

Summary
Monoacylglycerol lipase (MGL) is a serine hydrolase involved in the biological deactivation of the endocannabinoid 2-arachidonoyl-sn-glycerol (2-AG). Previous efforts to design MGL inhibitors have focused on chemical scaffolds that irreversibly block the activity of this enzyme. Here, we describe two naturally occurring terpenoids, pristimerin and euphol, which inhibit MGL activity with high potency (median effective concentration, IC50 = 93 nM and 315 nM, respectively) through a reversible mechanism. Mutational and modeling studies suggest that the two agents occupy a common hydrophobic pocket located within the putative lid domain of MGL, and each reversibly interact with one of two adjacent cysteine residues (Cys201 and Cys208) flanking such pocket. This previously unrecognized regulatory region may offer a novel molecular target for potent and reversible inhibitors of MGL.
Introduction
2-Arachidonoyl-sn-glycerol (2-AG), an endogenous lipid agonist for cannabinoid (CB) receptors (Sugiura et al., 1995; Mechoulam et al., 1995; Stella et al., 1997), is thought to modulate excitatory neurotransmission in the central nervous system by acting as a synaptic messenger (Katona and Freund, 2008). Electrophysiological, pharmacological and neuroanatomical evidence suggests that 2-AG is produced in dendritic spines of glutamatergic synapses and crosses the synaptic cleft in a retrograde direction to activate presynaptic CB1 receptors and inhibit glutamate release (Katona and Freund, 2008). CB1-bearing axon terminals also contain the enzyme monoacylglycerol lipase (MGL) (Gulyas et al., 2004), a cytosolic serine hydrolase (Karlsson et al., 1997) that cleaves 2-AG and presumably terminates its transsynaptic actions (Stella et al., 2007; Dinh et al., 2002).
MGL belongs to the ‘α/β hydrolase fold’ family of enzymes, whose conserved three-dimensional structures consist of a central β-sheet surrounded by a variable number of α-helices (Ollis et al., 1992). The active sites of these proteins are located in turns between α-helices and β-sheets and harbor a classic catalytic triad composed of a serine, a carboxylic acid and a histidine (Ollis et al., 1992). Structure-alignment and site-directed mutagenesis studies have identified residues Ser122, Asp239 and His269 as the catalytic triad in rodent MGL (Karlsson et al., 1997) Moreover, mass spectrometry experiments have shown that the substituted tetrazole AM6701 (5-((biphenyl-4-yl)methyl)-N,N-dimethyl-2H-tetrazole-2-carboxamide) inhibits human MGL by covalently modifying the enzyme's catalytic serine (Zvonok et al., 2008).
In addition to the Ser-Asp-His triad, three cysteine residues appear to contribute in important ways to the regulation of MGL function. Computational models position one of these residues, Cys242, in close proximity of the catalytic triad, while mutational analyses indicate that its replacement with glycine causes a substantial loss of catalytic activity (King et al., 2009). Two additional regulatory cysteines, Cys201 and Cys208, are found on the putative lid domain of MGL, a flexible structure that is thought to function as an entry gate to the active site of this and other lipases (Jaeger et al., 1999). Though mutations affecting Cys201 and Cys208 produce only a moderate decrease in MGL activity, they markedly reduce the ability of cysteine-reacting agents to inhibit such activity (King et al., 2009).
The presence of MGL in glutamatergic axon terminals (Gulyas et al., 2004) suggests that this enzyme may be involved in the termination of 2-AG-mediated retrograde transmission at excitatory synapses. This idea is supported by studies showing that virally induced MGL over-expression reduces receptor-dependent 2-AG accumulation in primary cultures of rat brain neurons (Dinh et al., 2002), and that siRNA-mediated MGL silencing increases 2-AG levels in HeLa cells (Dinh et al., 2004). Pharmacological experiments in vivo have provided further support for a role of MGL in 2-AG deactivation. Microinjections of the carbamate-based MGL inhibitor URB602 (biphenyl-3-ylcarbamic acid cyclohexyl ester) into the periaqueductal grey region of the midbrain increased 2-AG levels and enhanced stress-induced analgesia in rats (Hohmann et al., 2005). Moreover, local or systemic URB602 administration reduced nocifensive responses elicited in rodents by experimental nerve injury (Desroches et al., 2008) and by injections of formalin (Guindon et al., 2007) or carrageenan (Comelli et al., 2007). More recently, the potent carbamate-based MGL inhibitor JZL-184 (4-(bis-benzo[1,3]dioxol-5-yl-hydroxy-methyl)-piperidine-1-carboxylic acid 4-nitro-phenyl ester) was shown to produce a classical set of cannabinoid-like responses – including decreases in pain behavior, body temperature and motor activity – when injected in mice (Long et al., 2009a). Administration of JZL-184 also elevates 2-AG levels in peripheral tissues, highlighting the important role of MGL in the hydrolysis of 2-AG outside the brain (Long et al., 2009b), where this endocannabinoid was originally discovered [Mechoulam, 1995].
All potent inhibitors of MGL activity reported thus far – including carbamates (Long et al., 2009; Muccioli et al., 2008), maleimides (Saario et al., 2005), tetrazoles (Zvonok et al., 2008) and isothiazolinones (King et al., 2009) – exert their effects by forming covalent bonds with reactive serine or cysteine residues. Since there are potential clinical disadvantages to this mode of action, in the present study we screened a commercial chemical library to identify scaffolds that might be utilized to design reversible MGL inhibitors. We describe the discovery of two bioisosteric terpenoids, pristimerin and euphol, which inhibit MGL with high potency by interacting in a reversible manner with a novel regulatory region of MGL.
Results and Discussion
Pristimerin inhibits MGL
The screening of the chemical library revealed the presence of multiple active compounds. Because of its high potency, we selected for further studies the friedelane triterpenoid, pristimerin (Table 1)(Dev et al., 1989). Pristimerin inhibited the activity of purified MGL with a median effective concentration (IC50) of 93±8 nM (mean ± SEM; n=3) and that of non-purified MGL (cell lysates of MGL-transfected HeLa cells) with an IC50 of 398±68 nM (n=4) (Figure 1A). The difference in potency observed between these two preparations may reflect the presence of competing reactions in the crude HeLa extract and/or inadequate post-translational modifications of the E. coli protein. Other friedelane triterpenoids represented in the library also inhibited MGL activity, albeit less potently than pristimerin did. These included celastrol, pristimerol, dihydrocelastrol, and dihydrocelastryl diacetate (Table 1). The inhibitory potencies of these compounds appeared to be particularly sensitive to chemical modifications at the two opposite ends of their rigid terpenoid scaffold: the esterification of the carboxyl group at carbon 29, which increased inhibitory activity, and the reduction of the quinone methide ring, which decreased such activity (Table 1).
TABLE 1. Inhibition of Purified Recombinant MGL by Friedelane-Based Pentacyclic Triterpenoids.
| Compound | Structure | IC50 |
|---|---|---|
| 1 Pristimerin | 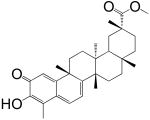 |
93 ± 8 nM |
| 2 Celastrol | 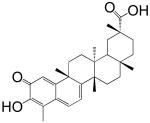 |
1.6 ± 0.4 μM |
| 3 Pristimerol | 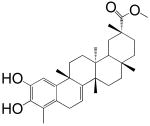 |
4 ± 0.5 μM |
| 4 Dihydrocelastrol | 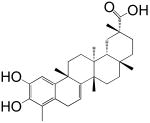 |
15 ± 4 μM |
| 5 Dihydrocelastryl Diacetate | 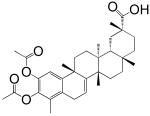 |
15 ± 3 μM |
Results are expressed as mean ± SEM (n = 3-4)
Figure 1. Pristimerin inhibits MGL.

(A) Concentration-dependent inhibition of purified MGL (squares, n=3) and HeLa MGL (circles, n=4) by pristimerin.
(B) Rapid dilution assays of HeLa-MGL in the presence of vehicle (squares, DMSO, final concentration 2%), pristimerin (triangles), or MAFP (circles). We measured the amount of reaction product (pmol/μg protein) generated over a 60-min period following a 20-min preincubation with inhibitor or vehicle (n=3).
(C) Time-course of MGL inhibition by pristimerin (300 nM, n=3).
(D) Michaelis-Menten analysis of the MGL reaction (measured as pmol/min/μg protein) in the presence of vehicle (squares, DMSO, 1%) (n=4) or pristimerin (100 nM, 300 nM, or 500 nM; n=3-4).
Enzyme activity is reported as percent of vehicle controls (DMSO, 1%). Results are expressed as mean ± SEM, with each assay performed in duplicate.
Pristimerin is a reversible MGL inhibitor
The quinone methide group tends to react with cysteine residues in proteins to form covalent adducts (Bolton et al., 1997). To determine whether pristimerin might act through a similar mechanism, we studied the interaction of this compound with purified MGL using a rapid dilution assay (King et al., 2009; Copeland 2005). Under the conditions of this test, incubation of MGL with a covalent inhibitor – for example, the serine-reactive agent methyl arachidonylfluorophosphonate (MAFP) – resulted in the formation of an enzyme-inhibitor complex that is resistant to dilution of the assay mixture (Figure 1B) (King et al., 2009). By contrast, rapid dilution of the MGL-pristimerin mixture resulted in a virtually complete recovery of catalytic activity, which is suggestive of a reversible inhibition (Figure 1B). Time-course experiments showed that addition of pristimerin to purified MGL produces an almost immediate blockade of MGL activity (Figure 1C). Additional kinetic studies using non-purified HeLa MGL demonstrated that pristimerin decreases the maximal catalytic velocity of this enzyme (Vmax in pmol/min/μg; vehicle, 200±20, n=11; 100 nM, 174±7, n=3; 300 nM, 104±7, n=4; 500 nM, 51±18, n=3; P<0.005) without influencing its Michaelis-Menten constant (Km in μM; vehicle, 28±4; 100 nM, 27±1; 300 nM, 24±5; 500 nM, 14±2; P>0.3) (Figure 1D). Together, the results indicate that pristimerin inhibits MGL through a mechanism that is rapid, reversible and non-competitive.
Euphol is a bioisoster of pristimerin
To further evaluate the role of the quinone methide group, we examined whether bioisosters of pristimerin that lack this moiety might also inhibit MGL activity. A computational analysis of structures represented in the chemical library revealed that the triterpene alcohol, euphol (Dev et al., 1989), takes a minimum energy conformation that is superimposable to the rigid scaffold of pristimerin, allowing the hydroxyl groups of the two molecules to overlap (Figure 2A). Enzyme assays showed that euphol inhibits purified MGL with an IC50 of 315±1 nM (n=3) and non-purified HeLa MGL with an IC50 of 882±78 nM (n=3) (Figure 2B). Moreover, rapid dilution and kinetic experiments demonstrated that euphol blocks MGL activity through a reversible and non-competitive mechanism, which is apparently identical to that of pristimerin (Figure 2C,D).
Figure 2. Euphol is a bioisoster of MGL.
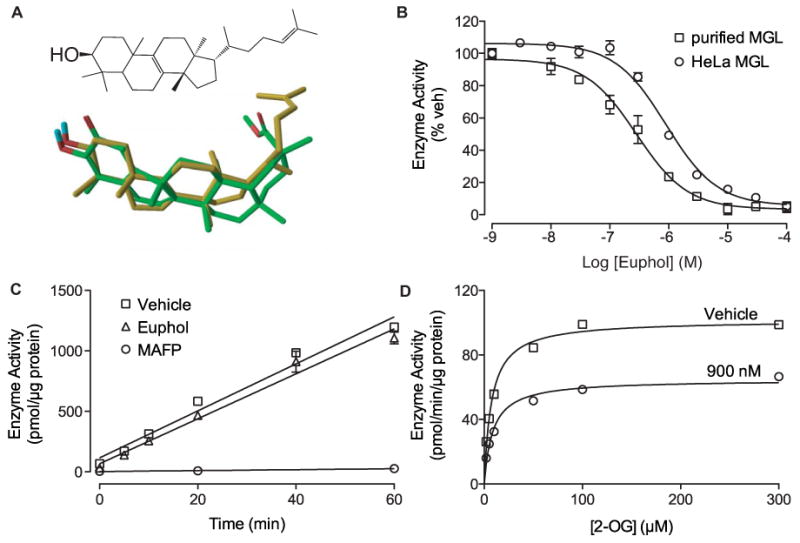
(A) Chemical structure of euphol (top) and 3-dimensional superposition of euphol (gold) and pristimerin (green). Oxygens, red; hydrogens, blue.
(B) Concentration-dependent inhibition of purified MGL (squares) and HeLa MGL (circles) by euphol. Enzyme activity is reported as percent of vehicle controls (DMSO, 1%). Results are expressed as mean ± SEM (n=3).
(C) Rapid dilution assays of HeLa-MGL in the presence of vehicle (squares, DMSO, final concentration 2%), euphol (triangles), or MAFP (circles). We measured the amount of reaction product (pmol/μg protein) generated over a 60-min period following a 20-min preincubation with inhibitor or vehicle. Results are expressed as mean ± SEM (n=3).
(D) Michaelis-Menten analysis of the MGL reaction in the presence of vehicle (squares, DMSO, 1%) (n=4) or euphol (900 nM). Results are expressed as mean ± SEM (n=3).
Interactions with regulatory cysteines: modeling studies
The structural and mechanistic similarities between pristimerin and euphol prompted us to examine whether the two compounds might interact with a common binding pocket in MGL. As a first test of this hypothesis, we utilized an induced-fit docking (IFD) protocol (Sherman et al., 2006) to compare the binding poses adopted by pristimerin and euphol in a refined computational model of MGL (King et al., 2009; Saario et al., 2005). Figure 3A illustrates a docking solution for pristimerin in which the lipophilic portion of the molecule lies on a pocket located within the lid domain of MGL and its 3-hydroxyl group faces the regulatory cysteines, Cys201 and Cys208 (King et al., 2009). This solution was further probed using molecular dynamics (MD) simulations. Starting from the orientation depicted in Figure 3A, pristimerin was found to oscillate around its initial conformation with a ligand atom root mean square distance (RMSD) of 0.36±0.08 Å. Average distances between the polar hydrogen of the 3-hydroxyl group of pristimerin and the sulfur atoms of Cys208 and Cys201 were 2.90±0.25 Å and 3.22±0.27 Å, respectively. This suggests that the binding of pristimerin to MGL might be strengthened by formation of a polar interaction with a regulatory cysteine, possibly Cys208.
Figure 3. Interaction of pristimerin and euphol with regulatory cysteines.
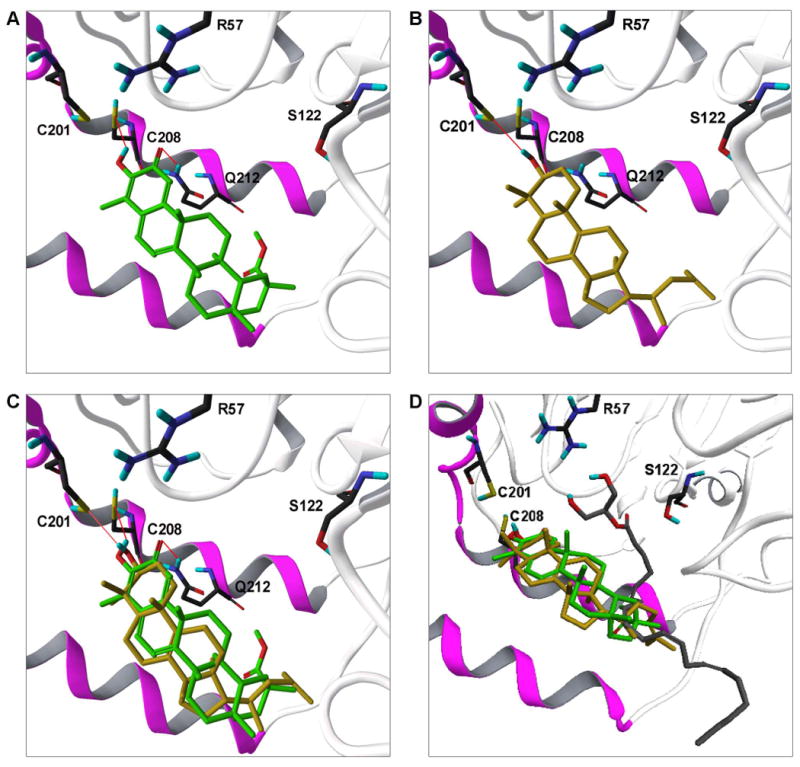
Predicted binding modes of pristimerin (A) and euphol (B) to the model of rat MGL, showing their possible interactions with Cys208 and Cys201. The docking pose adopted by pristimerin predicts an additional interaction with a neighboring glutamine residue (Gln212). Panel (C) shows a superposition of the two models. Panel (D) illustrates a docking pose for 2-AG in the presumptive active site of MGL [10] along with the putative region of overlap with pristimerin and euphol. MGL carbons black; pristimerin, green; euphol, gold; 2-AG, gray; oxygens, red; hydrogens, blue; sulfurs, yellow; nitrogens, dark blue; lid domain, magenta; α/β core, white.
A subsequent set of IFD analyses showed that euphol can be readily superposed on pristimerin so that its 3-hydroxyl group faces Cys208 and Cys201 (Figure 3B,C). Notably, however, the interaction of euphol with Cys208 was lost in the early phase of the simulation (average hydrogen-sulfur distance = 4.00±0.31 Å), whereas the interaction with Cys201 turned out to be significantly more stable (average hydrogen-sulfur distance, 3.13±0.39 Å). During MD simulations, euphol had a higher RMSD compared to pristimerin (0.96±0.09 Å), mainly owing to a change in the conformation of its tail at C17. A plausible interpretation of these results is that pristimerin and euphol occupy a common hydrophobic pocket within the lid domain of MGL, and each inhibitor engages through polar interactions one of two adjacent cysteine residues, Cys201 and Cys208, which flank this pocket. The limited overlap of the region occupied by pristimerin and euphol with the presumptive active site of MGL (Figure 3D) is consistent with the non-competitive kinetic behavior displayed by these inhibitors. However, there are other possible explanations for such behavior, including stabilization of a conformation of MGL that is inaccessible to the substrate.
Interaction with regulatory cysteines: mutational analyses
A prediction of our theoretical model is that mutations affecting either Cys208 or Cys201 should differentially affect the inhibitory potencies of pristimerin and euphol. To test this possibility, we mutated the two cysteines into glycines, expressed the mutants in HeLa cells, and determined the inhibitory effects of pristimerin and euphol on MGL activity in cell extracts. The replacement of Cys208 caused a marked rightward shift in the concentration-response curve for pristimerin (Figure 4A) (IC50; wt = 350±37 nM; C208G = 1182±174 nM; n=6; P<0.0001), whereas it had no effect on euphol (Figure 4B). Conversely, a mutation affecting Cys201 significantly reduced the inhibitory potency of euphol (Figure 4B) (IC50; wt = 513±9 nM; C201G = 2601±200 nM; n=4; P<0.0001) without affecting the potency of pristimerin (Figure 4A). Mutating a cysteine residue that is distant from both active site and lid domain, Cys32, did not influence the activity of either agent (data not shown). The results support the validity of the computational model illustrated in Figure 3D, suggesting that reversible interactions of pristimerin and euphol with Cys208 and Cys201, respectively, contribute to the inhibitory potencies of these two agents.
Figure 4. Effect of cysteine mutations on MGL inhibition by pristimerin and euphol.

Effects of mutating Cys201 or Cys208 on MGL inhibition by pristimerin (A) and euphol (B). Concentration-response curves for the inhibition of wild-type MGL (wt, squares) and MGL mutants containing cysteine-to-glycine mutations at either Cys201 (C201G, triangles) or Cys208 (C208G, circles). Enzyme activity is reported as percent of vehicle controls (DMSO, 1%). Results are expressed as mean ± SEM (n = 4-9). P<0.0001, ANOVA, followed by Dunnett's test.
Effects of pristimerin in intact rat brain neurons
We next asked whether reversible inhibition of MGL activity, such as that produced by pristimerin and euphol, might effectively protect 2-AG from degradation in intact brain neurons, which contain relatively high concentrations of 2-AG and other monoacylglycerols that are substrates for MGL. Incubation of primary cultures of rat cortical neurons with the cysteine-reactive MGL inhibitor, N-arachidonylmaleimide (1 μM, 25 min) (Saario et al., 2005), increased the levels of 2-AG and palmitoylethanolamide (PEA), a non-cannabinoid fatty-acid ethanolamide that is primarily hydrolyzed, in neurons, by fatty-acid amide hydrolase (FAAH) (Figure 5) (Desarnaud et al., 1995; Cravatt et al., 1996). Under the same experimental conditions, incubation with pristimerin (1 μM, 30 min) caused an elevation in cellular 2-AG concentrations (1.6±0.1 fold change, n=3), which was not accompanied by any significant changes in PEA (Figure 5). As expected from its weaker inhibitory potency, euphol (10 μM, 30 min) caused a small, non-significant change in the levels of 2-AG (1.2±0.2 fold change, n=3) and PEA (data not shown).
Figure 5. Pristimerin protects 2-AG from degradation in neurons.

Effects of pristimerin (1 μM), NAM (1 μM) or vehicle (0.1% DMSO in DMEM) in primary cultures of rat cortical neurons. Levels of 2-AG and PEA are reported as fold change over vehicle-treated neurons. Results are expressed as mean ± SEM (n=5). ****P<0.0001, ***P<0.001, *P<0.05, ns = not significant, unpaired Student's t-test.
The results described above suggest that pristimerin preferentially inhibits 2-AG hydrolysis in intact brain neurons. Consistent with this conclusion, a survey of endocannabinoid-metabolizing enzymes showed that pristimerin had no significant effect, in vitro, on the activities of rat brain diacylglycerol lipase (a serine hydrolase responsible for the production of 2-AG) (Bisogno et al., 2003), rat brain FAAH and rat recombinant N-acylethanolamide acid amidase (a cysteine amidase involved in the degradation of PEA and other fatty-acid ethanolamides) (Table S1) (Cravatt et al., 1996; Ueda et al., 2001). Though pristimerin did not affect the activity of the 2-AG hydrolase, ABHD12, it was potent at inhibiting the activity of another 2-AG-hydrolyzing enzyme, ABHD6 (IC50 = 98±8 nM, n=3) (Blankman et al., 2007). ABHD6 displays a closer sequence homology with MGL than ABHD12 does (in sequence similarity; MGL and ABHD6, 33%; ABHD6 and ABHD12, 26.3%). Euphol produced a similar, albeit weaker inhibitory effect on ABHD6 (IC50 = 9±1 μM, n=6).
Significance
MGL is localized to axon terminals throughout the brain and is thought to participate in the physiological elimination of 2-AG at central synapses. Consistent with this view, pharmacological blockade of MGL activity exerts multiple effects in live animals, including profound anti-nociception in models of acute and chronic pain. The irreversible mechanism of action of all potent MGL inhibitors described so far presents, however, possible clinical issues (e.g. the formation of antigenic adducts with plasma proteins) that might eventually hinder the therapeutic use of these agents. To overcome these potential limitations, in the present study we screened a commercial chemical library to identify structural motifs that might be utilized as a starting point for the design of reversible MGL inhibitors. Our screen led to the discovery of two bioisosteric terpenes, pristimerin and euphol, which inhibit MGL activity through a reversible mechanism and with high potency in vitro. Investigations into the inhibitory properties of these agents – utilizing a combination of biochemical, molecular modeling and mutagenesis approaches – suggest that they occupy a common hydrophobic pocket located within the α-helical lid domain of MGL, and each interact reversibly with either of two neighboring cysteines present at the border of such pocket: Cys201 and Cys208. Notably, prior studies had already implicated these residues in the covalent inhibition of MGL by isothiazolinone derivatives (King et al., 2009). The identification of this regulatory region of MGL provides a novel molecular target for the generation of potent and reversible inhibitors.
Experimental Procedures
Chemicals
Compounds 1-6 were part of the Spectrum Collection, purchased from MicroSource Discovery Systems Inc. (Gaylordsville, CT). MAFP and NAM were obtained from Cayman Chemical (Ann Arbor, MI), 2-oleoyl-sn-glycerol (2-OG) from Indofine Chemical (Hillsborough, NJ), and 2-[2H8]-AG from Cayman Chemical; [2H4]-PEA was synthesized as described (Fegley et al., 2005).
Screening for MGL Inhibitors
The Spectrum Collection (Microsource Discovery Systems, Inc, Gaylordsville, CT) is composed of 2,000 compounds, supplied in a 10 mM dimethyl sulfoxide (DMSO) solution. We pooled groups of 10 compounds and performed a primary screen at a concentration of 1 μM. Individual compounds from positive groups (≥50% MGL inhibition) were subjected to a secondary screen at 10 μM. Full concentration-inhibition curves were generated using new lots of dry compounds.
Enzyme Assays
Purified recombinant rat MGL was prepared and enzyme activity was assayed as described previously (King et al., 2007). Full-length rat ABHD6 or ABHD12 was subcloned into a pEF-V5/His vector by TOPO cloning (Invitrogen, Carlsbad, CA) and verified by DNA sequencing. HeLa cells were transiently transfected with pEF6 vector, ABHD6-V5-pEF6 or ABHD12-V5-pEF6 using Superfect reagent (Qiagen, Valencia, CA). 48 hours after transfection, cells were harvested and the homogenates were prepared in 50 mM Tris-Cl, pH 8.0, containing 0.32 M sucrose. ABHD activity was measured using a modified MGL assay procedure (pH 7.5, 3 μg protein per reaction, 30 min at 37°C) (King et al., 2007). Samples were extracted (King et al., 2007) and analyzed by liquid chromatography/mass spectrometry (LC/MS) (see below). 2-AG-hydrolase activity (in pmol/min/mg protein) in mock-transfected HeLa cells was 1.4±0.05, in pEF6-transfected (vector only) was 1.3±0.01, and in ABHD6-transfected cells was 6.9±0.2 (n = 3). FAAH and DGL activities were measured in rat brain homogenates as described [32]. Recombinant NAAA protein was prepared from HEK293 cells stably transfected with pCMV-Flag-rNAAA using SuperFect reagent (Qiagen) and screened with G418 (0.3 mg/ml). Cells were harvested and sonicated in 20 mM Tris-HCl (pH 7.5) with 0.32 M sucrose, centrifuged at 800 × g for 15 min at 4°C, and supernatant was centrifuged at 12,000 × g for 30 min at 4°C. The pellet was suspen ded in phosphate-buffered saline (PBS) and subjected to 2 freeze-thaw cycles at -80°C. The suspension was centrifuged at 105,000 × g for 1 hr at 4°C, and the supernatant containing rNAAA was kept at -80°C until use. NAAA activity was measured by combining substrate (50 μM heptadecenoylethanolamide) with protein (10 μg) in assay buffer (50 mM sodium hydrogen phosphate buffer, pH 5.0, 0.1% Triton X-100, 3 mM DTT) in a final volume of 0.2 ml for 30 min at 37°C. Reactions were stopped by addition of 0.2 ml cold methanol containing 1 nmol of heptadecanoic acid (NuChek Prep, Elysian, MN). For MGL, ABHD and NAAA assays samples were analyzed by LC/MS on an XDB Eclipse C18 column (2.1×30 mm i.d., 1.8 μm,) at 0.6 ml/min for 0.6 min with a solvent mixture of 95% methanol and 5% water, both containing 0.25% acetic acid and 5 mM ammonium acetate. The column temperature was 50°C. Samples were analyzed by electrospray ionization in the negative mode. Capillary voltage was 4 kV, fragmentor voltage was 100 V, nebulizer pressure was 60 psi. N2 was used as drying gas at a flow rate of 13 liters/min and a temperature of 350°C. We monitored the appropriate enzyme activity product (MGL and ABHD, m/z = 281, NAAA, m/z = 267) in the selected ion monitoring (SIM) mode using heptadecanoic acid as standard (m/z = 269).
Site-directed Mutagenesis
Generation of cysteine-to-glycine point mutants by site-directed mutagenesis and subsequent transfections in HeLa cells were performed as previously described (King et al., 2009).
Neuron Cultures
Primary cortical neuron cultures were prepared from embryonic day 18-20 Wistar rats (Stella et al., 2001). Cultures were maintained for 10 days at 37°C with 5% CO2 before treatment with pristimerin (1 μM), euphol (10 μM), NAM (1 μM) or vehicle (0.1% DMSO in Dulbecco's Modified Eagle Medium (DMEM)) for 30 min at 37°C. Reactions were stopped by washing with ice-cold PBS and cells were harvested in 2 ml 50% methanol. Lysates were vortexed for 10 s, protein concentrations were measured by bicinchoninic acid (BCA) protein assay (Pierce, Rockford, IL), and samples were extracted in 4 ml of ice-cold methanol/chloroform/water (1:2:1, vol:vol:vol) containing 0.5 nmol of 2-[2H8]-AG, and 10 pmol each of [2H4]-PEA, added as internal standard. Organic phases were recovered, evaporated under N2, reconstituted in 50 μl chloroform/methanol (1:3, vol:vol) and analyzed by LC/MS as described (Astarita and Piomelli, 2009).
Computational Modeling
Docking studies were carried out using the induced-fit docking (IFD) approach [23] based on Prime 1.7 (Schrödinger, LLC, New York, NY) and Glide 5.0 (Schrödinger, LLC, New York, NY) molecular modeling software. Coordinates of rat MGL were taken from a previously published model of the enzyme in complex with 2-AG (King et al., 2009). Inhibitors were built using Maestro 8.5 (Schrödinger, LLC, New York, NY) and their geometries were optimized to an energy gradient of 0.01 kcal/(mol·Å)with the OPLS2005 force field (Jorgensen et al., 1996). In turns, the inhibitors were placed in an arbitrary position within a region centered on residues Cys201, Cys208 or Cys242 of MGL using enclosing and bounding boxes of 20 and 10 Å on each side, respectively. For the IFD protocol, an initial softened-potential docking of the ligand to the rigid receptor was performed, with van der Waals radii scaling of 0.5 for both MGL and ligand non-polar atoms. Sampling of the protein conformation for each of the top 20 ligand poses (ranked by Glide) was performed using Prime. In this phase, residues within 5 Å of the ligand pose were refined by side-chain conformational search followed by full minimization of the residues and the ligand. Complexes within 30.0 kcal/mol of minimum energy structure were taken forward for re-docking. The related ligand was re-docked into each low-energy, induced-fit structure with default Glide settings (van der Waals radii scaling of 1.0 for MGL and 0.8 for the ligand). Each complex was then ranked according to the IFD score.
Molecular dynamics (MD) simulations of the IFD complexes were performed using OPLS2005 force field in combination with the surface generalized Born (SGB) continuum model for solvent (water) representation. SHAKE function was used and a time step of 2 fs was applied for 2 ns of simulation at 300 K, after an equilibration phase of 200 ps at the same temperature. Backbone atoms were frozen during the simulations to preserve MGL tertiary structure. Interactions between the 3-hydroxyl group of the inhibitors and cysteine sulfur atoms were monitored during the simulations. Distances are reported as average values over 200 snapshots extracted from MD trajectory. Pristimerin and euphol were aligned by a rigid fit procedure superposing the carbon atoms of ring A, B and C. The minimum energy conformation of euphol giving the best fit in terms of RMSD was selected for modeling purpose.
Supplementary Material
Acknowledgments
This work was supported by the National Institute on Drug Abuse (R01 DA-012447 to DP), the NIH Training Program in Cellular and Molecular Neuroscience (T32NS007444-7), and the Italian Ministry of University and Research (2005032713_002 to MM).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Astarita G, Piomelli D. Lipidomic analysis of endocannabinoid metabolism in biological samples. J Chromatogr B Analyt Technol Biomed Life Sci. 2009 doi: 10.1016/j.jchromb.2009.01.008. Advanced online publication, Jan 14, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bisogno T, Howell F, Williams G, Minassi A, Cascio MG, Ligresti A, Matias I, Schiano-Moriello A, Paul P, Williams EJ, et al. Cloning of the first sn1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J Cell Biol. 2003;163:463–468. doi: 10.1083/jcb.200305129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blankman JL, Simon GM, an Cravatt BF. A comprehensive protile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem Biol. 2007;14:1347–1356. doi: 10.1016/j.chembiol.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bolton JL, Turnipseed SB, Thompson JA. Influence of quinone methide reactivity on the alkylation of thiol and amino groups in proteins: studies utilizing amino acid and peptide models. Chem Biol Interact. 1997;107:185–200. doi: 10.1016/s0009-2797(97)00079-3. [DOI] [PubMed] [Google Scholar]
- 5.Comelli F, Giagnoni G, Bettoni I, Colleoni M, Costa B. The inhibition of monoacylglycerol lipase by URB602 showed an anti-inflammatory and anti-nociceptive effect in a murine model of acute inflammation. Br J Pharmacol. 2007;152:787–794. doi: 10.1038/sj.bjp.0707425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Copeland RA. In: Evaluation of Enzyme Inhibitors in Drug Discovery. Copeland RA, editor. Hoboken, NJ: John Wiley and Sons, Inc.; 2005. pp. 56–63. [Google Scholar]
- 7.Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- 8.Désarnaud F, Cadas H, Piomelli D. Anandamide amidohydrolase activity in rat brain microsomes. Identification and partial characterization. J Biol Chem. 1995;270:6030–6035. doi: 10.1074/jbc.270.11.6030. [DOI] [PubMed] [Google Scholar]
- 9.Desroches J, Guindon J, Lambert C, Beaulieu P. Modulation of the anti-nociceptive effects of 2-arachidonoyl glycerol by peripherally administered FAAH and MGL inhibitors in a neuropathic pain model. Br J Pharmacol. 2008;155:913–924. doi: 10.1038/bjp.2008.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dev S, Gupta AS, Patwardhan SA. In: Handbook of Terpenoids. Dev S, editor. Boca Raton, FL: CRC Press, Inc.; 1989. pp. 7–62. [Google Scholar]
- 11.Dinh TP, Carpenter D, Leslie FM, Freund TF, Katona I, Sensi SL, Kathuria S, Piomelli D. Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc Natl Acad Sci U S A. 2002;99:10819–10824. doi: 10.1073/pnas.152334899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dinh TP, Kathuria S, Piomelli D. RNA interference suggests a primary role for monoacylglycerol lipase in the degradation of the endocannabinoid 2-arachidonoylglycerol. Mol Pharmacol. 2004;66:1260–1264. doi: 10.1124/mol.104.002071. [DOI] [PubMed] [Google Scholar]
- 13.Fegley D, Gaetani S, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D. Characterization of the fatty acid amide hydrolase inhibitor cyclohexyl carbamic acid 3′-carbamoyl-biphenyl-3-yl ester (URB597): effects on anandamide and oleoylethanolamide deactivation. J Pharmacol Exp Ther. 2005;313:352–358. doi: 10.1124/jpet.104.078980. [DOI] [PubMed] [Google Scholar]
- 14.Guindon J, Desroches J, Beaulieu P. The antinociceptive effects of intraplantar injections of 2-arachidonoyl glycerol are mediated by cannabinoid CB2 receptors. Br J Pharmacol. 2007;150:693–701. doi: 10.1038/sj.bjp.0706990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gulyas AI, Cravatt BF, Bracey MH, Dinh TP, Piomelli D, Boscia F, Freund TF. Segregation of two endocannabinoid-hydrolyzing enzymes into pre- and postsynaptic compartments in the rat hippocampus, cerebellum and amygdala. Eur J Neurosci. 2004;20:441–458. doi: 10.1111/j.1460-9568.2004.03428.x. [DOI] [PubMed] [Google Scholar]
- 16.Hohmann AG, Suplita RL, Bolton NM, Neely MH, Fegley D, Mangieri R, Krey JF, Walker M, Holmes PV, Crystal JD, et al. An endocannabinoid mechanism for stress-induced analgesia. Nature. 2005;435:1108–1112. doi: 10.1038/nature03658. [DOI] [PubMed] [Google Scholar]
- 17.Jaeger KE, Dijkstra BW, Reetz MT. Bacterial biocatalysts: molecular biology, three-dimensional structures, and biotechnological applications of lipases. Annu Rev Microbiol. 1999;53:315–351. doi: 10.1146/annurev.micro.53.1.315. [DOI] [PubMed] [Google Scholar]
- 18.Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J Am Chem Soc. 1996;118:11225–11236. [Google Scholar]
- 19.Justinova Z, Mangieri RA, Bortolato M, Chefer SI, Mukhin AG, Clapper JR, King AR, Redhi GH, Yasar S, Piomelli D, et al. Fatty acid amide hydrolase inhibition heightens anandamide signaling without producing reinforcing effects in primates. Biol Psychiatry. 2008;64:930–937. doi: 10.1016/j.biopsych.2008.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karlsson M, Contreras JA, Hellman U, Tornqvist H, Holm C. cDNA cloning, tissue distribution, and identification of the catalytic triad of monoglyceride lipase. Evolutionary relationship to esterases, lysophospholipases, and haloperoxidases. J Biol Chem. 1997;272:27218–27223. doi: 10.1074/jbc.272.43.27218. [DOI] [PubMed] [Google Scholar]
- 21.Katona I, Freund TF. Endocannabinoid signaling as a synaptic circuit breaker in neurological disease. Nat Med. 2008;14:923–930. doi: 10.1038/nm.f.1869. [DOI] [PubMed] [Google Scholar]
- 22.King AR, Duranti A, Tontini A, Rivara S, Rosengarth A, Clapper JR, Astarita G, Geaga J, Luecke H, Mor M, et al. URB602 inhibits monoacylglycerol lipase and selectively blocks 2-arachidonoylglycerol degradation in intact brain slices. Chem Biol. 2007;14:1357–1365. doi: 10.1016/j.chembiol.2007.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.King AR, Lodola A, Carmi C, Fu J, Mor M, Piomelli D. A critical cysteine residue in monoacylglycerol lipase is targeted by a new class of isothiazolinone-based enzyme inhibitors. Br J Pharmacol. 2009;157:974–983. doi: 10.1111/j.1476-5381.2009.00276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, Pavon FJ, Serrano AM, Selley DE, Parsons LH, Lichtman AH, et al. Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat Chem Biol. 2009a;5:37–44. doi: 10.1038/nchembio.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Long JZ, Nomura DK, Cravatt BF. Characterization of monoacylglycerol lipase inhibition reveals differences in central and peripheral endocannabinoid metabolism. Chem Biol. 2009b;16:744–753. doi: 10.1016/j.chembiol.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mechoulam R, Ben-Shabbat S, Hanus L, Ligumsky M, Kaminski N, Schatz AR, Gopher A, Almog S, Martin BR, Compton DR, et al. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol. 1995;50:83–90. doi: 10.1016/0006-2952(95)00109-d. [DOI] [PubMed] [Google Scholar]
- 27.Microsource Discovery Systems, Inc. The Spectrum Collection. Retrieved May 20, 2009, from Web site: http://www.msdiscovery.com/home.html.
- 28.Muccioli GG, Labar G, Lambert DM. CAY10499, a novel monoglyceride lipase inhibitor evidenced by an expeditious MGL assay. Chembiochem. 2008;9:2704–2710. doi: 10.1002/cbic.200800428. [DOI] [PubMed] [Google Scholar]
- 29.Ollis DL, Cheah E, Cygler M, Dijkstra B, Frolow F, Franken SM, Harel M, Remington SJ, Silman I, Schrag J, et al. The α/β hydrolase fold. Protein Eng. 1992;5:197–211. doi: 10.1093/protein/5.3.197. [DOI] [PubMed] [Google Scholar]
- 30.Saario SM, Salo OMH, Nevalainen T, Poso A, Laitinen JT, Järvinen T, Niemi R. Characterization of the sulfhydryl-sensitive site in the enzyme responsible for hydrolysis of 2-arachidonoyl-glycerol in rat cerebellar membranes. Chem Biol. 2005;12:649–656. doi: 10.1016/j.chembiol.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 31.Sherman W, Day T, Jacobson MP, Friesner RA, Farid R. Novel Procedure for Modeling Ligand/Receptor Induced Fit Effects. J Med Chem. 2006;49:534–553. doi: 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- 32.Stella N, Schweitzer P, Piomelli D. A second endogenous cannabinoid that modulates long-term potentiation. Nature. 1997;388:773–778. doi: 10.1038/42015. [DOI] [PubMed] [Google Scholar]
- 33.Stella N, Piomelli D. Receptor-dependent formation of endogenous cannabinoids in cortical neurons. Eur J Pharmacol. 2001;425:189–196. doi: 10.1016/s0014-2999(01)01182-7. [DOI] [PubMed] [Google Scholar]
- 34.Sugiura T, Kondo S, Sukagawa A, Nakane S, Shinoda A, Itoh K, Yamashita A, Waku K. 2-Arachidonoylglycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem Biophys Res Commun. 1995;215:89–97. doi: 10.1006/bbrc.1995.2437. [DOI] [PubMed] [Google Scholar]
- 35.Ueda N, Yamanaka K, Yamamoto S. Purification and characterization of an acid amidase selective for N-palmitoylethanolamine, a putative endogenous anti-inflammatory substance. J Biol Chem. 2001;276:35552–35557. doi: 10.1074/jbc.M106261200. [DOI] [PubMed] [Google Scholar]
- 36.Zvonok N, Pandarinathan L, Williams J, Johnston M, Karageorgos I, Janero DR, Krishnan SC, Makryiyannis A. Covalent inhibitors of human monoacylglycerol lipase: ligand-assisted characterization of the catalytic site by mass spectrometry and mutational analysis. Chem Biol. 2008;15:854–862. doi: 10.1016/j.chembiol.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.