydride or a six member ring intermediates before cleavage. The hydrolysis at D method was later incorporated into a bottom-up proteomic workflow implementing 2D-gel protein separation and peptide mass fingerprinting for protein identification19. This approach provided similar results as the enzymatic (trypsin) method in identifying unknown proteins from Saccharomyces cerevisiae. The subsequent incorporation of microwave heating into this approach reduced the time for complete digestion down to 5 min20–24. To further increase the speed of analysis of this method and the sequence coverage, a microwave heating flow cell was developed and the reagent dithiothreitol (DTT) was added to the solution in order to simultaneously reduce disulfide bonds (i.e., cleave disulfide bonds) during the protein digestion at D25. This approach, termed microwave D-cleavage, made possible the digestion of proteins in 5 min and online with LC and/or ESI-MS/MS. Subsequent work confirmed the requirement of disulfide bond cleavage during the microwave acid hydrolysis process (by the addition of DTT) as well as characterized the effect of microwave heating under low pH conditions on carbohydrate side groups in model glycoproteins26.
In the echem WY-cleavage method, electrochemical oxidation of proteins induces cleavage at the amino acid residues tyrosine (Y) and tryptophan (W)11–14. Recently, this non-enzymatic and reagentless digestion approach was performed on peptides27 and proteins28 online with ESI-MS detection. Efficient electrochemical oxidative cleavage of peptides at the C-terminus of tyrosine residues was performed at potentials between 0.5 and 1.5 V (vs. quasi palladium (Pd) reference electrode)27. A recent detailed study of parameters governing the echem WY-cleavage of a series of tripeptides found that cleavage was efficient at low pH’s (1.9–3.1) and that the nature of the adjacent amino acid to either W or Y influenced the cleavage efficiency29. However, overall efficient electrochemical oxidative and cleavage of proteins has only been observed for small molecular mass proteins (< 14 kDa), while no digestion products were observed for several larger proteins including lysozyme and serum albumin28, 30. This shortcoming was attributed to problems with accessibility of W and Y residues to the electrode surface and/or to competing oxidation reactions. This trend of decreasing digestion efficiency with increasing protein molecular weight was also observed in our laboratory (unpublished results) as electrochemical digestions were performed readily only on peptides with molecular weights less than about 3,400 Da (vide infra).
Because of the low frequency of aspartic acid (5.45%) (versus a combined frequency of 11.38% for R and K)31 2010, peptides produced by the microwave D-cleavage approach have an average amino acid length of about 16. These larger peptides generally produce ions of high charge state (> 3+) when ionized by ESI because of their size and also because of the inclusion of one or more basic amino acids within their sequences25. As a result, these CID mass spectra are not as informative in terms of amino acid sequence because the localization of charges on basic groups along the backbone of the peptide does not allow for sufficiently random fragmentation. In fact, the high charge state peptides produced by the microwave D-cleavage method have been shown to be better suited for Electron Transfer Dissociation (ETD) than for CID, the former technique favoring ions with charge states higher than +2, and thus providing a high percentage of sequence coverage32. Conversely, direct comparison of CID tandem mass spectra of peptides generated by microwave D-cleavege and trypsin digestion showed improved peptide sequence coverage for the tryptic peptide over the microwave-generated peptide32. This behavior is expected to be similar for the echem WY-cleavage derived peptides since tryptophan and tyrosine have a combined frequency in protein sequences of 3.99 %31. However, reliable protein identification from peptides produced by the microwave acid hydrolysis step has been demonstrated using a combination of LC-ESI-LTQ-Orbitrap MS and MALDI-TOF/TOF-MS, the latter technique used to analyze peptides with masses above 3000 Da22 in order to generate singly charged ions. Finally, as mentioned earlier, the echem WY-cleavage technique has also yielded inconsistent results in the digestion of proteins with molecular masses above 3.4 kDa and as a result, its utility as a standalone digestion technique is limited.
These limitations, however, can be overcome by coupling these two non-enzymatic digestion techniques, microwave D-cleavage and echem WY-cleavage to induce an overall cleavage at D, W and Y. By performing the digestion with microwave D-cleavage first, the problem of digestion efficiency with increasing protein molecular mass of the echem WY-cleavage technique is circumvented. Large proteins are initially digested at the amino acid D, yielding peptides, albeit large, but amenable for reproducible and consistent echem WY-cleavage. Subsequent digestion of these peptides by echem WY-cleavage further decreases the peptide size. As a result, this combination of non-enzymatic techniques provides a means to produce peptides that are better suited for MS/MS analysis implementing CID, while consistently digesting proteins of any MW. For example, the in silico digestion of about 250 proteins from E. coli carried out with a combination of both microwave D-cleavage first, followed by echem WY-cleavage (for an overall cleavage at D, W, and Y) resulted in peptides with an average amino acid length of 10. This peptide length is much closer to the average length of peptides generated by trypsin digestion (9 amino acids), and as a result, the combined non-enzymatic approach is expected to create peptides that are better suited (in length and charge state) for CID than either microwave D-cleavage or echem WY-cleavage alone.
In the present study, the use of a tandem non-enzymatic digestion approach incorporating microwave D-cleavage digestion followed by echem WY-cleavage is explored. The combined approach, termed microwave-echem cleavage, is demonstrated with a series of standard peptides and proteins, as well as with proteins extracted from E. coli.
Experimental
Chemicals
Angiotensins I and II (human), glucagon (19–29, human), dynorphin (1–9, porcine), and [Ile-Ser]-bradykinin (human) were purchased from Anaspec Inc. (San Jose, CA). Oxidized insulin chain B, insulin, α-lactalbumin (bovine milk), lysozyme (chicken egg white), cytochrome c (horse heart), myoglobin (horse skeletal muscle), carbonic anhydrase (bovine erythrocytes), albumin (bovine serum), iodoacetamide, dithiothreitol (DTT), sodium dodecyl sulphate (SDS), and octyl β-D-glucopyranoside were all purchased from Sigma (St. Louis, MO). HPLC grade acetonitrile (ACN) and deionized water were purchased from Burdick and Jackson (Muskegon, MI). Reagent grade formic acid was obtained from Sigma-Aldrich (St. Louis, MO).
Bacteria
Escherichia coli (E. coli; ATCC 15597) were purchased from American Type Culture Collection (ATCC, Manassas, VA). Bacteria were grown under optimum laboratory conditions using tryptic soy agar (TSA; BD Science, Sparks, MD) at 37°C for 12–15 h. Bacterial cells were transferred to 1 mL of water using a sterile tungsten loop inoculator. The cells were vortexed for 10 sec and centrifuged for 5 min at 12,000 rpm to remove the media. The supernatant was removed and the pellet was re-suspended in 1 mL of water and the washing step was repeated. After three washing steps the cells were lysed by ultrasonication on ice. The cells were subjected to a sonication pulse (pulse strength 7) duration of 10 sec, followed by an idle 10 sec, and repeated 10 times. The sample was then centrifuged at 5,000 rpm for 15 min to remove any cell material. A 100 μL aliquot of the supernatant was diluted to 1 mL in 10% FA and 100 mM DTT just before being digested by the combined microwave-echem cleavage approach.
Sample Preparation
For digestion with echem WY-cleavage method alone, all peptide solutions were prepared in 50/50 (vol/vol) acetonitrile/water to a concentration of 5μM. Formic acid was added to a final composition of 47.5/47.5/5 (vol/vol/vol) acetonitrile/water/formic acid. The addition of 5% formic acid also served as the supporting electrolyte in the solution. Disulfide bonds in proteins were broken by the conventional method; a 100 μM solution of protein solution in water was reduced by adding DTT to a concentration of 2 mM and incubated at 37°C for 30 minutes. The protein sample was then alkylated by adding iodoacetamide to a concentration of 2 mM and then incubated in the dark at room temperature for another 30 minutes. The protein samples were then diluted to a concentration of 5μM in 47.5/47.5/5 (vol/vol/vol) acetonitrile/water/formic acid. All protein and peptide solutions were stored at −20° C.
For the online microwave-echem cleavage digestion (microwave D-cleavage/electrochemical oxidatative cleavage combination) with direct ESI-MS/MS or HPLC-MS/MS analysis, all protein solutions were prepared in 90/10 (vol/vol) water/formic acid to a concentration of 1 mg/mL. The addition of formic acid serves two purposes. For microwave D-cleavage, the acid keeps the pH below 2 for optimum hydrolysis, and for the electrochemical cleavage it acts as the supporting electrolyte in the solution as well as increasing hydrolysis efficiency29. To break disulfide bonds, DTT was added to a concentration of 100 mM. For online HPLC-MS/MS analysis, the sample was flowed through the combination digestion cells at 0.5 μL/min.
Online Digestion Instrumentation and Mass Spectrometry
The electrochemical system used as a standalone digestion technique, based on a three electrode cell, was an electrochemical flow cell (5021A Conditioning Cell, ESA Biosciences, Chelmsford, MA). A syringe pump (Harvard Apparatus, Holliston, MA) delivered an isocratic flow of 0.5–2 μL/min to the electrochemical cell. The working electrode was made from porous graphite and all potentials were versus a quasi Pd reference electrode. A positive voltage of 1.5 V was applied to the solution flowing through the cell for the digestion. Cell potential was controlled with a potentiostat (Bio-Analytical Systems, BAS, West Lafayette, IN; model CV-27 potentiostat).
For online analysis with the microwave D-cleavage/electrochemical oxidative cleavage combination, microwave heating was achieved using a CEM Focused Microwave Synthesis System (Model: Discovery; CEM, Matthews, NC) and electrochemical oxidation was implemented using the electrochemical flow cell. The modified microwave flow cell is described in detailed in a previous publication25 and was used here with no further modifications. Figure 1 shows a schematic of the overall microwave D-cleavage/electrochemical oxidative cleavage combination flow injection analysis system used in this study. Conditions were first optimized for each digestion technique. However, when combined, the experimental conditions of the first digestion technique (in this case, microwave D-cleavage) became also the default conditions (in terms of solution composition) for the online electrochemical digestion. This coupling led to different experimental conditions than when operated alone, making a direct comparison of the electrochemical process versus when combined with microwave D-cleavage difficult. However, the operational conditions of the microwave digestion step were found to be compatible with the electrochemical digestion step, which overly simplified the experimental setup.
Figure 1.

Top: Diagram of the flow injection analysis system used for the microwave/electrochemical (echem) cleavage combination. Bottom: Time sequence shows when each device is either ON (1) or OFF (0).
For protein digestion by the combination method, the samples were first digested by delivering the sample into the microwave flow cell at a rate of 0.5 μL/min. A 2.5 μL internal volume microwave reaction loop was made with 54.5 cm of fused silica capillary (Polymicro, Phoenix, AZ) with an outer diameter of 360 μm and an inner diameter of 76 μm. The residence time for the sample inside the microwave flow cell was 5 min at this flow rate. The temperature of the system was brought up to 130°C over a 2 minute interval with the microwave oven using a power between 50–150 Watts, and then held at 130°C for 60 minutes, for a total of 58 minutes of microwave heating. The sample continued to flow directly through the electrochemical cell held at +1.5V (vs. quasi palladium reference electrode), which was turned on 10 minutes after the microwave digestion was started to account for the dead volume in the microwave loop. The sample was either injected directly into a nESI source for analysis or into a 20 μL loop on a 6-port valve connected to the HPLC system (Ultimate™, LC Packings, Sunnyvale, CA). The total collection time was 50 minutes (to account for dead volume between the cells and the loop). After the digestion reactions were carried out, the electrochemical cell was turned off to allow for a flushing solution (90/10 % by volume: water/FA) to be loaded into the syringe pump. The cleavage cells were flushed at a flow rate of 2 μL/min with the voltage on (+1.5V) using the flushing solvent for 60 minutes (perform during the LC separation). The flushing of the cells could occur at the same time as the separations because the 6-port valve was switched to the position that only allowed flow from the HPLC to go to the column, while the flow from the cells went to waste. The products were separated by reversed phase LC (RP-LC) using a homemade C18 column (5 μm bead size, 100 Å pore size, Microchrom Bioresources, Auburn, CA) with dimensions of 100 mm length, 360 μm OD, and 76 μm ID. A 20 minute loading time using 95% solvent A (0.1% formic acid, 1 μL/min flow rate) followed by a jump to 15% solvent B (80% ACN) and a 35 minute linear gradient of 15% to 50% solvent B was used with a 0.5 μL/min flow rate (separation flow) into the nanoelectrospray (nESI) source of a quadrupole ion trap MS (LCQ™ Classic, ThermoFinnigan, San Jose, CA). A flush of 95% B for 10 minutes followed. For the separation of E. coli products, after the 20 min load onto the column, a jump to 15% B was followed by a linear gradient from 15% B to 65% B in 85 mins. To ensure that the dead volume of the 20 μL loop did not affect the separation, the 6-port valve was switched to bypass the loop. Tandem MS/MS experiments with the ion trap MS were performed by acquiring a full-scan mass spectrum between m/z 300 and 2000 followed by three data dependent product ion mass-spectral scans of the most intense precursor ions (a.k.a., “big-three” scan). MALDI-Time-of-Flight-(ToF) MS (Voyager DE-Pro, Applied Biosystems, Foster City, CA) analyses were conducted in the linear mode using sinapic acid as the matrix or in the reflectron mode using α-cyano-hydroxycinnamic acid as the matrix. In these experiments, peptides were digested in the electrochemical flow cell and fractions were collected for off-line MALDI-MS analyses.
Database Search
The SEQUEST software (ThermoFinnigan, San Jose, CA) was used to carry out all database searches. An in silico “enzyme” was created within the BioworksBrowser™ to perform the digestion at the C-terminus of D residues. Even though cleavage can occur at the N-terminus to D residues, the favored reaction is for the C-terminus cleavage. The parameters used in the search include up to 10 missed cleavage sites, group scan of 50, minimum group count of 1, minimum ion count of 12, and the charge state option set to ‘auto’.
Results and Discussion
Online Microwave/Electrochemical Digestion of Angiotensin I
The effect of the combined online microwave D-cleavage and the echem WY-cleavage technique on the peptide angiotensin I (sequence: DRVYIHPFHL) was tested (several examples of peptides digested only with the electrochemical method, including a peptide mixture, a negative control and the largest peptide successfully digested with this method in our laboratory, insulin chain B, can be found in the Supporting Material section, Figures S-1 to S-5). Figure 2 illustrates the effect of each digestion component as well as the combined effect. Figure 2(a) represents the control ESI-mass spectrum of angiotensin I with both the microwave and electrochemical cells turned off. Shown in Figure 2(b) is the ESI-mass spectrum of angiotensin I with the microwave heating on, but no voltage applied on the electrochemical cell. Under these conditions, the peak corresponding to cleavage at the C-terminus of D is observed at m/z’s 1181.8 (+1) and 591.4 (+2), while no peaks can be assigned to electrochemical oxidative cleavage(s). Assuming equal detection sensitivities between angiotensis I and the digestion product (since both product and precursor peptides contain arginine in its sequence), a cleavage yield of 40–50% is estimated for the microwave D-cleavage step. Figure 2(c) shows the resulting ESI-mass spectrum when applying a +1.5 V potential to the electrochemical cell, but with the microwave oven off. Under these digestion conditions, peaks corresponding to only echem WY-cleavage are observed at m/z 763.5 ((Y)IHPFHL) and m/z 550.5 (DRVY). Figure 2(d) shows the ESI-mass spectrum for angiotensin I when the microwave heating and the electrochemical cell voltage are both turned on. Under these conditions, signals corresponding to products from both digestions are observed. Signals observed at m/z 1181.4 and m/z 591.4 correspond to the +1 and +2 ion of the microwave D-cleavage products, with a missed echem WY-cleavage. Similarly, signals observed at m/z 550.5 and m/z 763.5 correspond to echem WY-cleavage with a missed microwave D-cleavage. Finally, the peak found at m/z 435.4 resulted from consecutive cleavages at both D and Y. Digestion products at m/z’s 763, 550 and 435 observed in Figure 2(d) were confirmed by MS/MS analysis and their tandem mass spectra can be found in the Supporting Information section, Figure S-6. The combined digestion processes of both microwave D-cleavage and echem WY-cleavage are not exhaustive, as signals corresponding to the intact peptide at m/z 1296.8 and 649.9 (+1 and +2 ion respectively) are still prominent. Oxidation of Y and W with no cleavage is also known to occur28; however, our results show that oxidation of Y occurred without cleavage (indicated by a mass change of +16 without cleavage). The cleavage efficiency of the echem WY-cleavage alone has been estimated between 40–80% in model tripeptides29. Hence, the estimated overall efficiency of the combined microwave-echem digestion method is between 16–40%. Moreover, the overall efficiency of this combined approach is limited by the efficiency of the initial step, the microwave D-cleavage, as it generates the smaller peptides that are suitable for the echem WY-digestion. From published studies on the microwave acid hydrolysis method by several groups5, 21, increasing digestion time and/or lowering the pH does not affect the efficiency of the protein digestion, but rather affects the site-specificity of the process, that is, more non-specific cleavages are observed. These observations point to the accessibility of the D groups being the limiting factor in increasing sequence coverage for protein digestion via acid hydrolysis at D.
Figure 2.

(a) Flow injection analysis of Angiotensin I (MW = 1295.6 Da) through the combination cells into nESI-MS without microwave heating and no voltage applied. (b) Only microwave heating (130°C), no voltage. (c) voltage applied (+1.5V) but no microwave heating, and (d) Microwave heating (130°C) and voltage applied (+1.5V).
Combined, these results show the ability of both non-enzymatic digestion techniques to work in tandem and online. In addition, results illustrate the advantage of tandem digestion as both techniques are prone to miss cleavage sites; however, the peptides showing missed cleavages by one technique were also digested by the other technique, and vice versa. The ability of the echem WY-cleavage to act as a back-up digestion when the microwave D-cleavage misses to cleave a site will depend of course on the size of the fragment, that is, the echem WY-cleavage will be effective only when the peptide fragment is 3.5 kDa or less.
Disulfide Bond Cleavage
In the microwave D-cleavage method25, DTT is added during the microwave heating step to reduce and cleave disulfide bonds simultaneously with hydrolysis of peptide bonds at aspartic acids. To test whether DTT interferes with the downstream online electrochemical oxidative process, insulin in the presence of DTT was delivered through the two digestion flow cells, first through the microwave flow cell and then through the electrochemical flow cell, and products directly detected by nESI-MS. Insulin was chosen for this test since it does not have aspartic acid groups and has four tyrosine groups (2 in each chain A and B), two interchain disulfide bonds and one intrachain disulfide bond (in chain A). Table 1 shows the structure of insulin and lists the peptides expected to be formed after microwave treatment alone and after the combination microwave/echem digestion.
Table 1.
Structure of insulin with expected and observed (bold) peptides that are produced after each stage of the process.
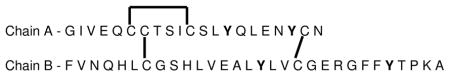 | ||
|---|---|---|
| Peptides | MW (Da) | |
| Before Microwave | – | 5740 |
| After Microwave | Chain A | 2340 |
| Chain B | 3400 | |
| After Microwave and Electrochemistry | A (G1-Y14) | 1470 |
| A (Q15-Y19) | 663 | |
| A (C20-N21) | 235 | |
| B (F1-Y16)* | 1826 | |
| B (L17-Y26)* | 1187 | |
| B (T27-A30) | 415 | |
denotes peptides observed with oxidation +48 Da occurring at cysteine residues.
Figure 3(b) shows the nESI-mass chromatogram of insulin flowing through both microwave and electrochemical flow cells. The electrochemical flow cell is maintained at +1.5V throughout the entire period. During the first 30 min, the intact insulin protein is only subjected to electrochemical oxidation conditions and in the presence of DTT with no microwave heating. The mass spectrum during this period (Figure 3(a)) shows signals corresponding to intact insulin and a small contribution from oxidized insulin (non-cleavage oxidation of tyrosine residues); however, no disulfide bond cleavage is observed. Moreover, no oxidative cleavage products are observed even though there are four Y residues in insulin. This implies that reduction of disulfide bond by DTT is not favorable at room temperature and under oxidative electrochemical conditions and that the electrochemical oxidation alone cannot induce bond cleavage in intact insulin. At 30 min into the chromatogram, the microwave oven is turned and after a 10 min delay (time for the temperature to reach the set point value of 130°C and dead volume in the system) many new signals are observed in the mass spectrum (Figure 3(c)). These signals correspond to products of disulfide bond cleavage (reduction) by DTT due to microwave heating as well as signals corresponding to echem WY-cleavage. These results clearly demonstrate that the presence of DTT in solution does not hinder the electrochemical cleavage step; in fact, this process is aided by the preceding cleavage of disulfide bonds in insulin. However, peaks corresponding to insulin Chain B and intact (no disulfide bond cleavage) insulin dominate the spectrum, again stemming from the low efficiency of the process when applied to large molecular mass proteins. Insulin Chain A is not detected as its signal is most likely suppressed by other products generated during the combined digestion process. Although low in intensity, the peaks observed at m/z’s 416.4, 937.8, and 1236.9 correspond to specific cleavages at both Y residues in insulin Chain B, and their sequences were confirmed by MS/MS analyses (see Supporting Information for MS/MS data, Figures S-7). It should be noted also in Figure 3 that along with a specific cleavage at Y, the cysteine-containing peptides show a +48 mass change due to further electrochemical oxidation of the thiol groups, as these are not alkylated after reduction by DTT during the microwave heating step. As a result, cysteine residues are oxidized at the electrode, causing the addition of three oxygen atoms to form a cysteic acid group.
Figure 3.

(a) There are no electrochemical oxidative cleavage products observed without microwave heating preceding the step (to cleave disulfide bonds). (b) The extracted ion chromatogram (XIC: m/z 851.1, 416.4, 937.8, 1236.9) of peaks of interest from the disulfide bond reduction and electrochemical digestion of insulin (5,740 Da) being introduced into the microwave-echem digestion cells. The voltage was applied continuously and microwave heating was applied at 30 min. (c) About 10 min after microwave heating is applied, peaks are observed corresponding to disulfide bond reduction and cleavage from electrochemical oxidation.
Identification of Myoglobin and Bovine Serum Albumin with Online Microwave/Echem Cleavage
Myoglobin (16 kDa) and Bovine Serum Albumin (BSA, 64 kDa) were successfully digested and identified with the combined microwave/echem system. For each test protein, digestion was performed by the combination online digestion technique, with eluent products collected into a 20 μL injection loop on a 6-port valve for subsequent HPLC-nESI-MS/MS analysis. Results are presented in the Supplementary Information section (Table S-2, Table S-3 and Figures S-8 and S-9), showing peptides formed, SEQUEST scores and tandem mass spectra of selected peptides from the digestion of BSA with the microwave/echem digestion technique.
Comparison of Collision Induced Dissociation of Online Microwave D-cleavage and Microwave/Echem Digestion Products
It has been shown that the digestion products formed from microwave D-cleavage are better suited for analysis by MS/MS using electron transfer dissociation (ETD) rather than collision induced dissociation (CID). This is a consequence of the low abundance of D in protein sequences which results in large peptides (~16 amino acids in length) with high charge states (>+2)32. However, when these D-cleavage products are further digested by electrochemical oxidation, the new products formed are more amenable to CID since their expected size is about 10 amino acids in length (vide supra). Figure 4a shows the CID-tandem mass spectrum of a peptide formed from only microwave D-cleavage of myoglobin at D(20) which contains 2 non-cleavage oxidized tryptophan residues. The parent ion has a 3+ charge (m/z 754.8) and only 11 of a possible 38 (~30%) y- and b-ions from this peptide are matched by a SEQUEST database search. Figures 4b and 4c show the CID-tandem mass spectra of the corresponding 1+ peptides formed from the cleavage at W(7) and W(14) residues (m/z 900.1 and m/z 781.1, respectively) and that overlap the peptide sequence in Figure 4a. These peptides show a much greater sequence coverage (9 out of 12 y- and b-ions in 4b, and 8 out of 12 in 4c) which is reflected in their overall SEQUEST Xcorr scores (Table S-2). The combined microwave/echem digestion leads to a greater fragmentation sequence coverage and thus higher confidence for peptide/protein identification using CID-MS/MS.
Figure 4.

Tandem mass spectra (CID) comparison of peptides from the same sequence region in myoglobin formed by (a) microwave D-cleavage only, (b) and (c) from microwave/echem cleavage combination. Greater fragmentation sequence coverage results from the microwave/echem cleavage method than for the microwave D-cleavage method alone.
Analysis of a Protein Extract from E. coli by Online Microwave/Echem Digestion and LC-MS/MS
To demonstrate the ability of the on-line microwave/echem digestion coupled with LC-MS/MS to detect and identify proteins from a complex mixture, a protein extract from an E. coli cell lysate was analyzed. Table S-4 in the Supplementary Information section lists the top 10 proteins identified in E. coli (using SEQUEST database search) along with the corresponding number of peptides derived from that protein with Xcorr-scores values above 1. An extended listing of all proteins identified in E. coli along with all peptide sequences and SEQUEST scores is available in the Supporting Information section (Table S-5).
Because of the use of online microwave/echem digestion, the CID-tandem mass spectra generated from these digested proteins allowed for their identification with a high degree of confidence. This technique produced peptides that were suitable for CID MS/MS, which led to more peptides to be identified than by online microwave D-cleavage alone. For example, when only using online microwave D-cleavage digestion for the analysis of E. coli protein extract25, the top ten proteins identified by SEQUEST database search had an average of 3 peptides identified per protein. On the other hand, the top ten proteins identified in this study when using the microwave/echem digestion had on average 6 peptides identified per E. coli protein (Table S-4). This led to an average sequence coverage of 16.5% for this study, an increase from the 12.0% average coverage for the microwave D-cleavage method alone. Based on the results, the combined microwave/echem digestion method is capable of digesting a complex mixture of proteins from a cell lysate and with a high degree of confidence, demonstrating a notable improvement over either non-enzymatic digestion technique alone.
Conclusions
Preliminary results presented in this study demonstrated the capability of the combined microwave/echem method for the non-enzymatic digestion of proteins in 6 min. The work here presented focused on overcoming the drawbacks of using CID for MS/MS analysis of peptides generated by the non-enzymatic microwave D-cleavage method. The strategy presented overcame this deficiency by combining two rapid non-enzymatic digestion techniques, the microwave D-cleavage and the electrochem WY-cleavage. The resulting microwave/echem digestion approach produces peptides that are similar in size to those produced by trypsin digestion of proteins, and thus, conducive for CID MS/MS in a quadrupole ion trap MS (e.g., LCQ). However, this study did not intend to provide a direct comparison between this approach and trypsin digestion, nor points to be a substitute to the digestion of proteins with the enzyme trypsin. The combined approach overcomes limitations of the microwave D-cleavage approach in that only large peptides are produced (resulting from the low frequency of aspartic acid in protein sequences). These large peptides, when subjected to ESI, generate high charge state ions that make mass assignment and peptide identification difficult when fragmented by CID. By incorporating a second online non-enzymatic digestion step based on electrochemical oxidation (echem WY-cleavage) following the microwave D-cleavage, two additional amino acid sites are accessible for cleavage (W and Y). As a result, these large peptides are further digested into smaller peptides that are comparable in size and charge to those obtained by trypsin digestion. Results presented also showed that the peptides produced by the combination microwave/echem digestion are better suited for CID than the larger peptides produced by microwave D-cleavage alone. The order in which these two processes are applied is important as the echem WY-cleavage digestion is not reproducible for digestion of proteins with molecular masses above 3.4 kDa, and thus the microwave D-cleavage technique serves as a pre-digestion step of large proteins and cleavage of disulfide bonds. The current lower digestion efficiency of the microwave/echem method when compared to trypsin digestion methods makes this approach better suited for high throughput analyses or sample screening. Efforts are currently underway in our laboratory to incorporate 2 dimensional (2D) LC with the combined microwave/echem digestion in order to increase the efficiency of the combined non-enzymatic digestion process and the number and dynamic range of proteins detected.
Supplementary Material
Acknowledgments
The authors are grateful for funding by a grant from the National Institutes of Health that made this work possible (NIH grant number R15-RR020354-01A1).
References
- 1.Aebersold R, Goodlett DR. Chemical Reviews. 2000 doi: 10.1021/cr990076h. [DOI] [PubMed] [Google Scholar]
- 2.Cox KA, Gaskell SJ, Morris M, Whiting A. J Am Soc Mass Spectrom. 1996;7:522–531. doi: 10.1016/1044-0305(96)00019-0. [DOI] [PubMed] [Google Scholar]
- 3.Wysocki VH, Tsaprailis G, Smith LL, Breci LA. J Mass Spectrom. 2000;35:1399–1406. doi: 10.1002/1096-9888(200012)35:12<1399::AID-JMS86>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 4.Huang Y, Triscari JM, Tseng GC, Pasa-Tolic L, Lipton MS, Smith RD, Wysocki VH. Anal Chem. 2005;77:5800–5813. doi: 10.1021/ac0480949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Inglis AS. Methods in Enzymology. 1983;91:324–332. doi: 10.1016/s0076-6879(83)91030-3. [DOI] [PubMed] [Google Scholar]
- 6.Carlos Rodriguez J, Wong L, Jennings PA. Protein Expression Purif. 2003;28:224–231. doi: 10.1016/s1046-5928(02)00700-3. [DOI] [PubMed] [Google Scholar]
- 7.Bornstein P, Balian G. Methods Enzymol. 1977;47:132–145. doi: 10.1016/0076-6879(77)47016-2. [DOI] [PubMed] [Google Scholar]
- 8.Fontana A, Dalzoppo D, Grandi C, Zambonin M. Methods Enzymol. 1983;91:311–318. doi: 10.1016/s0076-6879(83)91028-5. [DOI] [PubMed] [Google Scholar]
- 9.Huang HV, Bond MW, Hunkapiller MW, Hood LE. Methods Enzymol. 1983;91:318–324. doi: 10.1016/s0076-6879(83)91029-7. [DOI] [PubMed] [Google Scholar]
- 10.Degani Y, Patchornik A. Biochemistry. 1974;13:1–11. doi: 10.1021/bi00698a001. [DOI] [PubMed] [Google Scholar]
- 11.Iwasaki H, Cohen LA, Witkop B. J Am Chem Soc. 1963;85:3701–3702. [Google Scholar]
- 12.Iwasaki H, Witkop B. J Am Chem Soc. 1964;86:4698–4708. [Google Scholar]
- 13.Isoe S, Cohen LA. Arch Biochem Biophys. 1968;127:522–527. doi: 10.1016/0003-9861(68)90257-9. [DOI] [PubMed] [Google Scholar]
- 14.Walton D, Richards PG, Heptinstall J, Coles B. Electrochimica Acta. 1997;42:2285–2294. [Google Scholar]
- 15.Kinter M, Sherman NE. Protein Sequencing and Identification Using Tandem Mass Spectrometry. Wiley-Interscience; NY: 2000. [Google Scholar]
- 16.van Montfort BA, Doeven MK, Canas B, Veenhoff LM, Poolman B, Robillard GT. Biochim Biophys Acta. 2002;1555:111–115. doi: 10.1016/s0005-2728(02)00264-5. [DOI] [PubMed] [Google Scholar]
- 17.Quach TTT, Li N, Richards DP, Zheng J, Keller BO, Li L. J Proteome Res. 2003;2:543–552. doi: 10.1021/pr0340126. [DOI] [PubMed] [Google Scholar]
- 18.Fischer F, Wolters D, Roegner M, Poetsch A. Mol Cell Proteomics. 2006;5:444–453. doi: 10.1074/mcp.M500234-MCP200. [DOI] [PubMed] [Google Scholar]
- 19.Li A, Sowder RC, II, Henderson LE, Moore SP, Garfinkel DJ, Fisher RJ. Anal Chem. 2001;73:5395–5402. doi: 10.1021/ac010619z. [DOI] [PubMed] [Google Scholar]
- 20.Hauser NJ, Basile F, Nashville TN. 2004 [Google Scholar]
- 21.Swatkoski S, Russell SC, Edwards N, Fenselau C. Anal Chem. 2006;78:181–188. doi: 10.1021/ac051521d. [DOI] [PubMed] [Google Scholar]
- 22.Swatkoski S, Gutierrez P, Ginter J, Petrov A, Dinman JD, Edwards N, Fenselau C. J Proteome Resh. 2007;6:4525–4527. doi: 10.1021/pr0704682. [DOI] [PubMed] [Google Scholar]
- 23.Lill Jennie R, Ingle Elizabeth S, Liu Peter S, Pham V, Sandoval Wendy N. Mass Spectrom Rev. 2007;26:657–671. doi: 10.1002/mas.20140. [DOI] [PubMed] [Google Scholar]
- 24.Swatkoski S, Gutierrez P, Wynne C, Petrov A, Dinman JD, Edwards N, Fenselau C. J Proteome Res. 2008;7:579–586. doi: 10.1021/pr070502c. [DOI] [PubMed] [Google Scholar]
- 25.Hauser NJ, Basile F. J Proteome Res. 2008;7:1012–1026. doi: 10.1021/pr700596e. [DOI] [PubMed] [Google Scholar]
- 26.Li J, Shefcheck K, Callahan J, Fenselau C. Int J Mass Spectrom. 2008;278:109–113. doi: 10.1016/j.ijms.2008.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Permentier HP, Jurva U, Barroso B, Bruins AP. Rapid Comm Mass Spectrom. 2003;17:1585–1592. doi: 10.1002/rcm.1090. [DOI] [PubMed] [Google Scholar]
- 28.Permentier HP, Bruins AP. J Am Soc Mass Spectrom. 2004;15:1707–1716. doi: 10.1016/j.jasms.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 29.Roeser J, Permentier HP, Bruins AP, Bischoff R. Anal Chem. 2010;82:7556–7565. doi: 10.1021/ac101086w. [DOI] [PubMed] [Google Scholar]
- 30.Permentier HP, Bruins AP, Nashville TN. 2004 May 23–27; [Google Scholar]
- 31.ExPASy. Swiss Institute of Bioinformatics; [accessed on June 17, 2010]. web site. [Google Scholar]
- 32.Hauser N, Han H, McLuckey SA, Basile F. J Proteome Res. 2008;7:1867–1872. doi: 10.1021/pr700671z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.