

Abstract
Aging of the human brain is associated with “normal” functional, structural, and molecular changes that underlie alterations in cognition, memory, mood and motor function, amongst other processes. Normal aging also imposes a robust constraint on the onset of many neurological diseases, ranging from late onset neurodegenerative diseases, such as Alzheimer’s (AD) and Parkinson’s diseases (PD), to early onset psychiatric disorders, such as bipolar disorder (BPD) and schizophrenia (SCZ). The molecular mechanisms and genetic underpinnings of age-related changes in the brain are understudied, and, while they share some overlap with peripheral mechanisms of aging, many are unique to the largely non-mitotic brain. Hence, understanding mechanisms of brain aging and identifying associated modulators may have profound consequences for the prevention and treatment of age-related impairments and diseases. Here we review current knowledge on age-related functional and structural changes, their molecular and genetic underpinnings, and discuss how these pathways may contribute to the vulnerability to develop age-related neurological diseases. We highlight recent findings from human postmortem brain microarray studies, which we hypothesize, point to a potential genetically-controlled transcriptional program underlying molecular changes and age-gating of neurological diseases. Finally, we discuss the implications of this model for understanding basic mechanisms of brain aging and for the future investigation of therapeutic approaches.
1. Introduction
Age-gated neurodegenerative and psychiatric diseases affect ~15% of people during their lifespan and are almost uniformly devastating. Current treatments for these diseases are not curative and have limited efficacy for symptom relief. Outside of the disease context, aging by itself is also associated with variable rates of cognitive and motor decline, which can be severely impairing for the most affected individuals. By definition, age is a constraint for the onset and progression of functional declines and age-associated diseases, and thus may represent a key entry point for understanding basic mechanisms of brain aging and for the future investigation of therapeutic approaches for age-related diseases.
Here, we use the term “normal brain aging” to describe aging of the central nervous system in the absence of clinically-diagnosed neurodegenerative or psychiatric diseases, or of related pathology. However, as described in this review, it is becoming increasingly clear that molecular changes occurring during normal brain aging substantially overlap with those observed in the context of many diseases. Thus “normal brain aging” actually refers to average successful aging. This somewhat circular parsing of “normal or average disease-free” from disease-associated age-related changes is currently an unavoidable caveat of the field, but is necessary for investigating the contribution of aging to disease processes. Similarly, the line between developmental and age-related changes is unclear. Subjects as young as 14 years of age display molecular changes in the brain on a continuum with age-related changes that extend throughout old age (Erraji-Benchekroun et al., 2005), and it is likely that molecular aging partially extends from developmental processes.
Normal aging of the brain (i.e., in subjects without age-related neurological disorder) is understudied compared with peripheral aging, and has many fundamentally different molecular mechanisms and modulators. Some mechanisms, such as those related to insulin signaling and cellular insult, are shared between the periphery and brain; however, mechanisms related to cellular turnover and depletion, such as telomere shortening and senescence, are not as pertinent in the largely non-dividing brain. Instead, progressive morphological and molecular changes within life-long existing neurons and glia likely underlie age-related cognitive, motor, and mood changes and disease susceptibility (Fig. 1). Brain aging is also subjected to many unique genetic modulators, such as neurotransmitters, neurotrophins, and neurological disease-related genes (Fig. 1). Here, we briefly review current knowledge on the functional correlates of brain aging and discuss their putative biological underpinnings, starting at the gross structural and functional, cellular, and molecular levels, including genetic modulators. We next introduce the overlap and potential contribution of normal brain aging to age-gated neurological diseases.
Fig. 1. “Normal” age-related molecular changes in neurons and glia, and putative modulators, mediators, and functional consequences.

(A) Known age-related cellular phenotypes in neurons (blue) and glia (red). Cells are depicted as a generic neuron and glia for simplicity, but some information is known about class-specific changes in neurons and glia. Also not shown are changes in brain white matter tract and blood vessel integrity. Many neuronal phenotypes (such as DNA damage) also occur in glia, but are not depicted here for clarity. In parentheses are single representative examples (amongst many) of age-regulated gene expression changes seen by human brain microarray that may contribute to/underlie a particular cellular phenotype. (B) Schematic of putative modulators, mediators, and consequences of brain aging.
Based on our recent findings (Glorioso et al., 2010), we hypothesize that a conserved molecular brain aging program under the control of longevity genes may contribute to functional decline and gating of age-related diseases (See schema in Fig. 9). While a great deal of further investigation on multiple fronts will be required to confirm our model, we propose it as a framework for future studies. This framework is worth discussing, not only as a contribution to basic knowledge, but because many feasible clinical applications would directly extend from the model if it turns out to be correct. We present the observations that led to the model, including the fact that molecular aging preferentially affects a large proportion of neurological disease-related genes, and that those genes are systematically affected during aging in pro-disease directions, together suggesting that aging may promote disease. This is consistent with other microarray studies showing significant overlap between gene transcript changes associated with AD and normal aging in the human brain (Avramopoulos et al., 2010; Miller et al., 2008). Additionally, based on our observation that a low-expressing polymorphism in a putative longevity gene, Sirtuin 5 (SIRT5prom2), associates with accelerated rates of molecular aging for selective disease-related genes (Glorioso et al., 2010), we hypothesize that age- (and disease-) related changes may be under differential control by longevity genes. Lastly, we discuss future directions for prevention, delay, and predicting onset of age-gated neurological diseases and functional declines.
Fig. 9. Proposed molecular/cellular pathways underlying molecular aging and its functional consequences.
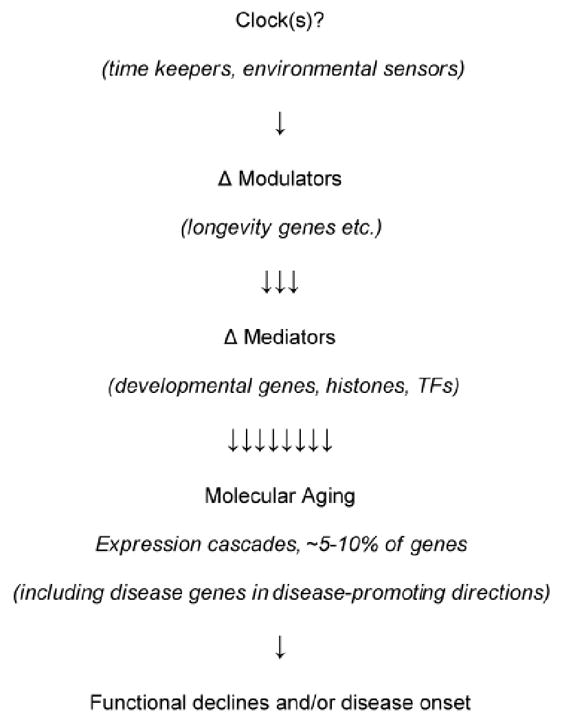
Modulation of this pathway by genetic and/or environmental factors is hypothesized to underlie molecular mechanism(s) behind the gating of disease onset and variable functional declines by normal brain aging.
2. Normal age-related changes in the human brain
2.1. Changes in cognition, motor function, and affect
“Normal” age-related declines in neurological functioning (i.e. not observed in the context of any age- or brain-related diseases) have been extensively studied, and are robust and consistent. Meta-analysis of cross-sectional and longitudinal datasets have demonstrated a ~40–60% decline in cognitive speed at age 80 compared to age 20 in non-demented adults (Salthouse, 1982, 1996). Aging differentially affects aspects of neurological functioning (Christensen, 2001; Ruffman et al., 2008; Salthouse, 1982; Schaie, 1996). For instance, “fluid abilities,” or those reliant on processing speed, problem solving, inhibitory function, working memory, long-term memory and spatial ability, decline with age (Fig. 2) (Park and Reuter-Lorenz, 2009; Salthouse, 1982, 1996). In fact, performance IQ (reliant primarily on fluid abilities) measured by the Welchsler test drops on average 30 points from age 20 to age 70 (Salthouse, 1996; Wechsler, 1981). In contrast, so-called “crystallized abilities” related to knowledge or expertise, such as vocabulary, world knowledge, general knowledge, implicit memory, understanding proverbs, and occupational expertise, do not decline or may even show improvement over the lifespan (Fig. 2; reviewed in (Christensen, 2001; Park and Reuter-Lorenz, 2009; Salthouse, 1982)). Consistent with this observation, verbal IQ measured in WAIS-R standardization sample only drops on average 5 points from age 20 to age 70 (Kaufman, 1990; Salthouse, 1996). Importantly, grey matter volume loss and molecular changes may underlie these functional changes in cognitive ability (Park et al., 2002).
Fig. 2. Differential effects of aging on cognitive processes.
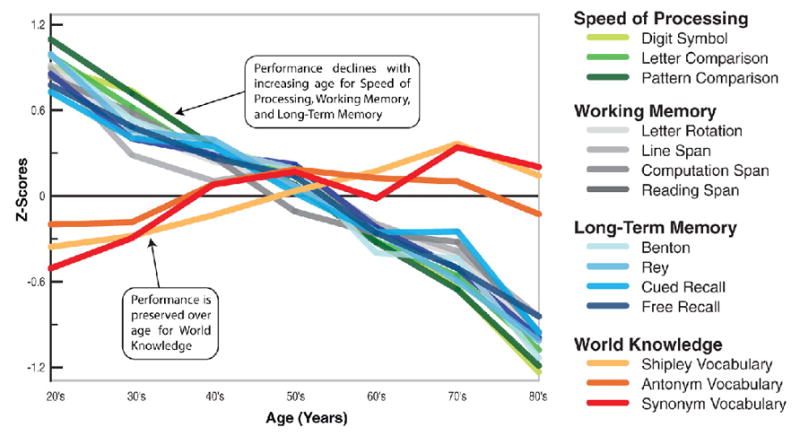
Adapted from (Park and Reuter-Lorenz, 2009). Z-score (y-axis) is a measure of distance from the mean in standard deviations ((value- mean)/stdev) in the respective cognitive ability listed at the right.
A variety of motor functions, including reaction time, speed of movement, as well as hand and foot coordination, consistently slow down with age (Era et al., 1986; Fozard et al., 1994; Kauranen and Vanharanta, 1996). This is likely mediated by aging of the central nervous system (CNS), as age-related decline in cortical dopamine D2 receptor level (Volkow et al., 1998; Volkow et al., 2000) correlates with motor speed and cognitive function (Volkow et al., 1998). Like cognitive aging, these changes follow a continuous and progressive pattern, as exemplified in the Baltimore longitudinal study, which demonstrated that simple and disjunctive reaction times (i.e. go no-go task) increased by 0.5 (~4.0%/decade) and 1.6 msec/year (3.9%/decade), respectively, starting at age 20 and continuing through the oldest ages (90+ years) (Fozard et al., 1994).
Meta-analyses show that changes in mood and affective perception also occur with age. Older adults are worse at recognizing sad or angry emotions compared to younger adults, an effect that is not accounted for by general cognitive decline, as older subjects perform equally as well in recognition of happiness and actually better in recognition of disgust (Ruffman et al., 2008). Generally, older adults show a positivity-bias in attention and memory, recover faster from negative events, and are less likely to engage in destructive interpersonal interactions, such as shouting during conflict (reviewed in (Mather and Carstensen, 2005)). Consistent with these reports, older adults have a lower prevalence of diagnosed major depression, with average onset of major depression peaking at age 30.5 and declining risk beginning in the early 40s (Hasin et al., 2005). This seems counterintuitive for two reasons. First, suicide rates are greatly increased in people over 65 compared to the general population (14.2 and 10.2 per 100,000 respectively), with highest rates in non-hispanic white males over 85 (48 per 100,000) (http://www.nimh.nih.gov/health/publications/suicide-in-the-us-statistics-and-prevention/index.shtml#CDC-Web-Tool). Second, the frontal brain areas required for cognition that decline functionally with age are also required for emotional control, which does not seem to decline with age.
One explanation is that increasing suicide rates with age are not due to increasing risk of major depression, but rather to higher ratios of completion to attempts. This appears specifically pertinent for men, especially those living alone, widowed, and/or terminally ill (De Leo et al., 2001; Hawton and van Heeringen, 2009). Alternatively, while mood disorder diagnoses do not necessarily increase with age, studies report increasing numbers of depressive symptoms in older individuals, potentially reflecting diagnostic structure and procedural issues (Fiske et al., 2009). Indeed, psychomotor retardation, anhedonia, decreased memory and slower cognitive speed are more prevalent in older depressed individuals, and are often misattributed to aging per se, leading to under-diagnosing depression (Alexopoulos, 2005; Alexopoulos et al., 2005). The converse also appears to be true, as adults with poorer performance on cognitive tests also show less positivity-bias, suggesting decreased emotional control (Mather and Carstensen, 2005), together leading to a cause-effect conundrum. Furthermore, low mood and major depression often occur in the context of neurodegenerative and dementia-related diseases, such as Alzheimer’s (AD) and Parkinson’s (PD) diseases, in people with poorer cardiovascular health, such as stroke (Fiske et al., 2009). Combined, these observations suggest that individuals adaptively learn and become more proficient at maintaining a positive affect during successful aging, but that this control declines with declining cognition and in the context of degenerative states. Thus, age-related loss of emotional regulation in people with more rapidly declining cognition may be an important risk factor for depression in old age.
It is important to note that there is large individual variability in the rates of age-related cognitive decline, ranging from little or no decline in cognition to more rapid decline (Fig. 3; (Wilson et al., 2002)). A potential caveat to many cross-sectional studies on this topic is that the largest predictor of cognitive capacity in old age is cognitive capacity in youth, accounting for ~50% of the variance (Deary et al., 2007). With this in mind, there is good longitudinal evidence for both genetic and environmental contributors to rates of cognitive decline. The APOE-ε4 allele, for example, which increases risk for and lowers onset age of AD, has been extensively studied and there is substantial evidence for association with accelerated cognitive decline, even in the absence of AD (Anstey and Christensen, 2000; Deary et al., 2007; Deary et al., 2002; Mattay et al., 2008). There is also some evidence for genetic contribution of COMT, BDNF, Klotho, PRNP, and DISC-1 (see also genetics section (Deary et al., 2007; Deary et al., 2004; Mattay et al., 2008)). Environmental factors associated with slower cognitive decline include exercise, healthy body mass index, higher education level, good cardiovascular health, being a non-smoker, and caloric restriction. Evidence also suggests protective effects for nutritional factors, such as antioxidant, omega-3 fatty acids, and red wine consumption (Anstey and Christensen, 2000; Deary et al., 2007; Fillit et al., 2002; Joseph et al., 2009; Muscari et al., 2009; Panza et al., 2004).
Fig. 3. Individual differences in rates of cognitive decline.
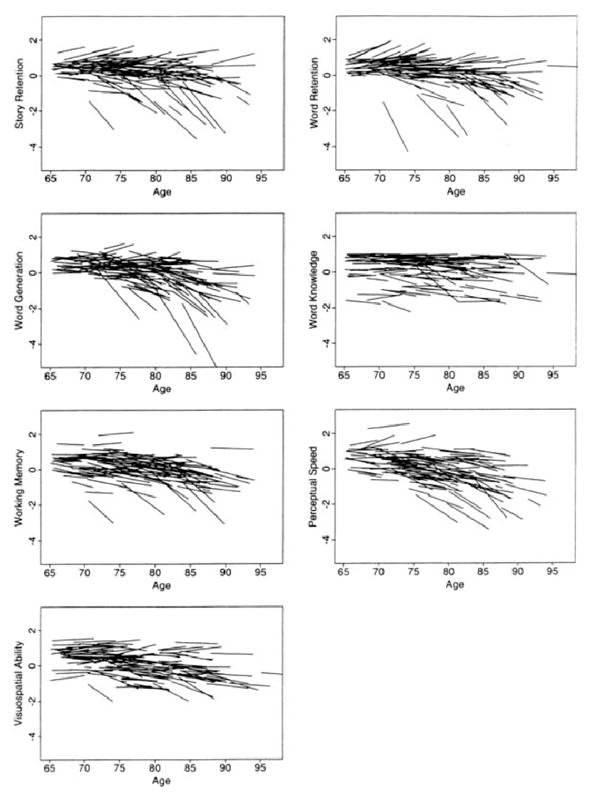
Adapted from (Wilson et al., 2002). Graphs are organized according to the age of the participant at each evaluation; the length of each line relative to the x-axis indicates the years of longitudinal observation for that individual. Values on the y-axis are z-scores. Overall, substantial heterogeneity is evident in each cognitive domain, with a few people exhibiting a precipitous decline and most people remaining stable, declining, or improving slightly. These are underestimates of change rates, as they are not corrected for practice effects, which were significant.
2.2. Functional/structural changes
What structural and functional changes potentially underlie age-related changes in cognition, motor function, and affect? Longitudinal and cross-sectional MRI studies have demonstrated significant grey matter loss with age at rates of 3.2–5.3 ml per year or approximately 0.5% per year (Fig. 4) (Good et al., 2001; Smith et al., 2007; Tisserand et al., 2002; Tisserand et al., 2004).
Fig. 4. Grey matter volume decreases linearly with age throughout adulthood.
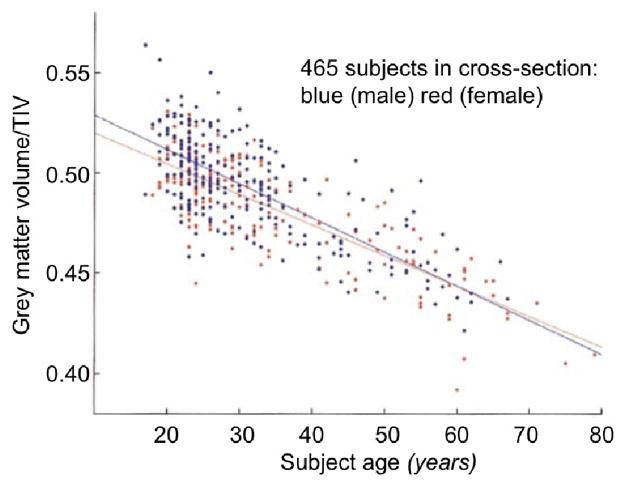
Adapted from (Good et al., 2001). Scatter plot of total grey matter volume versus subject age. Grey matter volumes are presented as fractions of total intracranial volume (TIV) to correct for differences in head size. Linear regression lines for males and females are superimposed. It is evident that large losses in grey matter volume (~20% from age 20 to 80) occur during adulthood. No large male/female differences are observed.
A striking example is a study conducted by Resnick et. al (2003), who followed longitudinally 92 non-demented adults (ages 59–85) at baseline, 2, and 4 years out and reported significant (p<0.001) decreases in total grey matter at each successive time point within individuals (Resnick et al., 2003). These changes were area-specific, as frontal cortex areas such as anterior cingulate cortex, prefrontal cortex, left and right insula, and left inferior frontal cortex, have the most consistently reported decreases in volume with age, whereas the hippocampus and amygdala display variable effects or unchanged volumes between studies (Gianaros et al., 2006; Good et al., 2001; Resnick et al., 2003; Seshadri et al., 2004; Tisserand et al., 2002; Tisserand et al., 2004). The area-specificity of these changes is consistent with age-related cognitive changes, i.e. the most robust changes are seen in frontal areas required for cognitive processing and working memory. Additionally, losses of white matter integrity and changes in brain activity in response to tasks on fMRI, such as hippocampal hypo-responsiveness and greater frontal bi-laterality in memory tasks, are seen with age (reviewed in (Park and Reuter-Lorenz, 2009)). Finally, substantial evidence indicates that structural and functional changes correlate with cognitive changes (reviewed in (Park and Reuter-Lorenz, 2009)).
2.3. Changes in cellular morphology
What underlies changes in grey-matter/white matter volumes? It was previously thought that neuronal death occurred in the brain with age. However, better stereological techniques and careful exclusion of subjects with neurodegenerative diseases has shown that little or no neuronal death occurs during normal aging (Gomez-Isla et al., 1997; Haug et al., 1984) (reviewed in: (Morrison and Hof, 1997)). Instead, grey matter volume losses appear to result from neuronal dendritic arbor shrinkage and synaptic losses (de Brabander et al., 1998; Scheibel et al., 1975) (reviewed in: (Dickstein et al., 2007)). For example, Duan et al (2003) demonstrated a ~43% and ~27% loss of apical and basal cortical neuron dendritic spines, respectively, in aged versus young macaque monkeys (Fig. 5) (Duan et al., 2003). These reductions in synapse density may underlie cognitive deficits, as they correlate with the degree of age-related reduction in cognitive functioning within individual primates (Peters et al., 2008; Peters et al., 1998).
Fig. 5. Age-related loss of dendritic spine density.
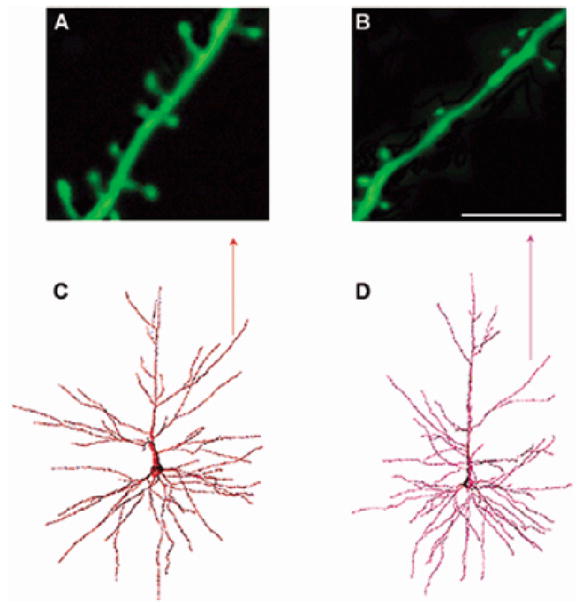
(A–D), adapted from (Dickstein et al., 2007). (C–D), originally adapted from (Duan et al., 2003). Representative images of spine densities on neocortical pyramidal neurons from young and aged rhesus monkeys are presented. Panels A and B show confocal laser scanning images of apical dendritic segments in a young (10–12 yrs) (A) and aged (24–25 yrs) (B) rhesus monkey (scale bar = 8 μm). Note the increased spine density in the young monkey compared to the old monkey. Panels C and D show examples of a retrogradely traced neuron, filled with Lucifer Yellow, and reconstructed in 3-dimensions using NeuroZoom and NeuroGL software applications. The neuron in (C) is from a young animal and the neuron in (D) is from an aged animal. The arrow points to the dendritic segments analyzed in A and B.
Similarly, for glial cells, older studies concluded that astrocytic gliosis and microgliosis, as visualized by MHC-II or GFAP (glial markers) staining in brain slices, was caused by increasing glial numbers with age, which has been shown by more recent stereological techniques to be largely inaccurate (Conde and Streit, 2006; Finch, 2003; Unger, 1998). Instead, glial processes thicken with age (and thus show greater glial marker staining, termed “glial dystrophy”), perhaps to preserve correct distances from shrinking neuronal processes for glial-neuronal exchange (Conde and Streit, 2006; Finch, 2003; Unger, 1998).
3. Molecular pathways of brain aging
In addition to cellular morphological changes, aging neurons display evidence for increasing DNA damage, accumulation of reactive oxygen species, calcium dysregulation, mitochondrial dysfunction, and inflammatory processes (reviewed in (Yankner et al., 2008)). Some of these processes are cell-type specific, such as the loss of specific neuropeptide markers of interneuron classes (Erraji-Benchekroun et al., 2005). Cell type dissection of these processes in the human and primate brain are still largely lacking, so we discuss changes in the context of a “generic” neuron and glia here. Underlying these cellular phenotypes, CNS molecular aging both shares and has unique molecular pathways with mechanisms involved in aging of peripheral somatic organs. Shared pathways include those related to metabolism and caloric restriction, as well as those related to cellular insult, including DNA damage, inflammation, and damage by reactive oxygen species (ROS) (see below; reviewed in (Yankner et al., 2008)). In peripheral tissues, aging is additionally driven by senescence-related mechanisms, such as telomere shortening and depletion of precursor cells, which are not as pertinent in the CNS, since cellular turnover is limited. Conversely, several molecular mechanisms may predominate in the nervous system, including those driven by neurotrophins (BDNF) and neurotransmitters (serotonin, dopamine, glutamate (Backman et al., 2006; Mattson, 2008; Mattson et al., 2004a, b); see below).
3.1. Metabolic changes
Caloric restriction is one of the most investigated mechanism leading to increased longevity across species, ranging from studies in yeast to primates (Narasimhan et al., 2009). Relevant to brain aging specifically, a primate caloric restriction model demonstrated delayed brain atrophy (Colman et al., 2009), while short-term caloric restriction is prospectively associated with improved memory in older humans (Witte et al., 2009). The effect of caloric restriction may be an evolutionarily conserved means of sensing poor nutritional environments and thus increasing reproductive lifespan to wait for richer environments. Its mechanism is tied into and shares signaling with ROS-related mechanisms, as caloric restriction -driven metabolic slowing results in fewer mitochondrial produced ROS and thus less DNA damage (reviewed in (Papaconstantinou, 2009)). Mediators of caloric restriction, including growth hormone, insulin, insulin-like growth factor (IGF-1), the circulating longevity hormone, Klotho, which binds to the IGF-1 receptor, as well as the Sirtuin family of longevity genes, have been shown to modulate both organismal lifespan and healthspan across species (Bartke, 2006; Kurosu et al., 2005; Narasimhan et al., 2009). Emerging evidence, including causal studies in model organisms and genetic association of human polymorphisms with brain aging (see genetics section), suggest that these mediators may represent key players in brain aging.
The Sirtuin family of longevity genes, encompassing seven homo logous NAD-dependent histone and protein deacetylases offer particularly promising targets for key brain aging modulators. Related to their varied subcellular localizations, they have pleiotropic anti-aging effects in neurons and glia, including abrogation of protein aggregates, accumulation of misfolded proteins, enhancement of stress response, improving DNA repair, as well as prevention of inflammatory processes in glia, cell death pathways, and mitochondrial dysfunction (reviewed in (Gan and Mucke, 2008)). In relation to brain aging specifically, SIRT1 has been shown to prevent Wallerian-type axonal degeneration after injury (Araki et al., 2004) and to be a key player in neurogenesis (Libert et al., 2008). Other promising targets in this pathway, which have been predominantly studied in model organisms and have yet to be studied in the CNS context, include FOXO, Daf16, p66, PTEN, m-TOR, and CLOCK-1, amongst many others (reviewed in (Narasimhan et al., 2009)), as the investigation of mechanisms underlying the metabolic control of brain aging are only beginning to be understood.
3.2. Cellular insult
Nuclear and mitochondrial DNA damage, predominantly single base mutations such as ROS-induced 8-oxoguanine DNA lesions, steadily accumulate in the aging brain across species (reviewed in (Droge and Schipper, 2007; Yankner et al., 2008)). There is considerable support for pathways leading to diseases that are downstream of increased DNA damage and ROS-mediated mechanisms, including accelerated brain aging and neurodegeneration in people with progeria syndromes caused by DNA repair gene mutations (reviewed in (Yankner et al., 2008). Moreover, the severity of age-related memory impairment is correlated with brain and plasma levels of antioxidants (reviewed in (Droge and Schipper, 2007)). Evidence also suggests a role for P53 in brain aging and centrally-driven lifespan regulation, as selective brain knock-down of P53 in Drosophila results in increased longevity and resistance to oxidative stress (Bauer et al., 2005). However, the pathways linking DNA damage to age-related neuronal/glial morphological changes remain poorly understood.
3.3. Neurotrophins
While there is a large literature relating neurotrophins, such as GDNF, NGF and bFGF, to treatment and prevention of neurodegenerative disease, such as AD and PD, BDNF is the better characterized modulator of normal brain aging (reviewed in (Tapia-Arancibia et al., 2008)). Particularly, there is a large literature connecting the human BDNF val66met (secretion deficient) polymorphism to brain aging in mice and humans (see genetics section below). Additionally, it is a logical modulator of molecular aging, as it is an activity-dependent secreted growth factor that declines steadily with age in the brain (Erraji-Benchekroun et al., 2005), is neuroprotective against a variety of insults, and is required for changes in spine density underlying learning and memory systems that decline with age (reviewed in (Tapia-Arancibia et al., 2008)). Indeed, infusion of BDNF can restore age-related impairment in long term potentiation (LTP) in middle-aged rats (Rex et al., 2006). BDNF is also required for neurogenesis in mice and its expression is induced by exercise and caloric restriction, which are known to be modulators of normal brain aging (reviewed in (Tapia-Arancibia et al., 2008)). Together, these results establish causality and biological relevance for BDNF in several brain aging measures.
The pathways downstream of BDNF that specifically modulate cellular age-related molecular changes are just beginning to be elucidated. Microarray experiments in conditional BDNF KO mice have shown that gene expression changes downstream from low BDNF correlate with normal age-related changes, but also provide evidence that BDNF is not a “global modulator,” as the correlation was not high enough to fully account for all age-regulated molecular changes (Glorioso et al., 2006; Sibille et al., 2007). Nevertheless, some of the most BDNF-regulated genes display robust age-related molecular changes (Glorioso et al., 2006), including RGS4, CCND2, APBAP2, SYN2, SST, NPY, and PENK1. This suggests that a subset of normal age-related molecular changes in both mouse and human are directly modulated by BDNF level and function. Together, this suggests that BDNF and its downstream molecular pathways may be attractive targets for therapeutic intervention, although much remains to be investigated as to the specificity of BDNF effects for age-related changes.
3.4. Neurotransmitters
There is increasing evidence for a role for neurotransmitters, particularly serotonin, glutamate and dopamine, in modulating normal brain aging. Because so many current psychiatric and neurological drugs target neurotransmitter receptors, this has potentially important implications for public health and for new treatment strategies aimed at aging and age-related disease. Serotonin has been hypothesized to play a role in normal brain aging, as its levels and selected receptor functions are age-regulated. Moreover, serotonin shares signaling pathways with other known age-regulatory molecules, such as BDNF and IGF-1 (reviewed in (Mattson et al., 2004a, b), and because the selective serotonin reuptake inhibitor antidepressants (implicitly thought of as anti-aging) can modulate neurogenesis (Dranovsky and Hen, 2006). Additionally, serotonin blocking drugs and mutations for serotonin synthesis (tph-1) or receptor function (ser-1, ser-4) in C. elegans delay age-related phenotypes and/or extend longevity (Murakami et al., 2008; Petrascheck et al., 2007, 2009; Sze et al., 2000). In mice, deletion of the serotonin 1B (HTR1B) receptor leads to cognitive changes (Wolff et al., 2003) (reviewed in (Buhot et al., 2003)) and to anticipated locomotor and molecular brain aging phenotypes (Sibille et al., 2007), providing causal evidence linking genetic disruption of serotonergic signaling and aging. However, lifespan/healthspan extension by serotonergic genetic or pharmacological modulation in higher order organisms has not been investigated. Also lacking are prospective studies of human age-related phenotypes in relation to exposure to serotonergic drugs.
Glutamate, the brain predominant excitatory neurotransmitter, is also a probable candidate for modulating brain aging, as it facilitates release of BDNF, is essential for LTP, synaptic plasticity, neurogenesis, activity-dependent neuronal survival, and neural outgrowth during development (reviewed in (Mattson, 2008)). Through elevation of intracellular Ca2+ and effects on CREB and NFκB mediated signaling, glutamate modulates both rapid changes in dendritic architecture and long term transcriptional changes on a variety of genes, including neurotrophins, through which it likely contributes to age-related morphological changes (reviewed in (Mattson, 2008)). Glutamate is also an important player because of its role in both protection in the context of ischemic preconditioning as well as facilitation of excitotoxic neuronal injury and death (reviewed in (Mattson, 2008)).
Dopamine has strong correlative and causative evidence for modulation of brain aging (Backman et al., 2006). It is also implicated in several age-gated diseases, including PD, HD, SCZ, and BPD. Components of the dopamine system also decline with age, including the dopamine transporter (DAT) as well as the D1 and D2 receptors (reviewed in (Backman et al., 2006)). Mice fed with Levodopa, a dopamine precursor, have extended longevity by 50% and improved fertility. This effect is potentially mediated by the caloric restriction pathway, as mice showed reduced growth despite the same amount of food intake as controls (Cotzias et al., 1974; Cotzias et al., 1977). This cross-talk with caloric restriction pathways is corroborated by studies in D2 receptor knock-out (KO) mice, which have decreased levels of GH and IGF-1 (Diaz-Torga et al., 2002). In humans, pharmacological studies in healthy volunteers have shown that D-amphetamine enhances performance on a variety of tasks and that the D2 receptor agonist, Bromocriptine, facilitates spatial working memory, suggesting that D2 receptor down-regulation with age may play a role in age-related memory and cognitive decline (Backman et al., 2006). Additionally, PET studies have demonstrated that in healthy adults, striatal D2 receptor binding correlates with executive and motor functioning as well as perceptual speed (both of which correlated with subject age), even when controlling for chronological age (Backman et al., 2000; Backman et al., 2006; Volkow et al., 1998). This suggests that D2 levels may be partially responsible for the effects of molecular aging on cognitive function. In addition to declining dopamine signaling having cross-talk with caloric restriction pathways and directly mediating age-related cognitive decline, dopamine pathways also have cross-talk with ROS age-related pathways. For example, Monoamine oxidase B, which acts to inactivate dopamine, robustly increases with age (Erraji-Benchekroun et al., 2005; Glorioso et al., 2010), generates large numbers of free radicals, and may thus contribute to increasing ROS with age (reviewed in (Luo and Roth, 2000)). Dopamine also regulates glial dystrophy-related pathways by regulating NFκB and GFAP levels (reviewed in (Luo and Roth, 2000)) and interacts with calcium-related pathways and circadian-rhythm pathways through interaction with Clock, amongst other genes (reviewed in (Rollo, 2009)).
4. Molecular underpinnings (gene expression correlates) of normal human brain aging
Several groups, including our own, have characterized the gene expression underpinnings of these age-related changes using human post-mortem brain microarray (Berchtold et al., 2008; Erraji-Benchekroun et al., 2005; Lu et al., 2004). Consistently, a selective portion of the genome (~5–10%) has been shown to have age-regulated changes in expression levels in the brain. The most affected genes are consistent with the age-related cellular phenotypes they likely underlie, i.e. decreasing neurotrophic factors (BDNF, IGF-1), calcium-related proteins (Calbindin), markers of synaptic density (SYN2), and neurotransmitter receptors (HTR2A, DRD2), as well as increasing markers of DNA damage (BCL-2) and glial dystrophy (GFAP, NFk-B). The mechanism(s) driving these selective age-regulated expression changes is largely unknown. Lu et al (2004) postulated a clock mechanism whereby accumulating DNA damage with age could selectively alter promoter regions of age-regulated genes (Lu et al., 2004). In support of this theory, they demonstrated that genes with age-regulated expression levels in human prefrontal cortex had promoter regions that were more vulnerable to DNA damage than genes that were not age-regulated (Lu et al., 2004).
At the same time, we showed that age-regulated gene expression changes were highly conserved across two areas of prefrontal cortex (Fig. 6). These changes were so robust and consistent that biological age (defined as molecular age and obtained from a composite score of age-regulated gene transcript levels) could be used to predict subject chronological age (Fig. 7; (Erraji-Benchekroun et al., 2005)). We further demonstrated that the gene expression correlates of aging were conserved across two separate cohorts, including two additional brain areas, amygdala and anterior cingulate cortex (Glorioso et al., 2010). These results were so remarkably robust between any two areas (i.e., average p-value of correlation <10−7) that they led us to hypothesize that they do not originate from random age-related wear and tear of the brain, but perhaps derive from a transcriptional program that is analogous to, or that extend extending from, brain development. In support of this hypothesis, nervous system development was consistently identified by unbiased large scale analyses as a biological pathway most over-represented within age-related affected genes and included numerous transcriptional regulators (transcription factors, histones, etc.) (Glorioso et al., 2010).
Fig. 6. Molecular aging is conserved in the prefrontal cortex.

Adapted from (Erraji-Benchekroun et al., 2005). An age-related expression cluster representation is depicted for 588 core age-affected genes. Note the continuous progression and similar effects in two areas of the prefrontal cortex (Brodman areas, BA9 and BA47). Each probeset is represented by a row, each array or brain area per subject by a column. Samples are organized left to right by brain area and increasing age. Green and red bars indicate decreased and increased gene expression, respectively, versus the averaged signal for these genes across all samples. Along the Y-axis, probesets are clustered according to similarities in expression profiles across age. In this cohort, a similar number of probesets were downregulated (n=291, upper panel) and upregulated (n=297, lower panel) throughout lifetime. Columns to the right indicate the distribution of genes with glial- (WM) or neuronal-enriched (GM) origin of gene transcripts. Notice the high concentration of glial-enriched genes with increased expression with age, while most, but not all, neuronal-enriched genes appeared to be downregulated with age.
Fig. 7. Molecular age predicts chronological age.
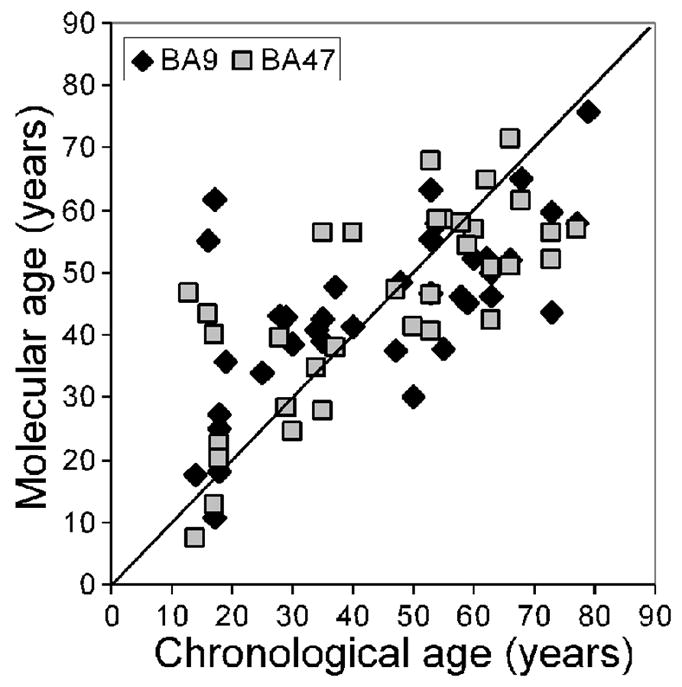
Adapted from (Erraji-Benchekroun et al., 2005). The “molecular” age represents a summary number for each individual that is defined by age-regression analysis of expression levels for core age-affected genes when compared to transcript changes in all other subjects. Overall, there is a high correlation between “molecular” and chronological ages (BA9: r=0.65; BA57, r=0.73). A few samples demonstrated larger deviation than average at both young and older ages. These subjects did not correspond to any identifiable clinical, demographic, or experimental parameters.
What are these age-regulated changes? Fitting with these expression changes underlying shrinking neuronal processes and glial dystrophy, age down-regulated transcripts are predominantly of neuronal origin, whereas age up-regulated transcripts are predominantly of glial origin ((Erraji-Benchekroun et al., 2005), Fig. 6). We further showed that genes associated with six age-gated neurological diseases were highly over-represented within the molecular correlates of aging. These disease-related genes almost unanimously changed in directions that were predicted to cause disease (Table 1). This is consistent with, and also extends using a different approach, two reports showing significant overlap between microarray changes associated with AD and normal aging in human brain (Avramopoulos et al., 2010; Miller et al., 2008). Particularly Miller et al. (2008) reported a significant overlap between AD-related changes in hippocampus and the normal age-related changes previously reported in PFC (Lu et al., 2004), despite differences in brain areas and methodologies in the compared studies (Miller et al., 2008). Avramopoulos et al. (2010) found a more extensive overlap between AD-related changes in temporal cortex and those PFC aging changes, concluding that at least some aspects of molecular aging are shared between normal aging and AD (Avramopoulos et al., 2010). These observations prompted the hypothesis that age-regulated loss or gain of disease-related gene expression may be the gating mechanism for age of onset across many diseases, which we further discuss below.
Table 1. Examples of agreement in directions of changes between age- and disease-related genes.
Adapted from (Glorioso et al., 2010). Red or green boxes indicate significant up-and down-regulations (p<0.05) of mRNA and/or protein levels in disease or during aging. Grey boxes indicate genetic associations with disease with unknown or unclear reports of directionality in disease.
| Gene | Direction of change in DISEASE | Direction of change with AGE | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AD | PD | HD | ALS | SCZ | BPD | MD | ACC | AMY | BA9 | BA47 | ||
| NF-kappa B | NF-kB | |||||||||||
| Amyloid beta precursor protein | APPB2/PAT1 | |||||||||||
| binding- GABA transaminase 2 | GABA-T | |||||||||||
| Period homolog-3 | PER3 | |||||||||||
| Clusterin/Apolipoprotein-J | CLU | |||||||||||
| Monoamine Oxidase B | MAOB | |||||||||||
| Valosin-containing protein | VCP | |||||||||||
| Microtubule-associated protein tau | MAPT | |||||||||||
| Amyloid beta precursor protein | Fe65 | |||||||||||
| Mitochondrial Complex 1 Subunit binding-1 | NDUSF3 | |||||||||||
| Mitochondrial Complex 1 Subunit | NDUFB5 | |||||||||||
| Mitochondrial Complex 4 Subunit | COX7B | |||||||||||
| Mitochondrial Complex 1 Subunit | NDUSF3 | |||||||||||
| Amyloid precursor-like protein 2 | APLP2 | |||||||||||
| Parkinson Disease-7 | DJ-1 | |||||||||||
| Parkinson Disease-5 | UCHL1 | |||||||||||
| Parkinson Disease-13 | HTRA2 | |||||||||||
| Parkinson Disease-2 | Parkin | |||||||||||
| α-synuclein_α-syn | α-SYN | |||||||||||
| Mitochondrial Complex 1 Subunit | NDUSF2 | |||||||||||
| Reelin | RELN | |||||||||||
| Cholecystokinin | CCK | |||||||||||
| Neuropeptide-Y | NPY | |||||||||||
| Cyclin-dependent Kinase-5 | CDK5 | |||||||||||
| Parkinson Disease-6 | Pink1 | |||||||||||
| Aryl hydrocarbon receptor nuclear | BMAL1 | |||||||||||
| Serotonin 5A Receptor translocator-like | HTR5A | |||||||||||
| Serotonin 2A Receptor | HTR2A | |||||||||||
| Regulator of G-protein signaling-4 | RGS4 | |||||||||||
| Somatostatin | SST | |||||||||||
| Brain-derived neurotrophic factor | BDNF | |||||||||||
| GABA receptor, alpha-5 subunit | GABRA5 | |||||||||||
| Dopamine Receptor D1 | DRD1 | |||||||||||
| Neuregulin-1 | NRG1 | |||||||||||
| Parvalbumin | PVALB | |||||||||||
| Glutamate decarboxylase 1 | GAD67 | |||||||||||
| Huntingtin | HD | |||||||||||
| Manganese Superoxide dismutase | SOD2 | |||||||||||
| Cannabanoid Receptor-1 | CB1 | |||||||||||
Additionally and consistent with the large individual variability seen in rates of age-related cognitive decline across individuals, we observed variability in cross-sectional rates of molecular brain aging (Fig. 7), which could not be attributed to cohort characteristics such as body mass index ((Erraji-Benchekroun et al., 2005). This led to the hypothesis that individual differences in molecular brain aging rates may be partially accounted for by genetic variation. We tested this hypothesis using a candidate gene approach and identified a single nucleotide polymorphism (SNP) in the promoter of the Sirtuin 5 gene, which correlates with reduced expression and associated with variable rates of molecular brain aging (Glorioso et al., 2010). See sections 5 and 6.5.
5. Genetics of normal brain aging
The genetics of brain aging has a vast associated literature, which is predominated by genetic studies of age-related neurological diseases. Here, we briefly present candidate genes with the most evidence for association with “normal” brain aging rates, although in many cases, these genes are also associated with diseases. We organize them into three broad categories: genes related to molecular pathways of aging, longevity genes, and genes associated with age-gated neurological disease, although substantial overlap can occur between these categories.
5.1. Genes related to molecular pathways of aging
5.1.1. BDNF
One of the most studied genetic variation in relation to brain aging is the BDNF Val66Met single amino acid substitution polymorphism, which results in decreased activity-dependent secretion of BDNF in neuronal cultures (Egan et al., 2003). The Met allele has been associated with poorer episodic memory, abnormal fMRI-assessed hippocampal function, accelerated age-related loss in verbal reasoning, and anticipated age-related grey matter volume losses by structural MRI (Egan et al., 2003; Harris et al., 2006; Nemoto et al., 2006; Sublette et al., 2008). Causal evidence for this polymorphism being associated with age-related functional and morphological phenotypes comes from human val66met met allele knock-in mice, which show memory deficits, decreased hippocampal volumes, and decreased dendritic complexity (Chen et al., 2006).
5.1.2. Serotonergic genes
The short allele of the serotonin transporter promoter tandem repeat polymorphism (SERT, SLC6A4), most famous for its association with depression in the context of stressful life events (Caspi et al., 2003), associates with worse delayed recall and smaller hippocampal volumes in older adults (O’Hara et al., 2007). Consistent with this, the long allele of this polymorphism was shown to be overrepresented in Japanese centenarians (Gondo et al., 2005). Another study linked a second well-characterized SERT polymorphism (VNTR2) with rates of cognitive decline, but not baseline cognition, on a variety of measures, although failed to find association with SLC6A4 (Payton et al., 2005). A serotonin 2A receptor (HTR2A) polymorphism has been linked with longitudinal decrease in delayed recall (Reynolds et al., 2006). These results are encouraging for a role of serotonin polymorphisms in brain aging, but require replication. Additionally, causal studies in model organisms linking these genes to brain aging are lacking, and the mechanism behind the contribution of these polymorphisms to altered rates of brain aging remain unknown.
5.1.3. Dopaminergic genes
Among dopaminergic polymorphisms, Catechol-O-methyl transferase (COMT) Val158Met Val allele, which is associated with decreased prefrontal cortex dopamine levels, has been most studied in relation to brain aging. It is consistently associated with accelerated rates age-related cognitive decline (de Frias et al., 2004; Houlihan et al., 2009; Starr et al., 2007) and differences in longitudinal loss of grey matter (Rowe et al., 2008). However, KO or knock-down studies of COMT establishing causation in model organisms are still lacking. One study has linked the tyrosine hydroxylase locus to human longevity (De Luca et al., 2001).
5.2. Longevity genes
5.2.1. Klotho
Klotho (KL), named after the Greek goddess who spun the thread of life, was first identified as a longevity gene in 1997 by analysis of a strain of mice with reduced longevity (lifespan around 2 months) and early onset of age-related disorders (ectopic calcification, osteopenia, arteriosclerosis, emphysema, and insulin resistance) (Kuro-o et al., 1997; Kurosu et al., 2005). This line of mutant mice was subsequently shown to have accelerated age-related neuronal pathology (Uchida et al., 2001) and premature cognitive aging (Thomson et al., 2005). KL is expressed in the distal tubule cells of the kidney and the choroid plexus of the brain and codes for a circulating hormone that binds to the insulin-like growth factor (IGF-1) receptor and represses intracellular insulin signaling (Kurosu et al., 2005). KL-hypermorphs, conversely, have an approximate 20% increase in longevity and delayed age-related disorders (Kurosu et al., 2005). The human KL-VS polymorphism results in a two amino acid change in the protein (Arking et al., 2002) and is associated with a heterozygous advantage for increased life span, increased risk for cardiovascular disease, osteoporosis, and stroke (Arking et al., 2005; Arking et al., 2003; Arking et al., 2002; Imamura et al., 2006; Ogata et al., 2002). It is also associated with decreased IQ at age 79, although the association was no longer significant after controlling for baseline IQ at age 11 (Deary et al., 2005).
5.2.2. Insulin signaling genes
In C. elegans and Drosophila, mutation in insulin-signaling genes can increase lifespan up to six-fold, and have thus been extensively studied (Bonafe et al., 2003; Kenyon, 2005). In CNS tissues specifically, knock-out of daf-2 (ortholog to the human IGF-I receptor (IGF-IR)) in only a small subset of neurons significantly extends C. elegans lifespan (reviewed in (Kenyon, 2005)). In humans, selected polymorphisms in the following genes were investigated: IGF-I receptor (IGF-IR), PIK3CB (phosphoinositol 3-kinase; C. elegans AGE-1 ortholog), IRS-1 (D. melanogaster CHICO gene ortholog), and FOXO1A (C. elegans DAF-16 ortholog). A significant association with human lifespan was reported for IGF-IR G/A 1013 polymorphism (Bonafe et al., 2003). Also, polymorphisms in AKT-1 and FOXO3A (Flachsbart et al., 2009) have subsequently been associated with human longevity, including in meta-analysis study (Pawlikowska et al., 2009). The large literature and extensive evidence linking insulin signaling polymorphisms and human lifespan is reviewed in (Bonafe and Olivieri, 2009), however, the relation of these polymorphisms specifically to brain aging in humans or in higher order model organisms has not been investigated.
5.2.3. P53
Reduced expression of Cep-1 (P53 ortholog) extends lifespan in C. elegans and overexpression of P53 reduces lifespan in mice (reviewed in (Bonafe and Olivieri, 2009)). An human arginine/proline polymorphism in codon 72 of P53 has some (albeit conflicting) evidence for association with human lifespan (reviewed in (Bonafe and Olivieri, 2009)), which has not been investigated in relation to human or model organism brain aging.
5.2.4. Sirtuins
Overexpression of sirtuins extends longevity in C. elegans and Drosophila (Bishop and Guarente, 2007). Additionally, several mouse KO models for members of the sirtuin gene family show decreased longevity and accelerated age-related phenotypes (reviewed in (Finkel et al., 2009)). However, the impact of sirtuin genetic or pharmacological manipulation on brain age-related phenotypes has remained largely unexplored. In humans, an association between two SIRT3 polymorphisms and longevity was reported (Bellizzi et al., 2005; Rose et al., 2003), as well as between a SIRT1 polymorphism and cognitive function in old age (Kuningas et al., 2007). Additionally, we showed that a low-expressing SIRT5 polymorphism is associated with accelerated human brain molecular aging using a microarray assay (Glorioso et al., 2010). However, the link between sirtuin polymorphisms and human brain aging has only begun to be investigated.
5.3. Genes related to neurological disease
5.3.1. APOE
APOE is a lipoprotein that binds to the LDL receptor family and has three human variants (E2, E3, and E4), each differing by a single amino acid. These polymorphisms have been extensively studied and have strong evidence for association with rates of morphological and functional aging. The E4 allele is consistently associated with greater risk and earlier onset of cardiovascular disease, stroke, and AD in a dose dependent manner, while the E2 allele has been shown to be protective (reviewed in (Deary et al., 2004)). Carriers of the E4 allele also show greater age-related decline in cognition (reviewed in (Deary et al., 2004)), as demonstrated by lower IQ at age 70 but not at age 11 (Deary et al., 2002). The molecular mechanism behind the E4 allele’s association with more rapid cognitive aging and greater risk of age-related disease appears to be threefold: an association with increased cholesterol levels, greater Aβ accumulation, and less ability for dendritic sprouting in E4 carriers, which has been causally related to E4 in animal models (reviewed in (Smith, 2002)).
5.3.2. PRNP
A prion protein gene (PRNP) variant met129val is associated with risk of Creutzfeldt-Jakob disease, AD, cognitive impairment, dementia, brain morphology, as well as normal cognitive aging, and it interacts with the Klotho KL-VS polymorphism (Berr et al., 1998; Kachiwala et al., 2005; Rujescu et al., 2003). The mechanism behind this effect potentially relates to ROS pathways, as PRNP functions neuroprotectively, likely as a super oxide dismutase (Rujescu et al., 2003).
5.3.3. DISC-1
Disrupted in schizophrenia 1 (DISC-1), mostly investigated for its association with SCZ and BPD, likely functions by influencing neurite extension, signal transduction, neurotransmission, and cytoskeleton re-modeling, although these mechanisms have not been fully elucidated (Thomson et al., 2005). A SNP in this gene that results in a single amino acid substitution has been associated with reduced hippocampal volume and accelerated age-related cognitive decline in humans (Thomson et al., 2005).
6. How may normal brain aging contribute to age-gated neurological disease?
Aging imposes a robust constraint on the onset of many neurological diseases, ranging from late onset neurodegenerative diseases, such as AD and PD (mean age at diagnosis ~70 (Van Den Eeden et al., 2003) and ~80 years (Nussbaum and Ellis, 2003), respectively), to earlier onset psychiatric disorders, such as SCZ and BPD (average onset 25 years) (Tsuang and Tohen, 2002). While many studies have focused on contrasting disease brains with chronologically age-matched controls, this strategy may be problematic, as it is becoming increasingly evident that normal aging is an integral aspect and modulator of disease onset and progression. Evidence for this comes from the sheer prevalence of diseases with increasing age, such as AD, for which prevalence increases exponentially from age 65 upward, reaching nearly 45% by age 95 (Fig. 8) (Nussbaum and Ellis, 2003).
Fig. 8. Increasing prevalence of Alzheimer’s disease (AD) by age.
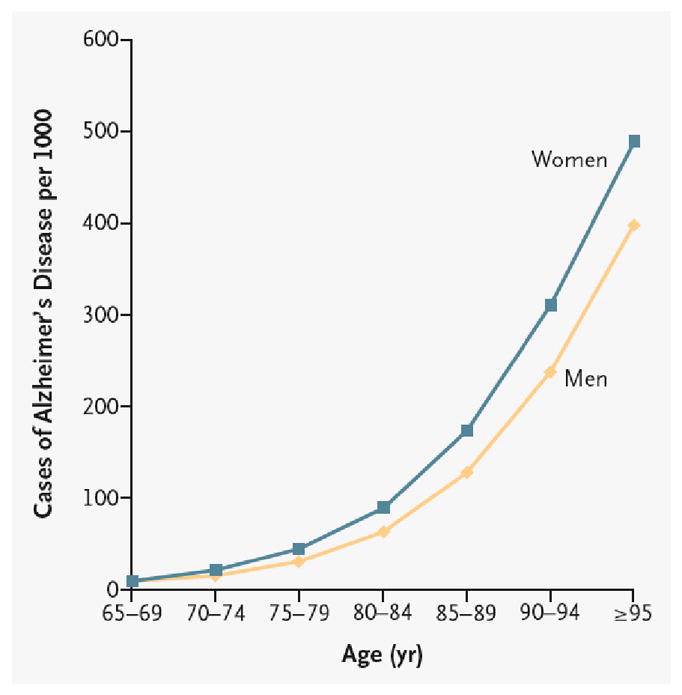
Adapted from (Nussbaum and Ellis, 2003). For unknown reasons, women demonstrate greater prevalence of AD than men at all ages after 75 years.
Also, genetic modulators and molecular pathways of normal brain aging share substantial overlap with those associated with age-gated neurological diseases. For example, BDNF Val66Met, COMT val158met, APOE4 and PRNP met129val have all been associated with risk for AD and PD (Bialecka et al., 2008; Feher et al., 2009; Mattay et al., 2008), and the BDNF val66met has additionally been associated with age of onset of SCZ (Numata et al., 2006). Epidemiological studies have shown that low calorie diets are associated with decreased risk of AD and PD (reviewed in (Joseph et al., 2009)). Caloric restriction also delays Aβ deposition in squirrel monkeys (Qin et al., 2006) and improves functional outcome in a monkey model of PD (Maswood et al., 2004). Additionally, BDNF administration can delay neurodegeneration in animal models of Huntington’s disease (HD) (reviewed in (Mattson, 2008)) and AD (reviewed in (Nagahara et al., 2009)). SIRT1 is also implicated in disease, as overexpression can protect against neurodegeneration in both AD and ALS model organisms (reviewed in (Kim et al., 2007)).
Additional evidence for normal aging modulating onset of age-gated disease comes from genetic models of increased longevity, which have consistently demonstrated delays in multiple age-related disorders. For example, mice hypermorphic for the longevity gene, Klotho, not only live ~20% longer, but also have a corresponding delay in onset of ectopic calcification, osteopenia, arteriosclerosis, emphysema, and insulin resistance (Kurosu et al., 2005; Masuda et al., 2005). There is increasing evidence for age-gated neurological diseases, as lifespan extension via reduction of insulin/insulin growth factor signaling (IIS pathway) resulted in delayed proteotoxicity in both C. elegans and mouse AD ease models (Cohen et al., 2006; Cohen et al., 2009).
While normal aging appears to modulate disease, it is important to point out that the converse does not hold, i.e., not all disease-susceptibility or disease-associated genes modulate aging. For example, the familial AD genes, PSEN1, PSEN2, and APP appear to have no association with cognitive aging (Deary et al., 2004; Mattay et al., 2008). Also, neuronal loss is a key feature of neurodegenerative disease, which, as previously discussed, does not occur during normal aging.
The modulation of a variety of neurological diseases by normal aging is potentially promising for the creation of novel “magic bullet” type anti-aging therapeutics for treatment of disease and age-related decline. However, understanding how aging gates neurological diseases and what elements contribute to individual variability in age of onset will be essential to move the field forward, but remains largely unknown. We hypothesize that a transcriptional program that progressively pushes disease genes in disease-promoting directions may represent the intrinsic component of normal brain aging that is necessary for disease onset.
6.1. Molecular brain aging: a longevity gene controlled transcriptional program necessary for age-related disease onset?
Recent studies from our group and others have led us to hypothesize that molecular brain aging is a bona fide functional decline and a disease-promoting program. This program appears to be analogous to or extending from development and to be under the control of longevity genes (amongst other genetic and environmental inputs). We propose a general mechanistic schema for how this model may work (Fig. 9). Evidence from human post-mortem brain microarray studies in support of this model are 1) the robustness of age-regulated gene expression changes in the human brain (Berchtold et al., 2008; Erraji-Benchekroun et al., 2005; Glorioso et al., 2010; Lu et al., 2004), 2) the over-representation of disease-related genes that change in disease promoting directions (Table 1), 3) the large number of developmental genes included in the gene expression correlates of aging (i.e., molecular aging), and 4) the acceleration of molecular brain aging rates in subjects with a low-expressing polymorphism of the putative longevity gene, SIRT5 (Glorioso et al., 2010). We sequentially discuss this evidence in the next sections. Notably, this evidence is still purely observational, correlative, and gathered in disease-free individuals. Additional studies, particularly in disease populations and animal models, will be required to test our hypothesis. Here, we present this hypothesis as a framework to generate hypotheses for future studies. We believe that these studies are worthwhile from a clinical as well as basic science perspective because if this mechanism proves to be correct, then tracking the rates of these changes (through peripheral blood-based samples for instance) and pharmacologically targeting this program could be a fairly straightforward approach to predict and treat many age-related neurological diseases and functional declines.
6.2. Brain age-regulated expression changes: a transcriptional program?
A few groups have investigated human molecular brain aging using microarray and found remarkably consistent results in number and identity of age-regulated changes (Berchtold et al., 2008; Erraji-Benchekroun et al., 2005; Lu et al., 2004). Most convincingly, we correlated age-regulated changes from separate cohorts, university brain donation programs, different microarray chip platforms, and brain areas, demonstrating a remarkable (R ~ 0.6 to 0.9; p<10−7) conservation of age-regulated changes (Glorioso et al., 2010). Furthermore, for the most age-regulated of genes (GFAP and CALB-1, for example), subject age and expression level were so tightly correlated that regression lines assessed using only ~35 subjects reach significance at the ~10−13 p-value levels. Together, this suggests that these highly consistent and selective gene expression changes may result from a tightly controlled age-regulated transcriptional program. Current alternative hypotheses, including wear and tear throughout lifespan, oxidative stress, etc., can only partially explain the selectivity of the observed changes.
6.3. This putative age-related transcriptional program has the potential to be the mechanism underlying how normal brain age may gate age-of-onset of diseases and functional declines
The evidence for this putative transcriptional program having the potential to gate the age-of-onset of several diseases comes first from the preferential regulation of neurological disease-related genes. In fact, we have shown that ~34% of age-regulated genes (>1,000) in the human brain have been somewhat associated throughout the literature to one or more of six age-gated neurological diseases (AD, PD, HD, ALS, BPD, SCZ) (Glorioso et al., 2010). Conversely, very few genes associated with these diseases have expression levels that do not vary significantly with chronological age. This is an impressive and unexpected contrast, given that only ~5–10% of the genome is age-regulated. Moreover, the direction of expression changes that occur with increasing age for genes otherwise associated with diseases is almost unanimously in the direction known to cause or promote disease (Table 1). This is consistent with two reports showing significant overlap between microarray changes associated with AD and normal aging in human brain (Avramopoulos et al., 2010; Miller et al., 2008). It would be interesting to combine both approaches and investigate whether AD subjects demonstrate older molecular brain ages compared to their chronologically age-matched controls, as would be predicted by a model of normal age by disease interaction.
By way of example, it seems very likely that the observed ~15–35% drops in expression of six different familial PD genes between age 20 to 80 in the human brain (Table 1) (Glorioso et al., 2010) would significantly contribute or even drive “the gating mechanism” for age-of-onset of PD in genetically vulnerable individuals. Framed differently, one could conceive that a loss of function mutation or a low-expressing promoter polymorphism in one copy of a protective disease-related gene, such as familial PD gene, PINK1, could be insufficient to cause disease in early adulthood. However, this genetic vulnerability may synergize with the significant loss of expression of the other wild-type PINK1 allele and other familial PD genes during the course of normal aging, and be sufficient to cause onset of symptoms or disease. From an evolutionary perspective, it may thus make sense to look at the preferential age-regulation of neurological disease genes from a different point of view, i.e., the genes underlying age-gated neurological diseases that are present in the population may represent age-modulated genes with insufficient or partial loss-of-function polymorphisms. Since the deleterious effects of these robust age-regulation or polymorphisms may only become evident after the aging process synergistically affect the expression of those genes, this would provide a mechanism for these polymorphisms to survive evolutionary selection forces. Conversely, this would also suggest that all age-regulated genes may be valid candidates for previously unidentified age-gated neurological disease genes.
6.4. This putative age-related transcriptional program may be analogous to or extend from development
Development-related functions were identified as top age-regulated pathways by unbiased Ingenuity software functional analysis in our human array datasets (Glorioso et al., 2010). It seems likely that genes associated with developmental transcriptional programs may have a dual role in promoting the aging process, especially as some developmental processes, such as synaptic pruning mirror aging phenotypes (synapse loss), and thus could extend from the same programs. Also, this may partially explain why so many genes related to diseases thought to be developmental (BPD, SCZ) are also age-regulated (Table 1). More specifically, we observed robust age-regulated expression changes in transcriptional regulators, such as histones (H1 and H3), methylation-related proteins (MECP2 and CHMP2), and transcription factors (DR1, TFEB, and FOXO1), which may function as potential mediators in this putative transcriptional program (Fig. 9). In support of this, FOXO1 is well established transcriptional regulator of longevity in C. elegans (Daitoku and Fukamizu, 2007), and histone targeting drugs (such as HDAC inhibitors) (Dang et al., 2009; Kawahara et al., 2009; Zhao et al., 2005) have recently been revealed as longevity modulators in model organisms. However, their causal modulation of brain aging remains to be investigated.
6.5. Longevity gene modulation of brain molecular aging: the case of SIRT5
If molecular aging were to reflect an underlying age-related transcriptional program, one could hypothesize that this program may itself be under genetic control. We have started the investigation of this hypothesis by testing whether rates of human brain molecular aging were associated with polymorphisms in candidate longevity genes (Glorioso et al., 2010). We selected a few polymorphisms in candidate genes and found the greatest association with a polymorphism (prom2) in a predicted promoter region of the putative longevity gene, SIRT5. As discussed above (section 3.1, 5.2.4) there is mounting evidence for a role for sirtuins in brain aging, however this has not been definitively established in either model organisms or in human association studies. This polymorphism was associated with lower SIRT5 expression levels in the anterior cingulate cortex (ACC) (Fig. 11), but not amygdala. In agreement with this brain-area specific lower SIRT5 expression, these same subjects had older molecular brain ages than predicted in ACC (Fig. 11) but not amygdala (data not shown). Together, this suggests that rates of molecular brain aging (at least in a cross-sectional manner), and thus perhaps disease susceptibility, age-of-onset, and functional decline, may associate with, or be regulated by, longevity gene polymorphisms.
Fig. 11. SIRT5 genetic variant and molecular aging in cingulate cortex.
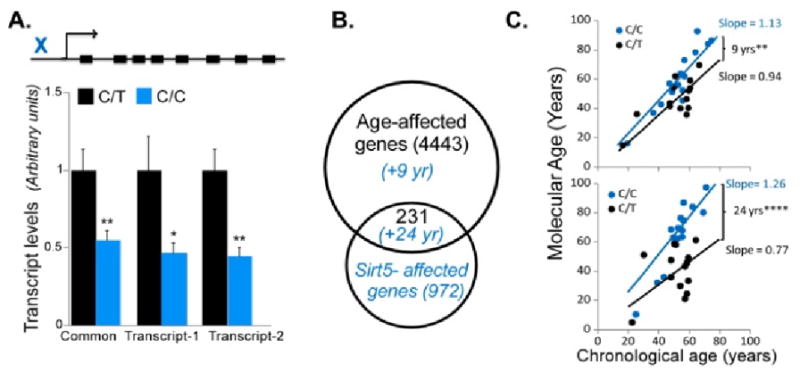
Adapted from (Glorioso et al., 2010). (A) Top: schematic representation of promoter localization of the C/C polymorphism (rs9382222). Decreased SIRT5 expression of both splice variants in C/C promoter polymorphism carriers was measured by qPCR. (B) Venn diagram of age- and SNP-related gene transcript changes. In blue are predicted changes in molecular aging for SIRT5 C/C allele carriers, based on the respective gene groups. (C) These changes predict increased molecular ages and cross-sectional age trajectories in C/C-allele carriers (top). This effect is greatly accelerated when considering only genes that display significant transcript changes in association with both age and the SIRT5 C/C risk allele (i.e., diagram intersection).
In this particular study, we further investigated which age-regulated changes were most affected in correlation with the presence of the SIRT5 risk-allele (Fig. 11, B). In agreement with SIRT5’s mitochondrial localization, the pathway most associated with both the SIRT5 risk allele and age-regulated changes was mitochondrial dysfunction. Indeed, many of these gene products are localized to the mitochondria, including familial PD genes, Pink-1 and DJ-1 (Fig. 12). We conjecture that these changes may increase risk or gate disease onset in the context of other genetic and/or environmental risk factors. Indeed, a study comparing array findings in striatum of PD brains to age-matched controls found decreases in many of the same genes that were affected in correlation with the presence of SIRT5prom2, including multiple components of the electron transport chain, PINK-1 and DJ-1, but often to a greater extent (1.2–8.6 fold decreases) than we observed (Simunovic et al., 2009). For example, they observed a ~2.2 and 8.6 fold decrease of PINK-1 and DJ-1 in striatum of PD subjects compared to age-matched controls (Simunovic et al., 2009), compared with our ~1.5 fold decreases seen in these genes at the oldest ages in SIRT5prom2 risk-allele subjects. Another consideration is that the ages of the subjects in our study only extended to age 71, approximately the mean onset age of PD, and thus some of these neurologically normal subjects may have developed PD if they had lived to older ages. Also, some of these subjects may have been protected from PD by other unidentified factors, despite the potential increased risk conferred by the SIRT5prom2 risk allele. Indeed, the SIRT5prom2 risk allele cannot be a sole determinant of PD, as it is too prevalent in the population (~43–50%) compared with the prevalence of PD (~1–3% in persons over 80 (Tanner and Goldman, 1996)). Thus, we speculate that it may only be one of many genetic and environmental factors that alters risk or age of onset of PD, congruent with the “common disease - common variant” hypothesis of disease.
Fig. 12. SIRT5 genotype accounts for age-related changes for two genes associated with familial Parkinson’s disease (PD).
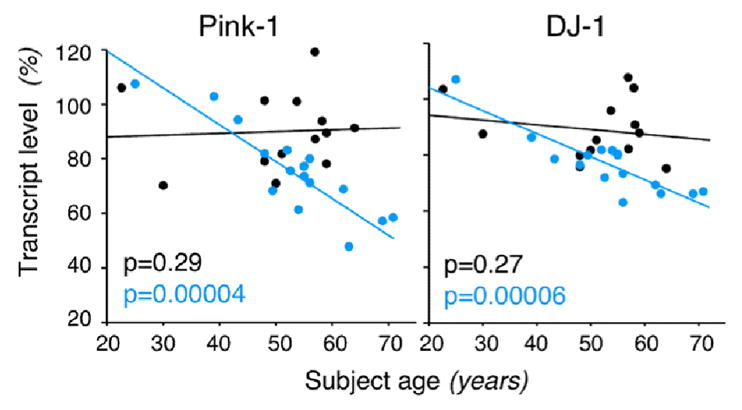
Adapted from (Glorioso et al., 2010). Expression of mitochondrial PD-related genes Pink-1 (left) and DJ-1 (right) vs. subject age in SIRT5prom2 C/C low-expressing subjects (blue) and C/T normal expressing subjects (black). Only Sirt5prom2 C/C subjects show age-related decreases in PD gene expression; p-values of age-regression lines are in lower left-hand corner.
While these results need to be replicated in independent cohorts and in brain areas most relevant to PD (i.e. in subtantia nigra), they suggest that causality studies should be performed in model systems and tested in at-risk and disease populations. Here, we conclude that SIRT5prom2 should be investigated as a potential risk factor or modulator of age-of-onset for PD and other mitochondrial-dysfunction related diseases, through this unique mechanism of accelerated age-regulated expression changes in mitochondrial genes. It more generally leads us to speculate that longevity genes may selectively modulate segments of molecular brain aging. This may contribute to the selective vulnerability seen in people for certain age-related neurological diseases but not others. We propose a model (derived from the case of SIRT5 as an example) depicting how this process may work in Fig. 13.
Fig. 13. Model of age by disease interaction.
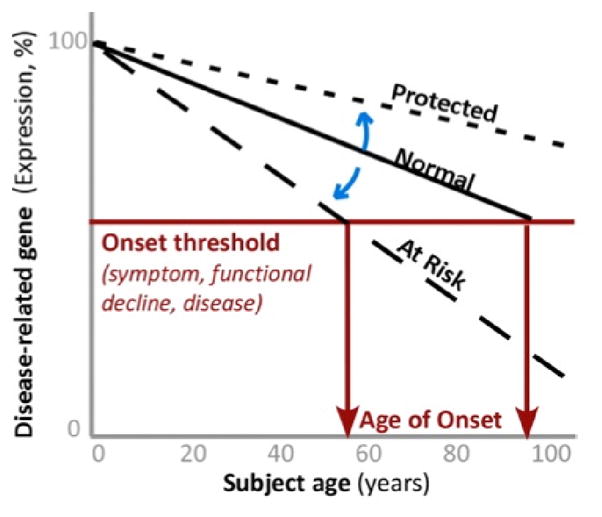
Adapted from (Glorioso et al., 2010). As loss of expression of disease-related genes below homeostatic threshold (horizontal red line) marks the onset of symptoms, we hypothesize that changes in the trajectory of age-related changes of disease-related genes (Y-axis) determine at what age (X-axis), or if, an individual may develop symptoms (vertical red arrows), manifested as functional declines. Modulators (blue arrows) may thus place individuals on “at risk” or “protected” trajectories. For instance, SIRT5 C/C-allele carriers are on an accelerated trajectory (i.e., older molecular ages) compared to C/T-allele carriers particularly for mitochondrial PD-related genes (Fig. 11, 12). The net result is that risk-allele carriers may cross threshold for disease onset and/or functional declines at earlier ages. Alternate trajectories not shown include lower expression levels of disease-related genes at all ages, or upward trajectories for genes with gain of function in disease and aging (i.e., upregulated Clusterin in aging and AD (Calero et al., 2000; Erraji-Benchekroun et al., 2005)).
6.6. Summary and further investigations needed in relation to an age-by-disease interaction hypothesis
We hypothesize that functional declines and age-gating of neurological diseases may commonly result from a genetically controlled transcriptional program extending from developmental programs. If true, then tracking rates of disease-related gene expression changes (in conjunction with genetic risk-allele testing) may allow prediction of disease onset in at-risk populations before irreversible changes occur (loss of dopaminergic neurons in PD, for example). It may also provide a read-out for determining whether new interventions are working. This may be possible by using peripheral blood expression levels as a proxy of central function; however in-depth comparison of blood and brain age-regulated expression profiles would first need to be performed. Also, the relationship between age-related expression level changes of disease-related genes and disease risk would need to be established. For example, is lower PINK-1 expression or faster rates of PINK-1 age-regulated expression decline a risk factor for PD or earlier age-of-onset of PD in living human cohorts? Additionally, investigating the breadth of genetic molecular brain aging modulators (such as SIRT5prom2) may identify many new targets for disease and age-related functional decline intervention, some of which may be fairly straightforward drug targets. Drugs targeting specific sirtuins, for example, are already in development (http://www.sirtrispharma.com/) and may be fairly easy targets for treatment of PD.
On a more basic level, many aspects of this proposed mechanism remain to be investigated. For example, causal links between changes in modulator levels, selected histones or transcription factors, and downstream changes in disease gene expression have not been established. These logical next steps will require more reductionist approaches in cell lines and animal models. Importantly, the upstream “clock input” (i.e., what counts days and years of life and translates them into declining function or levels of a molecular aging modulator/longevity genes) is largely unknown and is perhaps the “holy grail” to understanding basic molecular brain aging mechanisms. Groups have hypothesized that this could be the result of steadily accumulating DNA damage in sensitive promoter regions (Lu et al., 2004), drifting developmental programs (Budovskaya et al., 2008), or even cell-cycle and circadian rhythm related mechanisms (Glorioso et al., 2010). Developmental drift (Budovskaya et al., 2008) is a particularly appealing clock mechanism for brain aging as so many developmental genes are age-regulated; and since continuation of developmental synaptic pruning programs could explain the deleterious losses of dendritic spines seen during aging.
7. Overall summary and conclusion
Normal brain aging is associated with robust progressive changes in select domains of cognition, emotion, and mood throughout adulthood. Mirroring these changes are progressive grey matter volume losses due to shrinking dendritic arbors, and cellular phenotypic changes, such as progressive DNA damage accumulation and calcium dysregulation. Mirroring these changes on a molecular level are progressive gene expression changes in a selected group of genes (~5–10% of the genome), which contain many neurological disease-related genes and developmental genes. Normal aging and its genetics have been shown to modulate and be necessary for disease onset and functional declines. We optimistically speculate that tracking and targeting our hypothesized “genetically-controlled age-related transcriptional program” represents an effective and fairly straightforward avenue to predicting, delaying, treating, and preventing many age-related diseases and declines. However, further studies are first required, specifically the molecular dissection of this putative program in model systems, as well as the establishment of association between expression changes and disease onset age, risk, and functional declines in living human cohorts.
Research highlights.
Molecular mechanisms and genetic underpinnings of normal age-related brain changes share some overlap with peripheral mechanisms, but are also unique to the largely non-mitotic brain
Overview of age-related functional and structural changes, their molecular and genetic underpinnings
Brain molecular aging is remarkably conserved across cohorts and brain areas
Neurological disease pathways largely overlap with molecular aging
Genetic control of molecular aging: Subjects carrying a low-expressing polymorphism in a putative longevity gene (Sirtuin5) have older brain molecular ages
Molecular aging and genetic/environmental influences suggests a putative “common mechanism” for age of onset across several neurological diseases
Molecular aging of the brain may contribute to age-related neurological disease vulnerability.
Fig. 10. Representative examples of age-regulated expression change trendlines (in disease-promoting directions) for two familial PD genes in four human brain areas.
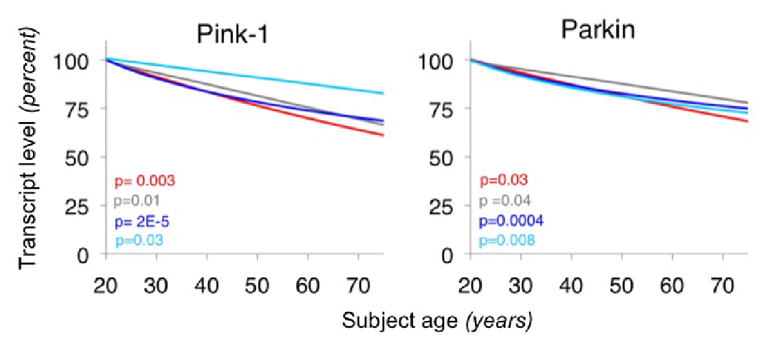
Adapted from (Glorioso et al., 2010). Regression lines of cross-sectional scatter plots of subject age versus gene expression (from human post-mortem array data) are depicted for two PD genes in four brain areas (anterior cingulate cortex (red), amygdala (grey), prefrontal cortex (PFC), BA9 (royal blue), and PFC BA47 (turquoise)). Individual subject data points were removed for visual clarity. Uncorrected p-values associated with regression lines (expression vs. age) are displayed in the lower left of graphs. Substantial ~15–35% drops in expression from age 20 to age 80 can be seen in each area and are directionally conserved across areas and studies. The direction of these changes are predicted to be promoting PD.
Acknowledgments
Support was provided by National Institutes of Aging (C.G.) and the National Institutes of Mental Health (E.S.), US, NIH. The funding agency had no role in the study design, data collection and analysis, decision to publish and preparation of the manuscript. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIMH or the National Institutes of Health. The authors declare no conflict of interest. We thank Dr. Marianne Seney for careful review of the manuscript.
Abbreviation list
(not including gene codes)
- AD
Alzheimer’s disese
- PD
Parkinson’s diseases
- BPD
bipolar disorder
- SCHZ
schizophrenia
- ROS
reactive oxygen species
- CR
caloric restriction
- PFC
prefrontal cortex
- SNP
single nucleotide polymorphism
- SIRT
Sirtuin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alexopoulos GS. Depression in the elderly. Lancet. 2005;365:1961–1970. doi: 10.1016/S0140-6736(05)66665-2. [DOI] [PubMed] [Google Scholar]
- Alexopoulos GS, Schultz SK, Lebowitz BD. Late-life depression: a model for medical classification. Biol Psychiatry. 2005;58:283–289. doi: 10.1016/j.biopsych.2005.04.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anstey K, Christensen H. Education, activity, health, blood pressure and apolipoprotein E as predictors of cognitive change in old age: a review. Gerontology. 2000;46:163–177. doi: 10.1159/000022153. [DOI] [PubMed] [Google Scholar]
- Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science. 2004;305:1010–1013. doi: 10.1126/science.1098014. [DOI] [PubMed] [Google Scholar]
- Arking DE, Atzmon G, Arking A, Barzilai N, Dietz HC. Association between a functional variant of the KLOTHO gene and high-density lipoprotein cholesterol, blood pressure, stroke, and longevity. Circ Res. 2005;96:412–418. doi: 10.1161/01.RES.0000157171.04054.30. [DOI] [PubMed] [Google Scholar]
- Arking DE, Becker DM, Yanek LR, Fallin D, Judge DP, Moy TF, Becker LC, Dietz HC. KLOTHO allele status and the risk of early-onset occult coronary artery disease. Am J Hum Genet. 2003;72:1154–1161. doi: 10.1086/375035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arking DE, Krebsova A, Macek M, Sr, Macek M, Jr, Arking A, Mian IS, Fried L, Hamosh A, Dey S, McIntosh I, Dietz HC. Association of human aging with a functional variant of klotho. Proc Natl Acad Sci U S A. 2002;99:856–861. doi: 10.1073/pnas.022484299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avramopoulos D, Szymanski M, Wang R, Bassett S. Gene expression reveals overlap between normal aging and Alzheimer’s disease genes. Neurobiol Aging. 2010 doi: 10.1016/j.neurobiolaging.2010.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backman L, Ginovart N, Dixon RA, Wahlin TB, Wahlin A, Halldin C, Farde L. Age-related cognitive deficits mediated by changes in the striatal dopamine system. Am J Psychiatry. 2000;157:635–637. doi: 10.1176/ajp.157.4.635. [DOI] [PubMed] [Google Scholar]
- Backman L, Nyberg L, Lindenberger U, Li SC, Farde L. The correlative triad among aging, dopamine, and cognition: current status and future prospects. Neurosci Biobehav Rev. 2006;30:791–807. doi: 10.1016/j.neubiorev.2006.06.005. [DOI] [PubMed] [Google Scholar]
- Bartke A. Long-lived Klotho mice: new insights into the roles of IGF-1 and insulin in aging. Trends Endocrinol Metab. 2006;17:33–35. doi: 10.1016/j.tem.2006.01.002. [DOI] [PubMed] [Google Scholar]
- Bauer JH, Poon PC, Glatt-Deeley H, Abrams JM, Helfand SL. Neuronal expression of p53 dominant-negative proteins in adult Drosophila melanogaster extends life span. Curr Biol. 2005;15:2063–2068. doi: 10.1016/j.cub.2005.10.051. [DOI] [PubMed] [Google Scholar]
- Bellizzi D, Rose G, Cavalcante P, Covello G, Dato S, De Rango F, Greco V, Maggiolini M, Feraco E, Mari V, Franceschi C, Passarino G, De Benedictis G. A novel VNTR enhancer within the SIRT3 gene, a human homologue of SIR2, is associated with survival at oldest ages. Genomics. 2005;85:258–263. doi: 10.1016/j.ygeno.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Berchtold NC, Cribbs DH, Coleman PD, Rogers J, Head E, Kim R, Beach T, Miller C, Troncoso J, Trojanowski JQ, Zielke HR, Cotman CW. Gene expression changes in the course of normal brain aging are sexually dimorphic. Proc Natl Acad Sci U S A. 2008;105:15605–15610. doi: 10.1073/pnas.0806883105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berr C, Richard F, Dufouil C, Amant C, Alperovitch A, Amouyel P. Polymorphism of the prion protein is associated with cognitive impairment in the elderly: the EVA study. Neurology. 1998;51:734–737. doi: 10.1212/wnl.51.3.734. [DOI] [PubMed] [Google Scholar]
- Bialecka M, Kurzawski M, Klodowska-Duda G, Opala G, Tan EK, Drozdzik M. The association of functional catechol-O-methyltransferase haplotypes with risk of Parkinson’s disease, levodopa treatment response, and complications. Pharmacogenet Genomics. 2008;18:815–821. doi: 10.1097/FPC.0b013e328306c2f2. [DOI] [PubMed] [Google Scholar]
- Bishop NA, Guarente L. Genetic links between diet and lifespan: shared mechanisms from yeast to humans. Nat Rev Genet. 2007;8:835–844. doi: 10.1038/nrg2188. [DOI] [PubMed] [Google Scholar]
- Bonafe M, Barbieri M, Marchegiani F, Olivieri F, Ragno E, Giampieri C, Mugianesi E, Centurelli M, Franceschi C, Paolisso G. Polymorphic variants of insulin-like growth factor I (IGF-I) receptor and phosphoinositide 3-kinase genes affect IGF-I plasma levels and human longevity: cues for an evolutionarily conserved mechanism of life span control. J Clin Endocrinol Metab. 2003;88:3299–3304. doi: 10.1210/jc.2002-021810. [DOI] [PubMed] [Google Scholar]
- Bonafe M, Olivieri F. Genetic polymorphism in long-lived people: cues for the presence of an insulin/IGF-pathway-dependent network affecting human longevity. Mol Cell Endocrinol. 2009;299:118–123. doi: 10.1016/j.mce.2008.10.038. [DOI] [PubMed] [Google Scholar]
- Budovskaya YV, Wu K, Southworth LK, Jiang M, Tedesco P, Johnson TE, Kim SK. An elt-3/elt-5/elt-6 GATA transcription circuit guides aging in C. elegans. Cell. 2008;134:291–303. doi: 10.1016/j.cell.2008.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhot MC, Wolff M, Benhassine N, Costet P, Hen R, Segu L. Spatial learning in the 5-HT1B receptor knockout mouse: selective facilitation/impairment depending on the cognitive demand. Learn Mem. 2003;10:466–477. doi: 10.1101/lm.60203. [DOI] [PubMed] [Google Scholar]
- Calero M, Rostagno A, Matsubara E, Zlokovic B, Frangione B, Ghiso J. Apolipoprotein J (clusterin) and Alzheimer’s disease. Microsc Res Tech. 2000;50:305–315. doi: 10.1002/1097-0029(20000815)50:4<305::AID-JEMT10>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H, McClay J, Mill J, Martin J, Braithwaite A, Poulton R. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science. 2003;301:386–389. doi: 10.1126/science.1083968. [DOI] [PubMed] [Google Scholar]
- Chen ZY, Jing D, Bath KG, Ieraci A, Khan T, Siao CJ, Herrera DG, Toth M, Yang C, McEwen BS, Hempstead BL, Lee FS. Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science. 2006;314:140–143. doi: 10.1126/science.1129663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen H. What cognitive changes can be expected with normal ageing? Aust N Z J Psychiatry. 2001;35:768–775. doi: 10.1046/j.1440-1614.2001.00966.x. [DOI] [PubMed] [Google Scholar]
- Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A. Opposing activities protect against age-onset proteotoxicity. Science. 2006;313:1604–1610. doi: 10.1126/science.1124646. [DOI] [PubMed] [Google Scholar]
- Cohen E, Paulsson JF, Blinder P, Burstyn-Cohen T, Du D, Estepa G, Adame A, Pham HM, Holzenberger M, Kelly JW, Masliah E, Dillin A. Reduced IGF-1 signaling delays age-associated proteotoxicity in mice. Cell. 2009;139:1157–1169. doi: 10.1016/j.cell.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colman RJ, Anderson RM, Johnson SC, Kastman EK, Kosmatka KJ, Beasley TM, Allison DB, Cruzen C, Simmons HA, Kemnitz JW, Weindruch R. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 2009;325:201–204. doi: 10.1126/science.1173635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conde JR, Streit WJ. Microglia in the aging brain. J Neuropathol Exp Neurol. 2006;65:199–203. doi: 10.1097/01.jnen.0000202887.22082.63. [DOI] [PubMed] [Google Scholar]
- Cotzias GC, Miller ST, Nicholson AR, Jr, Maston WH, Tang LC. Prolongation of the life-span in mice adapted to large amounts of L-dopa. Proc Natl Acad Sci U S A. 1974;71:2466–2469. doi: 10.1073/pnas.71.6.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotzias GC, Miller ST, Tang LC, Papavasiliou PS. Levodopa, fertility, and longevity. Science. 1977;196:549–551. doi: 10.1126/science.850799. [DOI] [PubMed] [Google Scholar]
- Daitoku H, Fukamizu A. FOXO transcription factors in the regulatory networks of longevity. J Biochem. 2007;141:769–774. doi: 10.1093/jb/mvm104. [DOI] [PubMed] [Google Scholar]
- Dang W, Steffen KK, Perry R, Dorsey JA, Johnson FB, Shilatifard A, Kaeberlein M, Kennedy BK, Berger SL. Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature. 2009;459:802–807. doi: 10.1038/nature08085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Brabander JM, Kramers RJ, Uylings HB. Layer-specific dendritic regression of pyramidal cells with ageing in the human prefrontal cortex. Eur J Neurosci. 1998;10:1261–1269. doi: 10.1046/j.1460-9568.1998.00137.x. [DOI] [PubMed] [Google Scholar]
- de Frias CM, Annerbrink K, Westberg L, Eriksson E, Adolfsson R, Nilsson LG. COMT gene polymorphism is associated with declarative memory in adulthood and old age. Behav Genet. 2004;34:533–539. doi: 10.1023/B:BEGE.0000038491.06972.8c. [DOI] [PubMed] [Google Scholar]
- De Leo D, Padoani W, Scocco P, Lie D, Bille-Brahe U, Arensman E, Hjelmeland H, Crepet P, Haring C, Hawton K, Lonnqvist J, Michel K, Pommereau X, Querejeta I, Phillipe J, Salander-Renberg E, Schmidtke A, Fricke S, Weinacker B, Tamesvary B, Wasserman D, Faria S. Attempted and completed suicide in older subjects: results from the WHO/EURO Multicentre Study of Suicidal Behaviour. Int J Geriatr Psychiatry. 2001;16:300–310. doi: 10.1002/gps.337. [DOI] [PubMed] [Google Scholar]
- De Luca M, Rose G, Bonafe M, Garasto S, Greco V, Weir BS, Franceschi C, De Benedictis G. Sex-specific longevity associations defined by Tyrosine Hydroxylase-Insulin-Insulin Growth Factor 2 haplotypes on the 11p15.5 chromosomal region. Exp Gerontol. 2001;36:1663–1671. doi: 10.1016/s0531-5565(01)00146-2. [DOI] [PubMed] [Google Scholar]
- Deary IJ, Gow AJ, Taylor MD, Corley J, Brett C, Wilson V, Campbell H, Whalley LJ, Visscher PM, Porteous DJ, Starr JM. The Lothian Birth Cohort 1936: a study to examine influences on cognitive ageing from age 11 to age 70 and beyond. BMC Geriatr. 2007;7:28. doi: 10.1186/1471-2318-7-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deary IJ, Harris SE, Fox HC, Hayward C, Wright AF, Starr JM, Whalley LJ. KLOTHO genotype and cognitive ability in childhood and old age in the same individuals. Neurosci Lett. 2005;378:22–27. doi: 10.1016/j.neulet.2004.12.005. [DOI] [PubMed] [Google Scholar]
- Deary IJ, Whiteman MC, Pattie A, Starr JM, Hayward C, Wright AF, Carothers A, Whalley LJ. Cognitive change and the APOE epsilon 4 allele. Nature. 2002;418:932. doi: 10.1038/418932a. [DOI] [PubMed] [Google Scholar]
- Deary IJ, Wright AF, Harris SE, Whalley LJ, Starr JM. Searching for genetic influences on normal cognitive ageing. Trends Cogn Sci. 2004;8:178–184. doi: 10.1016/j.tics.2004.02.008. [DOI] [PubMed] [Google Scholar]
- Diaz-Torga G, Feierstein C, Libertun C, Gelman D, Kelly MA, Low MJ, Rubinstein M, Becu-Villalobos D. Disruption of the D2 dopamine receptor alters GH and IGF-I secretion and causes dwarfism in male mice. Endocrinology. 2002;143:1270–1279. doi: 10.1210/endo.143.4.8750. [DOI] [PubMed] [Google Scholar]
- Dickstein DL, Kabaso D, Rocher AB, Luebke JI, Wearne SL, Hof PR. Changes in the structural complexity of the aged brain. Aging Cell. 2007;6:275–284. doi: 10.1111/j.1474-9726.2007.00289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dranovsky A, Hen R. Hippocampal neurogenesis: regulation by stress and antidepressants. Biol Psychiatry. 2006;59:1136–1143. doi: 10.1016/j.biopsych.2006.03.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Droge W, Schipper HM. Oxidative stress and aberrant signaling in aging and cognitive decline. Aging Cell. 2007;6:361–370. doi: 10.1111/j.1474-9726.2007.00294.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan H, Wearne SL, Rocher AB, Macedo A, Morrison JH, Hof PR. Age-related dendritic and spine changes in corticocortically projecting neurons in macaque monkeys. Cereb Cortex. 2003;13:950–961. doi: 10.1093/cercor/13.9.950. [DOI] [PubMed] [Google Scholar]
- Egan MF, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, Bertolino A, Zaitsev E, Gold B, Goldman D, Dean M, Lu B, Weinberger DR. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell. 2003;112:257–269. doi: 10.1016/s0092-8674(03)00035-7. [DOI] [PubMed] [Google Scholar]
- Era P, Jokela J, Heikkinen E. Reaction and movement times in men of different ages: a population study. Percept Mot Skills. 1986;63:111–130. doi: 10.2466/pms.1986.63.1.111. [DOI] [PubMed] [Google Scholar]
- Erraji-Benchekroun L, Underwood MD, Arango V, Galfalvy H, Pavlidis P, Smyrniotopoulos P, Mann JJ, Sibille E. Molecular aging in human prefrontal cortex is selective and continuous throughout adult life. Biol Psychiatry. 2005;57:549–558. doi: 10.1016/j.biopsych.2004.10.034. [DOI] [PubMed] [Google Scholar]
- Erraji-BenChekroun L, Underwood MD, Arango V, Galfalvy HC, Pavlidis P, Smyrniotopoulos P, Mann JJ, Sibille E. Molecular aging in human prefrontal cortex is selective and continuous throughout adult life. Biological Psychiatry. 2005;57:549–558. doi: 10.1016/j.biopsych.2004.10.034. [DOI] [PubMed] [Google Scholar]
- Feher A, Juhasz A, Rimanoczy A, Kalman J, Janka Z. Association between BDNF Val66Met polymorphism and Alzheimer disease, dementia with Lewy bodies, and Pick disease. Alzheimer Dis Assoc Disord. 2009;23:224–228. doi: 10.1097/WAD.0b013e318199dd7d. [DOI] [PubMed] [Google Scholar]
- Fillit HM, Butler RN, O’Connell AW, Albert MS, Birren JE, Cotman CW, Greenough WT, Gold PE, Kramer AF, Kuller LH, Perls TT, Sahagan BG, Tully T. Achieving and maintaining cognitive vitality with aging. Mayo Clin Proc. 2002;77:681–696. doi: 10.4065/77.7.681. [DOI] [PubMed] [Google Scholar]
- Finch CE. Neurons, glia, and plasticity in normal brain aging. Neurobiol Aging. 2003;24(Suppl 1):S123–127. doi: 10.1016/s0197-4580(03)00051-4. discussion S131. [DOI] [PubMed] [Google Scholar]
- Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460:587–591. doi: 10.1038/nature08197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiske A, Wetherell JL, Gatz M. Depression in older adults. Annu Rev Clin Psychol. 2009;5:363–389. doi: 10.1146/annurev.clinpsy.032408.153621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flachsbart F, Caliebe A, Kleindorp R, Blanche H, von Eller-Eberstein H, Nikolaus S, Schreiber S, Nebel A. Association of FOXO3A variation with human longevity confirmed in German centenarians. Proc Natl Acad Sci U S A. 2009;106:2700–2705. doi: 10.1073/pnas.0809594106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fozard JL, Vercryssen M, Reynolds SL, Hancock PA, Quilter RE. Age differences and changes in reaction time: the Baltimore Longitudinal Study of Aging. J Gerontol. 1994;49:179–189. doi: 10.1093/geronj/49.4.p179. [DOI] [PubMed] [Google Scholar]
- Gan L, Mucke L. Paths of convergence: sirtuins in aging and neurodegeneration. Neuron. 2008;58:10–14. doi: 10.1016/j.neuron.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianaros PJ, Greer PJ, Ryan CM, Jennings JR. Higher blood pressure predicts lower regional grey matter volume: Consequences on short-term information processing. Neuroimage. 2006;31:754–765. doi: 10.1016/j.neuroimage.2006.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glorioso C, Sabatini M, Unger T, Hashimoto T, Monteggia LM, Lewis DA, Mirnics K. Specificity and timing of neocortical transcriptome changes in response to BDNF gene ablation during embryogenesis or adulthood. Mol Psychiatry. 2006;11:633–648. doi: 10.1038/sj.mp.4001835. [DOI] [PubMed] [Google Scholar]
- Glorioso CA, Oh S, Sibille E. Brain Molecular Aging, Promotion of Neurological Disease and and Modulation by Sirtuin5 Longevity Gene Polymorphism. Neurobiol Dis, Revisions under resubmission. 2010 doi: 10.1016/j.nbd.2010.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, Parisi JE, Hyman BT. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann Neurol. 1997;41:17–24. doi: 10.1002/ana.410410106. [DOI] [PubMed] [Google Scholar]
- Gondo Y, Hirose N, Arai Y, Yamamura K, Shimizu K, Takayama M, Ebihara Y, Nakazawa S, Inagaki H, Masui Y, Kitagawa K. Contribution of an affect-associated gene to human longevity: prevalence of the long-allele genotype of the serotonin transporter-linked gene in Japanese centenarians. Mech Ageing Dev. 2005;126:1178–1184. doi: 10.1016/j.mad.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Good CD, Johnsrude IS, Ashburner J, Henson RN, Friston KJ, Frackowiak RS. A voxel-based morphometric study of ageing in 465 normal adult human brains. Neuroimage. 2001;14:21–36. doi: 10.1006/nimg.2001.0786. [DOI] [PubMed] [Google Scholar]
- Harris SE, Fox H, Wright AF, Hayward C, Starr JM, Whalley LJ, Deary IJ. The brain-derived neurotrophic factor Val66Met polymorphism is associated with age-related change in reasoning skills. Mol Psychiatry. 2006;11:505–513. doi: 10.1038/sj.mp.4001799. [DOI] [PubMed] [Google Scholar]
- Hasin DS, Goodwin RD, Stinson FS, Grant BF. Epidemiology of major depressive disorder: results from the National Epidemiologic Survey on Alcoholism and Related Conditions. Arch Gen Psychiatry. 2005;62:1097–1106. doi: 10.1001/archpsyc.62.10.1097. [DOI] [PubMed] [Google Scholar]
- Haug H, Kuhl S, Mecke E, Sass NL, Wasner K. The significance of morphometric procedures in the investigation of age changes in cytoarchitectonic structures of human brain. J Hirnforsch. 1984;25:353–374. [PubMed] [Google Scholar]
- Hawton K, van Heeringen K. Suicide. Lancet. 2009;373:1372–1381. doi: 10.1016/S0140-6736(09)60372-X. [DOI] [PubMed] [Google Scholar]
- Houlihan LM, Harris SE, Luciano M, Gow AJ, Starr JM, Visscher PM, Deary IJ. Replication study of candidate genes for cognitive abilities: the Lothian Birth Cohort 1936. Genes Brain Behav. 2009;8:238–247. doi: 10.1111/j.1601-183X.2008.00470.x. [DOI] [PubMed] [Google Scholar]
- Imamura A, Okumura K, Ogawa Y, Murakami R, Torigoe M, Numaguchi Y, Murohara T. Klotho gene polymorphism may be a genetic risk factor for atherosclerotic coronary artery disease but not for vasospastic angina in Japanese. Clin Chim Acta. 2006 doi: 10.1016/j.cca.2006.02.021. [DOI] [PubMed] [Google Scholar]
- Joseph J, Cole G, Head E, Ingram D. Nutrition, brain aging, and neurodegeneration. J Neurosci. 2009;29:12795–12801. doi: 10.1523/JNEUROSCI.3520-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kachiwala SJ, Harris SE, Wright AF, Hayward C, Starr JM, Whalley LJ, Deary IJ. Genetic influences on oxidative stress and their association with normal cognitive ageing. Neurosci Lett. 2005;386:116–120. doi: 10.1016/j.neulet.2005.05.067. [DOI] [PubMed] [Google Scholar]
- Kaufman AS. Assessing adolescent and adult intelligence. Allyn & Bacon; Boston: 1990. [Google Scholar]
- Kauranen K, Vanharanta H. Influences of aging, gender, and handedness on motor performance of upper and lower extremities. Percept Mot Skills. 1996;82:515–525. doi: 10.2466/pms.1996.82.2.515. [DOI] [PubMed] [Google Scholar]
- Kawahara TL, Michishita E, Adler AS, Damian M, Berber E, Lin M, McCord RA, Ongaigui KC, Boxer LD, Chang HY, Chua KF. SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell. 2009;136:62–74. doi: 10.1016/j.cell.2008.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon C. The plasticity of aging: insights from long-lived mutants. Cell. 2005;120:449–460. doi: 10.1016/j.cell.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Kim D, Nguyen MD, Dobbin MM, Fischer A, Sananbenesi F, Rodgers JT, Delalle I, Baur JA, Sui G, Armour SM, Puigserver P, Sinclair DA, Tsai LH. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J. 2007;26:3169–3179. doi: 10.1038/sj.emboj.7601758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuningas M, Putters M, Westendorp RG, Slagboom PE, van Heemst D. SIRT1 gene, age-related diseases, and mortality: the Leiden 85-plus study. J Gerontol A Biol Sci Med Sci. 2007;62:960–965. doi: 10.1093/gerona/62.9.960. [DOI] [PubMed] [Google Scholar]
- Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Iida A, Shiraki-Iida T, Nishikawa S, Nagai R, Nabeshima YI. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390:45–51. doi: 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- Kurosu H, Yamamoto M, Clark JD, Pastor JV, Nandi A, Gurnani P, McGuinness OP, Chikuda H, Yamaguchi M, Kawaguchi H, Shimomura I, Takayama Y, Herz J, Kahn CR, Rosenblatt KP, Kuro-o M. Suppression of aging in mice by the hormone Klotho. Science. 2005;309:1829–1833. doi: 10.1126/science.1112766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libert S, Cohen D, Guarente L. Neurogenesis directed by Sirt1. Nat Cell Biol. 2008;10:373–374. doi: 10.1038/ncb0408-373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T, Pan Y, Kao SY, Li C, Kohane I, Chan J, Yankner BA. Gene regulation and DNA damage in the ageing human brain. Nature. 2004;429:883–891. doi: 10.1038/nature02661. [DOI] [PubMed] [Google Scholar]
- Luo Y, Roth GS. The roles of dopamine oxidative stress and dopamine receptor signaling in aging and age-related neurodegeneration. Antioxid Redox Signal. 2000;2:449–460. doi: 10.1089/15230860050192224. [DOI] [PubMed] [Google Scholar]
- Masuda H, Chikuda H, Suga T, Kawaguchi H, Kuro-o M. Regulation of multiple ageing-like phenotypes by inducible klotho gene expression in klotho mutant mice. Mech Ageing Dev. 2005;126:1274–1283. doi: 10.1016/j.mad.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Maswood N, Young J, Tilmont E, Zhang Z, Gash DM, Gerhardt GA, Grondin R, Roth GS, Mattison J, Lane MA, Carson RE, Cohen RM, Mouton PR, Quigley C, Mattson MP, Ingram DK. Caloric restriction increases neurotrophic factor levels and attenuates neurochemical and behavioral deficits in a primate model of Parkinson’s disease. Proc Natl Acad Sci U S A. 2004;101:18171–18176. doi: 10.1073/pnas.0405831102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mather M, Carstensen LL. Aging and motivated cognition: the positivity effect in attention and memory. Trends Cogn Sci. 2005;9:496–502. doi: 10.1016/j.tics.2005.08.005. [DOI] [PubMed] [Google Scholar]
- Mattay VS, Goldberg TE, Sambataro F, Weinberger DR. Neurobiology of cognitive aging: insights from imaging genetics. Biol Psychol. 2008;79:9–22. doi: 10.1016/j.biopsycho.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Glutamate and neurotrophic factors in neuronal plasticity and disease. Ann N Y Acad Sci. 2008;1144:97–112. doi: 10.1196/annals.1418.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Maudsley S, Martin B. A neural signaling triumvirate that influences ageing and age-related disease: insulin/IGF-1, BDNF and serotonin. Ageing Res Rev. 2004a;3:445–464. doi: 10.1016/j.arr.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Maudsley S, Martin B. BDNF and 5-HT: a dynamic duo in age-related neuronal plasticity and neurodegenerative disorders. Trends Neurosci. 2004b;27:589–594. doi: 10.1016/j.tins.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Miller JA, Oldham MC, Geschwind DH. A systems level analysis of transcriptional changes in Alzheimer’s disease and normal aging. J Neurosci. 2008;28:1410–1420. doi: 10.1523/JNEUROSCI.4098-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison JH, Hof PR. Life and death of neurons in the aging brain. Science. 1997;278:412–419. doi: 10.1126/science.278.5337.412. [DOI] [PubMed] [Google Scholar]
- Murakami H, Bessinger K, Hellmann J, Murakami S. Manipulation of serotonin signal suppresses early phase of behavioral aging in Caenorhabditis elegans. Neurobiol Aging. 2008;29:1093–1100. doi: 10.1016/j.neurobiolaging.2007.01.013. [DOI] [PubMed] [Google Scholar]
- Muscari A, Giannoni C, Pierpaoli L, Berzigotti A, Maietta P, Foschi E, Ravaioli C, Poggiopollini G, Bianchi G, Magalotti D, Tentoni C, Zoli M. Chronic endurance exercise training prevents aging-related cognitive decline in healthy older adults: a randomized controlled trial. Int J Geriatr Psychiatry. 2009 doi: 10.1002/gps.2462. [DOI] [PubMed] [Google Scholar]
- Nagahara AH, Merrill DA, Coppola G, Tsukada S, Schroeder BE, Shaked GM, Wang L, Blesch A, Kim A, Conner JM, Rockenstein E, Chao MV, Koo EH, Geschwind D, Masliah E, Chiba AA, Tuszynski MH. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat Med. 2009;15:331–337. doi: 10.1038/nm.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narasimhan SD, Yen K, Tissenbaum HA. Converging pathways in lifespan regulation. Curr Biol. 2009;19:R657–666. doi: 10.1016/j.cub.2009.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemoto K, Ohnishi T, Mori T, Moriguchi Y, Hashimoto R, Asada T, Kunugi H. The Val66Met polymorphism of the brain-derived neurotrophic factor gene affects age-related brain morphology. Neurosci Lett. 2006;397:25–29. doi: 10.1016/j.neulet.2005.11.067. [DOI] [PubMed] [Google Scholar]
- Numata S, Ueno S, Iga J, Yamauchi K, Hongwei S, Ohta K, Kinouchi S, Shibuya-Tayoshi S, Tayoshi S, Aono M, Kameoka N, Sumitani S, Tomotake M, Kaneda Y, Taniguchi T, Ishimoto Y, Ohmori T. Brain-derived neurotrophic factor (BDNF) Val66Met polymorphism in schizophrenia is associated with age at onset and symptoms. Neurosci Lett. 2006;401:1–5. doi: 10.1016/j.neulet.2006.02.054. [DOI] [PubMed] [Google Scholar]
- Nussbaum RL, Ellis CE. Alzheimer’s disease and Parkinson’s disease. N Engl J Med. 2003;348:1356–1364. doi: 10.1056/NEJM2003ra020003. [DOI] [PubMed] [Google Scholar]
- O’Hara R, Schroder CM, Mahadevan R, Schatzberg AF, Lindley S, Fox S, Weiner M, Kraemer HC, Noda A, Lin X, Gray HL, Hallmayer JF. Serotonin transporter polymorphism, memory and hippocampal volume in the elderly: association and interaction with cortisol. Mol Psychiatry. 2007;12:544–555. doi: 10.1038/sj.mp.4001978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata N, Matsumura Y, Shiraki M, Kawano K, Koshizuka Y, Hosoi T, Nakamura K, Kuro OM, Kawaguchi H. Association of klotho gene polymorphism with bone density and spondylosis of the lumbar spine in postmenopausal women. Bone. 2002;31:37–42. doi: 10.1016/s8756-3282(02)00786-x. [DOI] [PubMed] [Google Scholar]
- Panza F, Solfrizzi V, Colacicco AM, D’Introno A, Capurso C, Torres F, Del Parigi A, Capurso S, Capurso A. Mediterranean diet and cognitive decline. Public Health Nutr. 2004;7:959–963. doi: 10.1079/phn2004561. [DOI] [PubMed] [Google Scholar]
- Papaconstantinou J. Insulin/IGF-1 and ROS signaling pathway cross-talk in aging and longevity determination. Mol Cell Endocrinol. 2009;299:89–100. doi: 10.1016/j.mce.2008.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park DC, Lautenschlager G, Hedden T, Davidson NS, Smith AD, Smith PK. Models of visuospatial and verbal memory across the adult life span. Psychol Aging. 2002;17:299–320. [PubMed] [Google Scholar]
- Park DC, Reuter-Lorenz P. The adaptive brain: aging and neurocognitive scaffolding. Annu Rev Psychol. 2009;60:173–196. doi: 10.1146/annurev.psych.59.103006.093656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlikowska L, Hu D, Huntsman S, Sung A, Chu C, Chen J, Joyner AH, Schork NJ, Hsueh WC, Reiner AP, Psaty BM, Atzmon G, Barzilai N, Cummings SR, Browner WS, Kwok PY, Ziv E. Association of common genetic variation in the insulin/IGF1 signaling pathway with human longevity. Aging Cell. 2009;8:460–472. doi: 10.1111/j.1474-9726.2009.00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payton A, Gibbons L, Davidson Y, Ollier W, Rabbitt P, Worthington J, Pickles A, Pendleton N, Horan M. Influence of serotonin transporter gene polymorphisms on cognitive decline and cognitive abilities in a nondemented elderly population. Mol Psychiatry. 2005;10:1133–1139. doi: 10.1038/sj.mp.4001733. [DOI] [PubMed] [Google Scholar]
- Peters A, Sethares C, Luebke JI. Synapses are lost during aging in the primate prefrontal cortex. Neuroscience. 2008;152:970–981. doi: 10.1016/j.neuroscience.2007.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A, Sethares C, Moss MB. The effects of aging on layer 1 in area 46 of prefrontal cortex in the rhesus monkey. Cereb Cortex. 1998;8:671–684. doi: 10.1093/cercor/8.8.671. [DOI] [PubMed] [Google Scholar]
- Petrascheck M, Ye X, Buck LB. An antidepressant that extends lifespan in adult Caenorhabditis elegans. Nature. 2007;450:553–556. doi: 10.1038/nature05991. [DOI] [PubMed] [Google Scholar]
- Petrascheck M, Ye X, Buck LB. A high-throughput screen for chemicals that increase the lifespan of Caenorhabditis elegans. Ann N Y Acad Sci. 2009;1170:698–701. doi: 10.1111/j.1749-6632.2009.04377.x. [DOI] [PubMed] [Google Scholar]
- Qin W, Chachich M, Lane M, Roth G, Bryant M, de Cabo R, Ottinger MA, Mattison J, Ingram D, Gandy S, Pasinetti GM. Calorie restriction attenuates Alzheimer’s disease type brain amyloidosis in Squirrel monkeys (Saimiri sciureus) J Alzheimers Dis. 2006;10:417–422. doi: 10.3233/jad-2006-10411. [DOI] [PubMed] [Google Scholar]
- Resnick SM, Pham DL, Kraut MA, Zonderman AB, Davatzikos C. Longitudinal magnetic resonance imaging studies of older adults: a shrinking brain. J Neurosci. 2003;23:3295–3301. doi: 10.1523/JNEUROSCI.23-08-03295.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rex CS, Lauterborn JC, Lin CY, Kramar EA, Rogers GA, Gall CM, Lynch G. Restoration of long-term potentiation in middle-aged hippocampus after induction of brain-derived neurotrophic factor. J Neurophysiol. 2006;96:677–685. doi: 10.1152/jn.00336.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds CA, Jansson M, Gatz M, Pedersen NL. Longitudinal change in memory performance associated with HTR2A polymorphism. Neurobiol Aging. 2006;27:150–154. doi: 10.1016/j.neurobiolaging.2004.12.009. [DOI] [PubMed] [Google Scholar]
- Rollo CD. Dopamine and aging: intersecting facets. Neurochem Res. 2009;34:601–629. doi: 10.1007/s11064-008-9858-7. [DOI] [PubMed] [Google Scholar]
- Rose G, Dato S, Altomare K, Bellizzi D, Garasto S, Greco V, Passarino G, Feraco E, Mari V, Barbi C, BonaFe M, Franceschi C, Tan Q, Boiko S, Yashin AI, De Benedictis G. Variability of the SIRT3 gene, human silent information regulator Sir2 homologue, and survivorship in the elderly. Exp Gerontol. 2003;38:1065–1070. doi: 10.1016/s0531-5565(03)00209-2. [DOI] [PubMed] [Google Scholar]
- Rowe JB, Hughes L, Williams-Gray CH, Bishop S, Fallon S, Barker RA, Owen AM. The val(158)met COMT polymorphism’s effect on atrophy in healthy aging and Parkinson’s disease. Neurobiol Aging. 2008 doi: 10.1016/j.neurobiolaging.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffman T, Henry JD, Livingstone V, Phillips LH. A meta-analytic review of emotion recognition and aging: implications for neuropsychological models of aging. Neurosci Biobehav Rev. 2008;32:863–881. doi: 10.1016/j.neubiorev.2008.01.001. [DOI] [PubMed] [Google Scholar]
- Rujescu D, Hartmann AM, Gonnermann C, Moller HJ, Giegling I. M129V variation in the prion protein may influence cognitive performance. Mol Psychiatry. 2003;8:937–941. doi: 10.1038/sj.mp.4001327. [DOI] [PubMed] [Google Scholar]
- Salthouse TA. Adult Cognition. Springer-Verlag; New York: 1982. [Google Scholar]
- Salthouse TA. Mechanisms of age-cognition relations in adulthood. Lawrence Earlbaum Associates; Hillsdale, NJ: 1996. [Google Scholar]
- Schaie KW. Intellectual development in adulthood. Cambridge University Press; Cambridge: 1996. [Google Scholar]
- Scheibel ME, Lindsay RD, Tomiyasu U, Scheibel AB. Progressive dendritic changes in aging human cortex. Exp Neurol. 1975;47:392–403. doi: 10.1016/0014-4886(75)90072-2. [DOI] [PubMed] [Google Scholar]
- Seshadri S, Wolf PA, Beiser A, Elias MF, Au R, Kase CS, D’Agostino RB, DeCarli C. Stroke risk profile, brain volume, and cognitive function: the Framingham Offspring Study. Neurology. 2004;63:1591–1599. doi: 10.1212/01.wnl.0000142968.22691.70. [DOI] [PubMed] [Google Scholar]
- Sibille E, Su J, Leman S, Le Guisquet AM, Ibarguen-Vargas Y, Joeyen-Waldorf J, Glorioso C, Tseng GC, Pezzone M, Hen R, Belzung C. Lack of serotonin1B receptor expression leads to age-related motor dysfunction, early onset of brain molecular aging and reduced longevity. Mol Psychiatry. 2007;12:1042–1056. 1975. doi: 10.1038/sj.mp.4001990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simunovic F, Yi M, Wang Y, Macey L, Brown LT, Krichevsky AM, Andersen SL, Stephens RM, Benes FM, Sonntag KC. Gene expression profiling of substantia nigra dopamine neurons: further insights into Parkinson’s disease pathology. Brain. 2009;132:1795–1809. doi: 10.1093/brain/awn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CD, Chebrolu H, Wekstein DR, Schmitt FA, Markesbery WR. Age and gender effects on human brain anatomy: a voxel-based morphometric study in healthy elderly. Neurobiol Aging. 2007;28:1075–1087. doi: 10.1016/j.neurobiolaging.2006.05.018. [DOI] [PubMed] [Google Scholar]
- Smith JD. Apolipoproteins and aging: emerging mechanisms. Ageing Res Rev. 2002;1:345–365. doi: 10.1016/s1568-1637(02)00005-3. [DOI] [PubMed] [Google Scholar]
- Starr JM, Fox H, Harris SE, Deary IJ, Whalley LJ. COMT genotype and cognitive ability: a longitudinal aging study. Neurosci Lett. 2007;421:57–61. doi: 10.1016/j.neulet.2007.05.023. [DOI] [PubMed] [Google Scholar]
- Sublette ME, Baca-Garcia E, Parsey RV, Oquendo MA, Rodrigues SM, Galfalvy H, Huang YY, Arango V, Mann JJ. Effect of BDNF val66met polymorphism on age-related amygdala volume changes in healthy subjects. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32:1652–1655. doi: 10.1016/j.pnpbp.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sze JY, Victor M, Loer C, Shi Y, Ruvkun G. Food and metabolic signalling defects in a Caenorhabditis elegans serotonin-synthesis mutant. Nature. 2000;403:560–564. doi: 10.1038/35000609. [DOI] [PubMed] [Google Scholar]
- Tanner CM, Goldman SM. Epidemiology of Parkinson’s disease. Neurol Clin. 1996;14:317–335. doi: 10.1016/S0733-8619(05)70259-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapia-Arancibia L, Aliaga E, Silhol M, Arancibia S. New insights into brain BDNF function in normal aging and Alzheimer disease. Brain Res Rev. 2008;59:201–220. doi: 10.1016/j.brainresrev.2008.07.007. [DOI] [PubMed] [Google Scholar]
- Thomson PA, Harris SE, Starr JM, Whalley LJ, Porteous DJ, Deary IJ. Association between genotype at an exonic SNP in DISC1 and normal cognitive aging. Neurosci Lett. 2005;389:41–45. doi: 10.1016/j.neulet.2005.07.004. [DOI] [PubMed] [Google Scholar]
- Tisserand DJ, Pruessner JC, Sanz Arigita EJ, van Boxtel MP, Evans AC, Jolles J, Uylings HB. Regional frontal cortical volumes decrease differentially in aging: an MRI study to compare volumetric approaches and voxel-based morphometry. Neuroimage. 2002;17:657–669. [PubMed] [Google Scholar]
- Tisserand DJ, van Boxtel MP, Pruessner JC, Hofman P, Evans AC, Jolles J. A voxel-based morphometric study to determine individual differences in gray matter density associated with age and cognitive change over time. Cereb Cortex. 2004;14:966–973. doi: 10.1093/cercor/bhh057. [DOI] [PubMed] [Google Scholar]
- Tsuang MT, Tohen M. Textbook in Psychiatric Epidemiology. Wiley: Weinheim, Germany; 2002. [Google Scholar]
- Uchida A, Komiya Y, Tashiro T, Yorifuji H, Kishimoto T, Nabeshima Y, Hisanaga S. Neurofilaments of Klotho, the mutant mouse prematurely displaying symptoms resembling human aging. J Neurosci Res. 2001;64:364–370. doi: 10.1002/jnr.1087. [DOI] [PubMed] [Google Scholar]
- Unger JW. Glial reaction in aging and Alzheimer’s disease. Microsc Res Tech. 1998;43:24–28. doi: 10.1002/(SICI)1097-0029(19981001)43:1<24::AID-JEMT4>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Van Den Eeden SK, Tanner CM, Bernstein AL, Fross RD, Leimpeter A, Bloch DA, Nelson LM. Incidence of Parkinson’s disease: variation by age, gender, and race/ethnicity. Am J Epidemiol. 2003;157:1015–1022. doi: 10.1093/aje/kwg068. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Gur RC, Wang GJ, Fowler JS, Moberg PJ, Ding YS, Hitzemann R, Smith G, Logan J. Association between decline in brain dopamine activity with age and cognitive and motor impairment in healthy individuals. Am J Psychiatry. 1998;155:344–349. doi: 10.1176/ajp.155.3.344. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Logan J, Fowler JS, Wang GJ, Gur RC, Wong C, Felder C, Gatley SJ, Ding YS, Hitzemann R, Pappas N. Association between age-related decline in brain dopamine activity and impairment in frontal and cingulate metabolism. Am J Psychiatry. 2000;157:75–80. doi: 10.1176/ajp.157.1.75. [DOI] [PubMed] [Google Scholar]
- Wechsler D. Manual for the Weschler Adult Intelligence Scale- Revised. The Psychological Corporation; New York: 1981. [Google Scholar]
- Wilson RS, Beckett LA, Barnes LL, Schneider JA, Bach J, Evans DA, Bennett DA. Individual differences in rates of change in cognitive abilities of older persons. Psychol Aging. 2002;17:179–193. [PubMed] [Google Scholar]
- Witte AV, Fobker M, Gellner R, Knecht S, Floel A. Caloric restriction improves memory in elderly humans. Proc Natl Acad Sci U S A. 2009;106:1255–1260. doi: 10.1073/pnas.0808587106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff M, Benhassine N, Costet P, Hen R, Segu L, Buhot MC. Delay-dependent working memory impairment in young-adult and aged 5-HT1BKO mice as assessed in a radial-arm water maze. Learn Mem. 2003;10:401–409. doi: 10.1101/lm.60103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yankner BA, Lu T, Loerch P. The aging brain. Annu Rev Pathol. 2008;3:41–66. doi: 10.1146/annurev.pathmechdis.2.010506.092044. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Sun H, Lu J, Li X, Chen X, Tao D, Huang W, Huang B. Lifespan extension and elevated hsp gene expression in Drosophila caused by histone deacetylase inhibitors. J Exp Biol. 2005;208:697–705. doi: 10.1242/jeb.01439. [DOI] [PubMed] [Google Scholar]