

Apo-B–Containing Lipoproteins and Atherosclerosis
High plasma levels of the apo-B–containing lipoproteins are casually implicated in the pathogenesis of atherosclerotic coronary heart disease (CHD) (1). This finding, which is backed by many decades of experimental animal studies and human clinical trial data, has sparked interest in defining which classes of apo-B–containing lipoprotein particles are most atherogenic (i.e., whether there are intrinsic differences in the atherogenicity of different classes of lipoprotein particles). There is overwhelming evidence that cholesterol-rich LDL, the smallest of the apo-B–containing particles, are atherogenic (1–3). Patients with familial hypercholesterolemia have a striking increase in the plasma levels of LDL and develop severe premature atherosclerosis. On the other hand, there is substantial evidence that larger lipoproteins, such as chylomicron and VLDL remnants, can also be atherogenic. For example, patients with Type III hyperlipidemia accumulate cholesterol-rich VLDL and chylomicron remnants and are susceptible to peripheral vascular disease and coronary artery disease (4, 5).
Although small LDL particles and larger remnant lipoproteins both appear to be atherogenic, it has been difficult to discern which particles are the most potent in causing atherosclerosis. Human epidemiologic studies do not provide unequivocal insights, in part because many patients with coronary artery disease have increased levels of both small LDL and large VLDL, obscuring the identity of the culprit lipoproteins. Studies of human genetic diseases also have not provided unequivocal answers. Humans with familial hypercholesterolemia and Type III hyperlipidemia both have high cholesterol levels and atherosclerosis. However, even if it were possible to assemble large groups of age- and sex-matched patients with familial hypercholesterolemia and Type III hyperlipidemia, and even if the subjects were perfectly matched for plasma cholesterol levels and other atherosclerosis risk factors, it might still be difficult to determine whether LDL particles or chylomicron/VLDL remnants are the most atherogenic. One of the reasons that it would be difficult is that Type III hyperlipidemia generally develops in young adults, whereas the elevated cholesterol levels in patients with familial hypercholesterolemia are lifelong.
Atherogenicity of Large, Triglyceride-Rich Lipoproteins
Most investigators have assumed that large triglyceride-rich lipoproteins are not as atherogenic as smaller cholesterol-rich lipoproteins. Humans with deficiencies in lipoprotein lipase or apolipoprotein CII have familial chylomicronemia, frequently maintaining plasma triglyceride levels >2,000 mg/dl (6, 7). These individuals also have high plasma cholesterol levels, generally more than 250 mg/dl, with nearly all of the cholesterol in large chylomicron/VLDL particles (the levels of cholesterol in the LDL and HDL fractions are typically quite low). Those who have studied patients with familial chylomicronemia have commented that these individuals are not particularly susceptible to coronary artery disease (6, 7). However, opinions about this issue are not uniform. Dr. Michael Hayden’s group at the University of British Columbia has challenged the concept that patients with familial chylomicronemia are protected from atherosclerosis (8). They reported four patients with familial chylomicronemia (from well characterized LPL mutations—mostly missense mutations yielding catalytically inactive protein) who had premature peripheral vascular disease or coronary artery disease or both. Like typical chylomicronemia patients, all four of these patients had markedly elevated plasma triglyceride levels (ranging from ~1800–4600 mg/dl), low LDL cholesterol levels (28 ± 16 mg/dl), and low HDL cholesterol levels (17 ± 7 mg/dl). Total plasma cholesterol levels were elevated (200–425 mg/dl), with nearly all of the cholesterol in the VLDL/chylomicron fraction (i.e., the d < 1.006 g/ml lipoproteins) (8). Whether the elevated cholesterol levels within the chylomicron/VLDL fraction were responsible for the atherosclerosis in these patients is not entirely clear, but it seems likely to us that this was the case.
The atherogenicity of very large, triglyceride-rich particles has also been examined in rabbit models. More than 50 years ago, Duff and McMillan (9) found that diabetic rabbits on a high-cholesterol diet had fewer atherosclerotic lesions than nondiabetic rabbits fed the same cholesterol-rich diet. While the diabetic and nondiabetic rabbits both had severe hypercholesterolemia, the diabetic rabbits also had severe hypertriglyceridemia (plasma triglyceride levels >5000 mg/dl). The observation that hypertriglyceridemia protects diabetic cholesterol-fed rabbits from atherosclerosis was verified and examined in more detail by the laboratory of Dr. Donald Zilversmit (10, 11). They proposed that the hypertriglyceridemic diabetic cholesterol-fed rabbits are protected from atherosclerosis because most of the cholesterol in these rabbits is carried in lipoproteins that are too large to enter the arterial wall. They found that the majority of the cholesterol in the diabetic rabbits was in “giant lipoproteins” with diameters >75 nm (10, 12). They also found that the ability of lipoproteins to enter the arterial intima depended on size, and that the “giant lipoproteins” were “practically excluded” from the arterial wall, with an intimal clearance rate less than 5% of LDL-sized particles (10, 11).
Lipoprotein Size and Atherosclerosis in Mouse Models
During the past 15 years, the mouse has become the predominant model for studying atherosclerosis. Mice are less expensive than rabbits or primates, and the ability to create transgenic and gene-knockout models has made it possible to investigate the relevance of dozens of genes in atherogenesis.
Very early after the onset of the “mouse era” in atherosclerosis research (13–16), gene-knockout models were established for both familial hypercholesterolemia (LDL receptor deficiency) (17, 18) and Type III hyperlipidemia (apo-E deficiency) (19–22). Chow-fed apo-E–deficient (Apoe−/−) mice manifest a dramatic accumulation of cholesterol-rich chylomicron/VLDL lipoproteins in the plasma, even on a low-fat chow diet, and develop advanced atherosclerotic lesions (22). LDL receptor–deficient (Ldlr−/−) mice have mild–moderate increases in LDL cholesterol levels on a chow diet and develop modest atherosclerotic lesions (22). Unfortunately, direct comparisons of Apoe−/− and Ldlr−/− mice shed little light on the relative atherogenicities of the large VLDL/LDL remnants and small LDL particles, simply because the total plasma cholesterol levels in the two mouse models are very different (~375–450 mg/dl in Apoe−/− mice versus only ~200–225 mg/dl in Ldlr−/− mice) (17–22). Another confounding factor is the overlapping lipoprotein sizes in the two models. Apoe−/− mice have elevated levels of LDL-sized particles, in addition to VLDL and chylomicron remnants (23–27), making it difficult to determine which lipoproteins are more important in contributing to atherosclerosis. Finally, there are significant differences in the apolipoprotein content of lipoproteins in Apoe−/− and Ldlr−/− mice. The vast majority of remnant lipoproteins in Apoe−/− mice contain apo-B48 (19–21, 28), while the LDL in Ldlr−/− mice contain apo-B100 (17, 28).
Modifying Lipid and Lipoprotein Phenotypes in Apoe−/− Mice with Apob Mutations
Lipoprotein size and susceptibility to atherosclerosis in Apoe−/− mice have been modified by rendering these mice homozygous for either the “apo-B48–only” or the “apo-B100–only” mutation in Apob (29). The laboratory of Dr. Stephen Young (24, 29) sought to answer a question that had vexed the lipoprotein/atherosclerosis field for several decades: Are the two different apo-B proteins, apo-B48 and apo-B100, intrinsically different in their abilities to cause atherosclerosis? The two apo-B isoforms differ in size by more than 2400 amino acids (98, 99), and it seemed plausible that they might differ in their metabolism within the arterial wall and differ in their ability to induce atherosclerosis. To test this issue, Dr. Young’s laboratory analyzed susceptibility to atherosclerosis in mice that synthesize exclusively apo-B48 (Apob48/48 mice) or apo-B100 (Apob100/100 mice) (27). The Apob48/48 mice were generated by using “pop-in, pop-out” gene targeting to insert a TGA stop codon into codon 2153 of the mouse apo-B gene. Apob100/100 mice were produced by inserting a CTA-leucine codon into codon 2153 of the mouse apo-B gene (28). Even when the CTA-leucine codon is edited to UTA by the apo-B RNA-editing machinery (30), no stop codon is generated, as UTA also specifies leucine.
The Apob48/48 and Apob100/100 mice were crossed with Apoe−/− mice to produce apo-E–deficient apo-B48–only mice (Apoe−/−Apob48/48) and apo-E–deficient apo-B100–only mice (Apoe−/−Apob100/100) (27). When the Apoe−/−Apob48/48 mice were fed a chow diet, their total and LDL cholesterol levels were somewhat higher than in Apoe−/−Apob+/+ mice (24). The total and LDL cholesterol levels in the Apoe−/−Apob100/100 mice were significantly lower than in Apoe−/−Apob+/+ mice (24). The lower LDL levels in Apoe−/−Apob100/100 mice are almost certainly due to the fact that the apo-B100–containing LDL can be removed from the plasma by the LDL receptor (27, 28), while apo-B48–containing lipoproteins in Apoe−/−Apob+/+ mice cannot be removed by this pathway. As illustrated in Figure 1, the vast majority of the plasma cholesterol in the Apoe−/−Apob100/100 mice is located in the VLDL fraction; consequently, these mice have been dubbed “VLDL-cholesterol mice” (24–26).
Figure 1.

Lipid and lipoprotein levels in Apoe−/−Apob100/100 (n = 44) and Ldlr−/−Apob100/100 (n = 42) mice. (A) Distribution of cholesterol within different lipoprotein fractions. (B) Mean plasma cholesterol levels in Apoe−/−Apob100/100 (n = 44) and Ldlr−/−Apob100/100 (n = 42) mice at 40 weeks of age. Reproduced from a paper by Véniant et al. (25) with permission from the American Society of Clinical Investigation.
We examined large groups of Apoe−/−Apob+/+, Apoe−/−Apob48/48, and Apoe−/−Apob100/100 mice (24). While the mean plasma cholesterol levels in the three groups of mice differed significantly, the cholesterol levels in individual mice actually overlapped considerably. We therefore were curious to determine whether, at a given cholesterol level, mice expressing exclusively apo-B48 (Apoe−/−Apob48/48 mice) had fewer (or more) atherosclerotic lesions than mice expressing exclusively apo-B100 (Apoe−/−Apob100/100 mice). Therefore, after 7 months on a regular chow diet, all of the Apoe−/−Apob+/+, Apoe−/−Apob48/48, and Apoe−/−Apob100/100 mice were euthanized and perfusion-fixed. Atherosclerosis was measured by pinning out the aorta and then using computer-assisted morphometric techniques to quantify the percentage of the aortic surface covered by atherosclerotic lesions. Most of the lesions in all three groups of mice were located in the ascending aorta and aortic arch. Atherosclerosis was more extensive in the Apoe−/−Apob48/48 mice, and less extensive in the Apoe−/−Apob100/100 mice, compared with the Apoe−/−Apob+/+ mice (27). Once again, however, a considerable amount of overlap existed. Importantly, the amount of atherosclerosis correlated with total plasma cholesterol levels in all three groups of mice. Also, all three groups appeared to fall on the same “total cholesterol vs. atherosclerosis curve.” In other words, the Apoe−/−Apob+/+, Apoe−/−Apob48/48, and Apoe−/−Apob100/100 mice had similar amounts of atherosclerosis when their total cholesterol levels were similar. From this study, Dr. Young’s group concluded that the total cholesterol level appeared to be the most important factor in the development of atherosclerosis and that there did not appear to be any significant differences between the intrinsic abilities of apo-B48– and apo-B100–containing lipoproteins to promote atherosclerosis (24). There was no relationship between plasma triglyceride levels and atherosclerotic lesions.
Lipoprotein Size and Atherosclerosis in Apoe−/− and Ldlr−/− Mice After “Evening Up” the Plasma Cholesterol Levels
Ldlr−/− mice do not develop severe hypercholesterolemia because their livers synthesize abundant amounts of apo-B48, and apo-B48–containing lipoproteins can be efficiently cleared independently of the LDL receptor (27, 28). We predicted that it would be possible to increase LDL levels in Ldlr−/− mice by eliminating apo-B48 synthesis. To test this idea, we bred Ldlr−/−Apob100/100 mice (27). As predicted, these mice had significantly higher plasma cholesterol levels than the Ldlr−/−Apob+/+ mice, and essentially all of the cholesterol in their plasma was located in the LDL, as judged by fractionation of the plasma by fast phase liquid chromatography (FPLC) (Figure 1). We have dubbed the Ldlr−/−Apob100/100 mice “LDL-cholesterol mice” (25–27). The distribution of cholesterol in these “LDL cholesterol mice” was strikingly different from those of the “VLDL cholesterol mice” (Apoe−/−Apob100/100) (Figure 1). Of note, the total plasma cholesterol levels in the Apoe−/−Apob100/100 and the Ldlr−/−Apob100/100 mice were virtually identical (25). HDL cholesterol levels were also indistinguishable (25). The triglyceride levels were somewhat higher in Apoe−/−Apob100/100 mice than in Ldlr−/−Apob100/100 mice (25).
While the total plasma cholesterol levels in Apoe−/−Apob100/100 and Ldlr−/−Apob100/100 mice were very similar, the size of the lipoprotein particles in the two models was not (25). Nearly all of the cholesterol in the plasma of Apoe−/−Apob100/100 mice was in the VLDL, whereas nearly all of the cholesterol in the Ldlr−/−Apob100/100 mice was in the LDL (Figure 1). The diameters of the lipoproteins were determined by dynamic light scattering analysis with a Microtrac Series 150 Ultrafine particle analyzer. We found that the lipoproteins in the plasma of Apoe−/−Apob100/100 mice were actually quite large (mean size of 63 nm) whereas the vast majority of lipoproteins in Ldlr−/−Apob100/100 mice were small (mean size of 24 nm) (Figure 2) (25). Indeed, there was almost no overlap in the sizes of the apo-B–containing lipoproteins in the plasma. Finding identical cholesterol levels but differences in lipoprotein size implied that the numbers of particles in the plasma of the two models would be different. Indeed, this was the case. Plasma apo-B100 levels were nearly fourfold higher in Ldlr−/−Apob100/100 mice than in Apoe−/−Apob100/100 mice (Figure 3) (25). Thus, we had created two hypercholesterolemic mouse models—both “apo-B100–only” and both with the same plasma cholesterol levels—but with striking differences in lipoprotein sizes and lipoprotein numbers (25).
Figure 2.

Lipoprotein sizes in Apoe−/−Apob100/100, Ldlr−/−Apob100/100, Ldlr−/−Apob+/+, and Apoe−/−Apob+/+ mice. The median diameter of lipoproteins in Apoe−/−Apob100/100 mice (n = 15) was 140% larger than in Ldlr−/−Apob100/100 (n = 21) mice, 90% larger than in Ldlr−/−Apob+/+ mice (n = 17), and 50% larger than in Apoe−/−Apob+/+ mice (n = 16). The size of VLDL (d < 1.006 g/ml) particles ranged from an average of 33.4 nm in Ldlr−/−Apob100/100 mice to 61 nm in Apoe−/−Apob100/100 mice; the size of IDL (d = 1.006–1.020 g/ml) particles ranged from an average of 27 nm in Ldlr−/−Apob100/100 mice to 38 nm in Apoe−/−Apob100/100 mice; the size of LDL (d = 1.020–1.052 g/ml) particles was 23 nm in Apoe−/−Apob100/100 plasma, 20 nm in Apoe−/−Apob+/+ plasma, 22 nm in Ldlr−/−Apob100/100 plasma, and 19 nm in Ldlr−/−Apob+/+ plasma. The difference in size between the bottom and top deciles of particles was 64.0 nm for Apoe−/−Apob100/100 mice, 27.2 nm for Apoe−/−Apob+/+ mice, 17.8 nm for Ldlr−/−Apob+/+ mice, and 17.0 nm for Ldlr−/−Apob100/100 mice. Reproduced from a paper by Véniant et al. (25) with permission from the American Society of Clinical Investigation.
Figure 3.

Apo-B100 levels in Apoe−/−Apob100/100 (n = 36) and Apoe−/−Apob+/+ mice (n = 34) at 16 and 32 weeks of age, as judged by a monoclonal antibody–based radioimmunoassay. Reproduced from a paper by Véniant et al. (25) with permission from the American Society of Clinical Investigation.
To obtain insights into the relative atherogenicities of two very different lipoprotein phenotypes, we examined susceptibility to atherosclerosis in Apoe−/−Apob100/100 and Ldlr−/−Apob100/100 mice (25). For controls, we analyzed Apoe−/− and Ldlr−/− mice that were homozygous for a wild-type apo-B allele (Apoe−/−Apob+/+ and Ldlr−/−Apob+/+ mice). All four groups of mice (n ≥ 40/group) were fed a chow diet for 40 weeks. At the 40-week time point, the total plasma cholesterol levels (mean ± S.D.) in Apoe−/−Apob100/100 and Ldlr−/−Apob100/100 were virtually identical (334 ± 12 and 336 ± 8 mg/dl, respectively). As expected, the total plasma cholesterol levels were higher in Apoe−/−Apob+/+ mice (497 ± 23 mg/dl) and lower in Ldlr−/−Apob+/+ mice (235 ± 12 mg/dl) (25).
Computer-assisted morphometric techniques were used to assess the percent of the aortic surface covered by atherosclerotic lesions at 40 weeks (25). The results were unequivocal. Mice with larger numbers of small lipoproteins (i.e., the Ldlr−/−Apob100/100 mice) had far more atherosclerosis than mice with smaller numbers of large lipoproteins (i.e., the Apoe−/−Apob100/100 mice) (14.0 ± 0.46% versus only 4.8 ± 0.37% of the aortic surface; P < 0.0001) (Figure 4) (25). Of note, Apoe−/−Apob+/+ mice actually had only about one-half as much atherosclerosis (7.47 ± 0.67% of the aortic surface) as Ldlr−/−Apob100/100 mice, despite having far higher plasma cholesterol levels. As expected, the Ldlr−/−Apob+/+ mice had minimal atherosclerosis (0.41 ± 0.16%, ~3% of that in Ldlr−/−Apob100/100 mice; P < 0.0001) (25). The Ldlr−/−Apob100/100 mice were far more susceptible to atherosclerosis than Apoe−/−Apob100/100 mice. Representative pinned-out aortas from all four genotypes of mice (Apoe−/−Apob+/+, Apoe−/−Apob100/100, Ldlr−/−Apob+/+, Ldlr−/−Apob100/100) are shown in Figure 5 (25). The microscopic appearance of the atherosclerotic lesions in the proximal aortas was similar in the different groups (Figure 6) (25).
Figure 4.

Morphometric assessment of atherosclerotic lesions in Apoe−/−Apob100/100 (n = 44), Apoe−/−Apob+/+ (n = 40), Ldlr−/−Apob100/100 (n = 42), and Ldlr−/−Apob+/+ (n = 40) mice. Differences between all groups were significant at the P < 0.0001 level with one exception: the lesions in Apoe−/−Apob+/+ mice were different from those in Apoe−/−Apob100/100 mice at P = 0.0004. Reproduced from a paper by Véniant et al. (25) with permission from the American Society of Clinical Investigation.
Figure 5.

Representative Sudan IV–stained thoracic aortas. The amount of atherosclerosis in these four aortas matched the mean level for each genotype of mice. Reproduced from a paper by Véniant et al. (25) with permission from the American Society of Clinical Investigation.
Figure 6.

Oil Red O–stained sections of proximal aortic roots for different mouse genotypes. Reproduced from a paper by Véniant et al. (25) with permission from the American Society of Clinical Investigation.
When we plotted atherosclerotic lesion measurements and total cholesterol levels in Ldlr−/−Apob100/100 and Apoe−/−Apob100/100 mice, we found that Ldlr−/−Apob100/100 mice had far more atherosclerosis than Apoe−/−Apob100/100 mice with similar total cholesterol levels (Figure 7) (25). A plot of atherosclerotic lesions and plasma apo-B100 levels (Figure 7) revealed that Ldlr−/−Apob100/100 mice—the group with the highest apo-B100 levels—had the most atherosclerosis.
Figure 7.

Relationship between atherosclerotic lesions and the total plasma cholesterol levels and plasma apo-B100 levels in Apoe−/−Apob100/100 (n = 41) and Ldlr−/−Apob100/100 (n = 40) mice. The top panel shows a plot of lesions, as assessed by morphometric techniques, versus total plasma cholesterol levels (mean of the five measurements). The bottom panel shows a plot of lesions versus the plasma apo-B100 levels (measured at 32 weeks). Reproduced from a paper by Véniant et al. (25) with permission from the American Society of Clinical Investigation.
We also measured the amount of free and esterified cholesterol in the aortas and the rate of aortic DNA synthesis. The Ldlr−/−Apob100/100 mice accumulated far more cholesterol in their aortas than the Apoe−/−Apob100/100 mice, despite almost identical plasma cholesterol levels (25) (Figure 8). We found a strong positive correlation between lesion size, measured morphometrically, and the aortic content of esterified cholesterol, both for all of the mice in the study (r = 0.858; P < 0.0001) and for most of the subgroups (Apoe−/−Apob+/+: r = 0.719, P < 0.0001; Apoe−/−Apob100/100: r = 0.705, P < 0.0001; and Ldlr−/−Apob100/100: r = 0.734, P < 0.0001) (25).
Figure 8.

Scoring of aortic pathology in the four groups of mice according to the aortic content of free and esterified cholesterol and aortic DNA synthesis rate. (A) Cholesterol ester content of aortas in Apoe−/−Apob100/100 (n = 43), Apoe−/−Apob+/+ (n = 38), Ldlr−/−Apob100/100 (n = 39), and Ldlr−/−Apob+/+ (n = 34) mice. (B) Free cholesterol content of aortas in the four different groups of mice (numbers of mice identical to those for panel A). (C) Aortic DNA synthesis rates in Apoe−/−Apob100/100 (n = 18), Apoe−/−Apob+/+ (n = 17), Ldlr−/−Apob100/100 (n = 25), and Ldlr−/−Apob+/+ (n = 14) mice. Reproduced from a paper by Véniant et al. (25) with permission from the American Society of Clinical Investigation.
To measure DNA synthesis within the aortas, we administered 2H2O to the mice and then assessed, by mass spectrometry, the incorporation of deuterium into newly synthesized DNA (25). The aortic DNA synthesis rate in the experimental animals was high; nearly 10–12% of the cells in the aorta had divided during the three weeks prior to euthanizing the mice (25). The correlation between lesions, as assessed by morphometry, and the aortic DNA synthesis rate was moderate (r = 0.574, P < 0.0001). The correlation between the aortic cholesterol content and the aortic DNA synthesis rate was lower (r = 0.482, P < 0.0001 for free cholesterol; r = 0.469, P < 0.0001 for esterified cholesterol). In line with the morphometric results and the cholesterol measurements, the aortic DNA synthesis in the Ldlr−/−Apob100/100 mice was almost twice as high as in Apoe−/−Apob100/100 mice (Figure 8) (25).
The total plasma cholesterol levels in Apoe−/−Apob100/100 and Ldlr−/−Apob100/100 mice are virtually identical, and the principal structural protein of lipoprotein particles, apo-B100, is the same in both models (25). While there are several potential explanations for these results, we believe that the most likely explanation is the differences in the lipoprotein phenotype, with large numbers of small lipoproteins (as in Ldlr−/−Apob100/100 mice) being more atherogenic than lower numbers of large lipoproteins (as in Apoe−/−Apob100/100 mice) (Figure 9) (25, 26). Of note, more than half of the cholesterol in Apoe−/−Apob100/100 mice was contained in “giant” lipoproteins with diameters greater than 70 nm—the types of particles that have little ability to penetrate the arterial wall (10).
Figure 9.

Mean extent of atherosclerotic lesions plotted against the mean total plasma cholesterol level. The atherosclerosis data (assessed by morphometric techniques) and the cholesterol data represent means calculated from all of the mice in each group. The steep increase in atherosclerosis in LDL receptor–deficient mice between total cholesterol concentrations of ~200 and ~300 mg/dl suggests that small LDL particles are particularly atherogenic. Reproduced from a paper by Véniant et al. (25) with permission from the American Society of Clinical Investigation.
Interpreting the Differences in Atherosclerotic Lesions in Apoe−/−Apob100/100 and Ldlr−/−Apob100/100 Mice
The total plasma cholesterol levels in Apoe−/−Apob100/100 and Ldlr−/−Apob100/100 mice were almost identical, but lipoprotein sizes were not. The striking differences in the sizes of cholesterol-carrying particles, given nearly equivalent plasma cholesterol levels, was explained by differences in the numbers of apo-B–containing lipoprotein particles in the plasma (25). Plasma levels of apo-B100 were almost fourfold higher in Ldlr−/−Apob100/100 mice.
Figure 9 shows the mean values for atherosclerotic lesions and cholesterol levels for each of the four genotypes of mice. These data suggest that susceptibility to atherosclerosis changes according to lipoprotein size. Thus, there appears to be a distinct “plasma cholesterol versus atherosclerosis” relationship for lipoproteins of different sizes. When most of the lipoproteins are small (i.e., in the LDL size range), as in the two groups of LDL receptor–deficient mice, changing the plasma cholesterol levels from ~200 to ~300 mg/dl resulted in a nearly 30-fold increase in amount of atherosclerotic lesions in the aorta (25). When most of the cholesterol was located within large lipoprotein particles (i.e., VLDL-sized particles), as in the Apoe−/−Apob100/100 and Apoe−/−Apob+/+ mice, increasing the plasma cholesterol level by ~150 mg/dl increased atherosclerotic lesions by less than twofold (25).
We believe that the very steep increase in lesions with relatively small changes in LDL levels is consistent with earlier findings in mouse models. Previously, we generated human apo-B transgenic mice (31) and examined their susceptibility to atherosclerotic lesions on a high-fat diet (32). The human apo-B transgene had little impact on the concentration of VLDL-IDL particles in the plasma, but increased LDL cholesterol levels (by ~80 mg/dl). This rather small increase in the concentration of LDL in the plasma was associated with a greater than 10-fold increase in the extent of atherosclerotic lesions (32). Similarly, increasing LDL levels in Apobec1 knockout mice (by rendering them homozygous for Ldlr deficiency) resulted in a striking increase in atherosclerosis (33). A steep “atherosclerosis versus LDL cholesterol slope” is probably not unique to mouse models. In humans, where most of the cholesterol is in LDL particles, doubling plasma cholesterol levels increases the risk of coronary disease sixfold (34).
It is possible, of course, that factors other than lipoprotein size and apo-B concentration could have affected atherogenesis in Ldlr−/−Apob100/100 and Apoe−/−Apob100/100 mice (25, 26). For example, apo-E in the lipoproteins of Ldlr−/−Apob100/100 mice might somehow increase the binding of lipoproteins to proteoglycans within the arterial wall, thereby accelerating atherogenesis. Similarly, the apo-AIV-enrichment of VLDL in Apoe−/−Apob100/100 mice might have had the opposite effect. Alternatively, perhaps the LDL in the Ldlr−/−Apob100/100 mice was more susceptible to oxidation because of a longer residence time in the plasma, leading to more atherosclerosis. Also, the macrophages in Ldlr−/−Apob100/100 mice obviously retain the capacity to produce apo-E, while those in Apoe−/−Apob100/100 mice do not. Bone marrow transplantation studies have suggested that the local production of apo-E within macrophages in the arterial wall can limit the progression of lesions (35). However, this “anti-atherogenic role of apo-E in the arterial wall” would seemingly not explain our findings, given that the mice with the capacity to synthesize apo-E (e.g., Ldlr−/−Apob100/100 mice) had more atherosclerosis, not less.
Studies with Ldlr−/−Apob100/100 and Apoe−/−Apob100/100 Mice That Are Also Homozygous for the Obesity (Ob) Mutation
Apoe−/− and Ldlr−/− mice are useful models for examining hyperlipidemia and atherosclerosis, but unlike many human patients with coronary artery disease, they are neither insulin-resistant nor diabetic. To develop mouse models that exhibit both insulin resistance and hyperlipidemia, we bred Apoe−/−Apob100/100ob−/− and Ldlr−/−Apob100/100ob−/− mice. As expected, the body weights of the Apoe−/−Apob100/100ob−/− and Ldlr−/−Apob100/100ob−/− mice were far greater than those of wild-type mice (Figure 10). Postprandial blood glucose levels were significantly elevated only in the Apoe−/−Apob100/100ob−/− mice (Figure 11A). However, both Apoe−/−Apob100/100ob−/− and Ldlr−/−Apob100/100ob−/− mice had marked hyperinsulinemia (P < 0.0001 vs. wild-type C57BL/6 mice, Figure 11B). The plasma insulin levels in Apoe−/−Apob100/100ob−/− were greater than those of Ldlr−/−Apob100/100ob−/− (>50 ng/ml in the former mice and 30–40 ng/ml in the latter; Figure 11B).
Figure 10.

Body weight in Apoe−/−Apob100/100ob−/− (n = 18–52) and Ldlr−/−Apob100/100 ob−/− (n = 22–52) mice. Body weights were significantly increased in Apoe−/−Apob100/100ob−/− and Ldlr−/−Apob100/100 ob−/− mice compared with wild-type C57BL/6 mice (P < 0.001 for both groups).
Figure 11.

Blood glucose and insulin levels in Apoe−/−Apob100/100ob−/− (n = 10–24) and Ldlr−/−Apob100/100 ob−/− (n = 5–17) mice. (A) Fed blood glucose levels were significantly higher in Apoe−/−Apob100/100ob− mice than in wild-type C57BL/6 mice (P values at different ages range from 0.05 to 0.001). (B) Fed insulin levels in Apoe−/−Apob100/100ob−/− and Ldlr−/−Apob100/100 ob−/− mice (n = 16–32). ++P < 0.01 vs. Ldlr−/−Apob100/100 ob−/− mice, +++P < 0.001 vs. Ldlr−/−Apob100/100 ob−/− mice, ***P < 0.001 vs. C57BL/6 mice. Shown are means ± SEM.
By 11–12 weeks of age, both Apoe−/−Apob100/100ob−/− and Ldlr−/−Apob100/100ob−/− mice were hypertensive, compared with wild-type C57BL/6 mice (data not shown). Previously, it has been demonstrated that Apoe−/− mice exhibit hypertension by 7.5 months of age, while no hypertension was found at 6 weeks of age (36). Also, hypertension has been observed in Ldlr−/−ob−/− mice when the blood pressure was measured with a telemetric system (37).
From 7–8 weeks to 15–16 weeks of age, the total cholesterol levels of Apoe−/−Apob100/100ob−/− and Ldlr−/−Apob100/100ob−/− mice oscillated between 850 and 1,100 mg/dl, almost tenfold higher than in wild-type C57BL/6 mice (Figure 12A, P <0.001). There were no significant differences in cholesterol levels between Apoe−/−Apob100/100ob−/− and Ldlr−/−Apob100/100ob−/− mice at any age; however, when the plasma lipoproteins were fractionated on an HPLC system, the distributions of VLDL, LDL, and HDL were quite different. HDL carried most of the cholesterol in wild-type C57BL/6 mice, consistent with published observations (38) (Figure 12B). Ldlr−/−Apob100/100ob−/− mice had more cholesterol in LDL than in VLDL or HDL; Apoe−/−Apob100/100ob−/− mice had more cholesterol in VLDL than in LDL or HDL (Figure 12B). HDL levels in Apoe−/−Apob100/100ob−/− and Ldlr−/−Apob100/100ob−/− mice were identical.
Figure 12.

Plasma lipid levels and lipoprotein profiles in Apoe−/−Apob100/100ob−/− and Ldlr−/−Apob100/100ob−/− mice. (A) Cholesterol levels in Apoe−/−Apob100/100ob−/−, Ldlr−/−Apob100/100 ob−/−, and C57BL/6 mice (n = 7–16). (B) Distribution of cholesterol in the plasma lipoproteins of Apoe−/−Apob100/100ob−/−, Ldlr−/−Apob100/100 ob−/−, and wild-type C57BL/6 mice. A total of 2–5 μl of fresh plasma was used for HPLC-fractionation of the plasma. (C) Triglyceride levels in Apoe−/−Apob100/100ob−/−, Ldlr−/−Apob100/100ob−/−, and C57BL/6 wild-type control mice (n = 7–18).
Plasma triglyceride levels in Apoe−/−Apob100/100ob−/− and Ldlr−/−Apob100/100ob−/− mice were three- and three- to five-fold higher, respectively, than those in wild-type C57BL/6 mice (Figure 12C, P < 0.0001 for both). The plasma free fatty acid levels were elevated in both Apoe−/−Apob100/100ob−/− and Ldlr−/−Apob100/100ob−/− mice (P < 0.0004 for both groups compared with wild-type C57BL/6 mice) (data not shown).
Quantitative measurements of atherosclerosis revealed that Ldlr−/−Apob100/100ob−/− mice (Figure 13A) had significantly more lesions Apoe−/−Apob100/100ob−/− mice (21.5 ± 5.4% of the surface of the aorta vs. 12.3 ± 3.7%), despite having virtually identical total cholesterol levels. Again, these findings are consistent with a greater atherogenicity of large numbers of LDL particles. Representative examples of lesions in Apoe−/−Apob100/100ob−/− and Ldlr−/−Apob100/100ob−/− mice are shown in Figure 13B.
Figure 13.

Atherosclerosis in Apoe−/−Apob100/100ob−/− and Ldlr−/−Apob100/100ob−/− mice. (A) The percentage of the aorta occupied by lesions was quantified in Apoe−/−Apob100/100ob−/− (n = 7) and Ldlr−/−Apob100/100ob−/− mice (n = 13) after 24 weeks on a chow diet. (B) Representative Sudan IV–stained aortas from a Apoe−/−Apob100/100ob−/− mouse and a Ldlr−/−Apob100/100ob−/− mouse.
A New Model That Might Ultimately Prove Useful to Analyze the Impact of Lipoprotein Size on Atherosclerosis—GPIHBP1 Knockout Mice
During the past year, Drs. Young, Fong, Bensadoun, and Beigneux investigated a new knockout mouse that could ultimately yield new insights into the relationship between lipoprotein size and atherogenesis. They found that mice lacking glycosylphosphatidylinositol-anchored high density lipoprotein-binding protein 1 (GPIHBP1) have chylomicronemia, with extraordinarily large apo-B–containing lipoprotein particles (39). It is now clear that GPIHBP1 plays a critical role in the lipolytic processing of apo-B–containing lipoproteins in the plasma. Gpihbp1-deficient (Gpihbp1−/−) mice on a chow diet have severe chylomicronemia, with grossly milky plasma (Figure 14) and plasma triglyceride levels of 2000–5000 mg/dl. Gpihbp1 is expressed at high levels in heart and adipose tissue and moderate levels in skeletal muscle—very similar to the pattern of expression for lipoprotein lipase (LpL). By immunohistochemistry, GPIHBP1 is located exclusively on the luminal face of endothelial cells, the site where lipolysis of triglyceride-rich lipoproteins is known to occur. Also, cultured cells that have been transfected with a Gpihbp1 cDNA bind both chylomicrons and LpL. Because GPIHBP1 binds both LpL and chylomicrons and because Gpihbp1−/− mice develop severe chylomicronemia, we suspect that GPIHBP1 is a physiologically significant “platform” for triglyceride hydrolysis within capillaries (39).
Figure 14.

Plasma samples after low-speed centrifugation. On the right, lipemic plasma from a Gpihbp1-deficient mouse. The two samples on the left were from unaffected littermates. Reproduced, with permission from Elsevier, from the article by Beigneux and coworkers (39).
Very preliminary observations suggest that chow-fed Gpihbp1−/− mice have an increased susceptibility to atherosclerosis (Mr. Michael Weinstein, Drs. Liya Yan, Loren Fong, unpublished observations). Should this preliminary finding hold up, this new model could prove to be useful in exploring the impact of lipoprotein size on atherogenesis.
Initial Identification of GPIHBP1 as a Molecule with Potential Relevance to Lipid Metabolism
Ioka et al. (40) used an expression cloning strategy to identify cDNAs (from an hepatic cDNA library) that rendered CHO cells capable of binding fluorescently labeled HDL. One of the cDNAs encoded scavenger receptor class B, type 1 (SR-BI)—a predictable result. The other cDNA encoded GPIHBP1. GPIHBP1 (228 amino acids in length) consists of an amino-terminal signal sequence, a domain rich in acidic amino acids (17 of 25 residues in the mouse sequence, and 21 of 25 in the human sequence, are glutamate or aspartate), an Ly-6 motif containing multiple cysteines, and a carboxyl-terminal hydrophobic domain that is ultimately replaced by a GPI anchor (40). The GPI anchor, which tethers GPIHBP1 to the surface of the cell, can be cleaved by a phosphatidylinositol-specific phospholipase C (PIPLC) (40). Like SR-BI (41, 42), GPIHBP1 was reported to mediate the selective uptake of lipids. Excess unlabeled HDL reduced the binding of 125I-HDL to GPIHBP1. The authors concluded that GPIHBP1 was probably involved in cellular cholesterol transport (40).
By northern blot, GPIHBP1 transcripts were reported to be most abundant in the heart (40). By in situ hybridization, Ioka et al. (40) found GPIHBP1 expression in cardiac muscle cells, hepatic Kupffer cells, the sinusoidal endothelium of the liver, the bronchial epithelium, and pulmonary alveolar macrophages. However, our immunohistochemical studies (39) have shown that Gpihbp1 is expressed exclusively in endothelial cells, mainly in heart, skeletal muscle, and adipose tissue. Moreover, chylomicronemia is the only obvious phenotype in Gpihbp1−/− mice (39).
Gpihbp1−/− Mice Are Severely Hypertriglyceridemic on a Chow Diet
Our group studied the functional importance of GPIHBP1 in lipoprotein metabolism by examining the phenotype of Gpihbp1−/− mice. Gpihbp1−/− mice were produced by eliminating all of the exons and introns of Gpihbp1; northern and western blots of tissues from Gpihbp1−/− mice showed a complete absence of Gpihbp1 expression (39).
Both male and female Gpihbp1−/− mice had milky plasma on a regular chow diet (Figure 14). This phenotype was invariably quite striking by 10 weeks of age. The plasma triglyceride levels were significantly elevated at all ages; nearly all mice had plasma triglyceride levels >1500 mg/dl at 7–10 weeks (Figure 15), and some had levels as high as 9000 mg/dl (39) (unpublished observations). Plasma cholesterol levels were also elevated in Gpihbp1−/− mice, with most ranging between 250 mg/dl and 500 mg/dl, with an occasional animal having a cholesterol level greater than 800 mg/dl (39). The plasma lipid levels in Gpihbp1+/− and Gpihbp1+/+ mice were indistinguishable. To define the distribution of lipids within the plasma lipoprotein fractions, plasma lipoproteins were fractionated by size on an FPLC column. The vast majority of the triglycerides and cholesterol in the Gpihbp1−/− mice was located in large lipoproteins (i.e., in the “chylomicron/VLDL” peak) (Figure 16). The diameters of the triglyceride-rich lipoproteins in Gpihbp1−/− mice were far larger than those of wild-type mice, as judged by a dynamic laser light scattering technique (Figure 17) (39). The median diameter of lipoproteins in Gpihbp1−/− mice was >150% larger than that of Gpihbp1+/+ mice, and >15% of the particles in the plasma of Gpihbp1−/− mice had diameters of >122 nm (Figure 17). The FPLC fractionation studies revealed that the HDL cholesterol levels in Gpihbp1−/− mice were low (Figure 16) (43, 44).
Figure 15.

Plasma triglyceride levels in Gpihbp1+/+, Gpihbp1+/−, and Gpihbp1−/− mice at different ages, showing higher triglyceride levels in Gpihbp1−/− mice (P < 0.0001 for each age group). Lipid levels in Gpihbp1+/+ and Gpihbp1+/− mice were not different. Reproduced, with permission from Elsevier, from the article by Beigneux and coworkers (39).
Figure 16.

Distribution of triglyceride and cholesterol in lipoproteins from Gpihbp1−/− mice. (A) Distribution of triglycerides in the plasma lipoproteins of Gpihbp1+/+ and Gpihbp1−/− mice. Plasma lipoproteins were separated by size on a Superose 6 FPLC column. (B) Distribution of cholesterol in the plasma lipoproteins of Gpihbp1+/+ and Gpihbp1−/− mice. Reproduced, with permission from Elsevier, from the article by Beigneux and coworkers (39).
Figure 17.

Distribution of lipoprotein diameters in the d < 1.022 g/ml lipoproteins from Gpihbp1−/− and Gpihbp1+/+ mice. (A) As judged by dynamic laser light scattering, the median diameter of lipoproteins was 157% larger in Gpihbp1−/− mice (n = 3) than in Gpihbp1+/+ mice (n = 6). 15.4% of the particles in Gpihbp1−/− mice had diameters of 122–289 nm. The smaller subpopulation of particles in Gpihbp1−/− mice had diameters of 39–111 nm. (B) Electron micrographs of negatively stained d < 1.006 g/ml lipoproteins from the plasma of Gpihbp1−/− and Gpihbp1+/+ mice, showing larger lipoproteins in Gpihbp1−/− mice. Reproduced, with permission from Elsevier, from the article by Beigneux and coworkers (39).
The phenotype of Gpihbp1−/− mice differs significantly from mice lacking lipoprotein lipase (Lpl−/−). Lpl−/− mice die within 24 h after birth (45, 46) with triglyceride levels as high as 20,000 mg/dl (45). The cause of death in newborn Lpl−/− pups is not known with certainty, but is presumed to be due to either ischemia (from chylomicrons clogging the circulation) or starvation (due to the inability to utilize lipid nutrients in milk). A small percentage of Lpl−/− pups can be rescued by injecting the pups with an adenovirus encoding LpL (44). Following the injection of the LpL adenovirus, LpL is expressed in the liver; however, the expression of LpL falls rapidly and is absent after a few weeks. This transient expression of LpL allows some Lpl−/− pups to survive the suckling phase, and those mice can then survive on a regular chow diet for up to two years. Interestingly, the adult Lpl−/− mice had plasma triglyceride levels of 2000–4000 mg/dl on a chow diet—very similar to the triglyceride concentrations in chow-fed Gpihbp1−/− mice (44). The similarity in plasma triglyceride levels between rescued Lpl−/− mice and Gpihbp1−/− mice has led us to suspect that very little lipolytic processing occurs in the absence of GPIHBP1 even though they have respectable levels of LpL in the plasma following an injection of heparin.
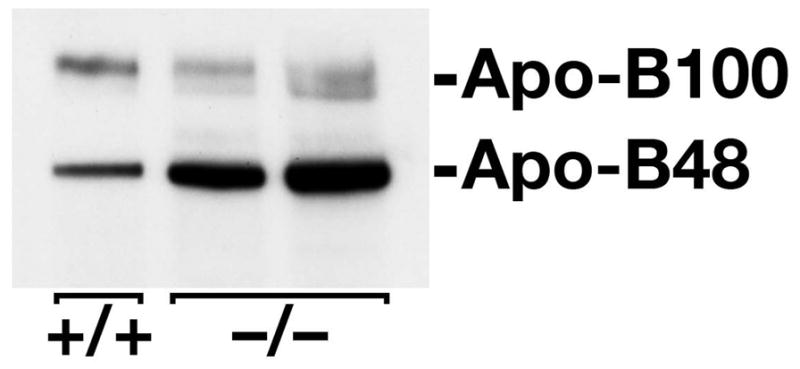
Gpihbp1−/− Mice Have High Levels of Apo-B48 in the Plasma
The plasma lipoproteins of Gpihbp1−/− mice contain high levels of apo-B48, as judged by SDS-polyacrylamide gel electrophoresis (47). Western blots on whole plasma samples confirmed this finding (Figure 18). Of note, plasma apo-B100 levels did not appear to be perturbed in Gpihbp1−/− mice.
Figure 18.

Delayed clearance of retinyl esters in Gpihbp1−/− mice. Retinyl palmitate (5000 IU in 50 μl of vegetable oil) was administered by gavage, and retinyl esters in the plasma were measured over the next 24 h. Inset: retinyl ester levels in Gpihbp1+/+ mice, plotted on a different scale, peaked between 1 and 2 h and had largely disappeared by 10 h. Reproduced, with permission from Elsevier, from the article by Beigneux and coworkers (39).
Thus far, the assessment of apo-B isoforms in the plasma of Gpihbp1−/− mice has been qualitative. We have not measured apo-B48 or apo-B100 concentrations, nor do we know how the amount of apo-B48 and apo-B100 in the plasma of these mice compares with those observed in Apoe−/− and Ldlr−/− mice. At this point, we do not know for sure whether the accumulation of apo-B48 is largely due to intestinal particles. Also, we do not know whether the apparently normal amounts of apo-B100 mean that apo-B100–containing particles can be processed without GPIHBP1.
There are several potential explanations for the striking increases in apo-B48 in the plasma of Gpihbp1−/− mice. One is that apo-B48–containing chylomicrons are very large lipoproteins, and it is mainly the large lipoproteins that accumulate when lipolysis is defective. Another is that GPIHBP1 plays a specialized role in the clearance of apo-B48–containing lipoproteins, either by binding directly to apo-B48 or indirectly by binding to other apolipoproteins on those particles. In the future, we hope to assess the impact of Gpihbp1 deficiency in mice that express exclusively apo-B100. If Gpihbp1−/−Apob100/100 mice do not exhibit hyperlipidemia or a striking accumulation of apo-B100 in the plasma, it would suggest that GPIHBP1 plays a special role in the metabolism of apo-B48–containing lipoproteins.
Mild Hypertriglyceridemia in Gpihbp1−/− Mice During the Suckling Phase
Gpihbp1−/− pups did not exhibit perinatal lethality (39), unlike the situation with newborn Lpl−/− pups (45). We recently examined the plasma lipid levels in suckling Gpihbp1−/− and Gpihbp1+/+ mice; the Gpihbp1−/− pups had far higher lipid levels (P < 0.001), but their triglyceride and cholesterol levels remained <250 mg/dl. In contrast, suckling Lpl−/− pups had plasma triglyceride levels >20,000 mg/dl (45). We do not yet understand the mechanisms for the low lipid levels in suckling Gpihbp1−/− mice, but we are intrigued by the fact that the suckling phase of mouse development is associated with high levels of Lpl expression in the liver (48). We suspect that Lpl expression in the liver underlies the milder-than-expected hyperlipidemia in Gpihbp1−/− mice during the suckling phase.
Markedly Delayed Clearance of Retinyl Palmitate in Gpihbp1−/− Mice
After oral administration of retinyl palmitate, retinyl esters are packaged into chylomicrons in the intestine and enter the circulation. Once the particles are taken up, the retinyl esters are not re-secreted. Thus, their disappearance from the plasma reflects the rate of chylomicron clearance. In wild-type mice, the retinyl esters peak quickly (within 1–3 h), and retinyl esters are almost absent from the plasma after 10 h. In Gpihbp1−/− mice, the plasma levels of retinyl esters are >10-fold higher than those in Gpihbp1+/+ mice, and the high levels of retinyl esters persist in the plasma for more than 24 h (Figure 19) (39). Given the profound delay in the clearance of retinyl palmitate in Gpihbp1−/− mice, we suspect that many other lipids would also have a prolonged circulation time in these mice. For example, we suspect that oxidized lipids in the diet would be absorbed by Gpihbp1−/− mice and then circulate for many hours or days. If so, they conceivably could contribute to the development of atherosclerotic lesions.
Figure 19.
Western blot of mouse plasma (1.0 μl) with a mouse apo-B–specific monoclonal antibody, showing increased amounts of apo-B48 in the plasma of Gpihbp1−/− mice. Reproduced, with permission from Elsevier, from the article by Beigneux and coworkers (39).
GPIHBP1 Is Located on the Luminal Face of the Capillary Endothelium of Tissues That Process Triglyceride-Rich Lipoproteins
When confronted with the finding of chylomicronemia in mice lacking a GPI-anchored cell-surface protein, we thought that there was only one plausible hypothesis—that GPIHBP1 is somehow involved in the LpL-mediated processing of triglyceride-rich lipoproteins along the luminal face of capillary endothelial cells. In support of this general concept, we found that GPIHBP1 is expressed highly in heart, adipose tissue, and skeletal muscle (39)—the very same tissues that express high levels of LpL and CD36, a fatty acid transporter (49). In addition, confocal immunofluorescence microscopy revealed that GPIHBP1 is found along the luminal surface of endothelial cells in both the heart and brown adipose tissue (39) (Figure 20). GPIHBP1 is also expressed within capillaries of skeletal muscle, but it is not expressed at significant levels in capillaries of the brain (39).
Figure 20.

GPIHBP1 is located within the lumen of the capillary endothelium of brown adipose tissue and heart. (A) Confocal microscopy showing the binding of antibodies against CD31 and GPIHBP1 to brown adipose tissue from a Gpihbp1+/+ mouse. Images were taken with a 100× objective. Arrows indicate a capillary shown at higher magnification in the insets (100× objective with 4× digital zoom). Insets: In cross-sections of a capillary, GPIHBP1 staining is particularly prominent on the luminal side. (B) Confocal microscopy showing the binding of antibodies against CD31 and GPIHBP1 to heart tissue from a Gpihbp1+/+ mouse. GPIHBP1 staining is particularly prominent on the luminal face of the capillary endothelium. Images were taken with a 100× objective. Reproduced, with permission from Elsevier, from the article by Beigneux and coworkers (39).
Reduced Levels of LpL in the Postheparin Plasma of Gpihbp1−/− Mice
We separated LpL and hepatic lipase (HL) in postheparin plasma by heparin-sepharose chromatography and then quantified HL and LpL activities. In four experiments, LpL activity was ~20–70% lower in Gpihbp1−/− mice than wild-type mice, and the LpL:HL ratio in the Gpihbp1−/− mice was reduced by ~35–90% (P = 0.03) (39). We also used a mouse LpL–specific ELISA to measure LpL mass in whole plasma (following an injection of heparin). The mean postheparin LpL protein mass was significantly lower in Gpihbp1−/− mice than in wild-type control mice (175 ± 111 vs. 581 ± 98 ng/ml; P < 0.001) (39). In contrast, the levels of LpL mass in the preheparin plasma samples were similar in wild-type and Gpihbp1−/− mice. At this point in our studies, we face a conundrum: Gpihbp1−/− mice have a respectable amount of enzymatically active LpL in the plasma after an injection of heparin, yet there is seemingly almost no lipolysis of the triglyceride-rich lipoproteins. How can this be explained? We do not know the answer to this question, but we speculate that the LpL that is released by heparin in Gpihbp1−/− mice is released from pools of LpL that are not directly relevant to lipoprotein lipolysis (39).
GPIHBP1 Binds Both LpL and Chylomicrons
Because Gpihbp1−/− mice exhibit striking chylomicronemia, we hypothesized that GPIHBP1 likely functions as a binding site—along the capillary endothelium—for LpL or chylomicrons or both (39). The fact that GPIHBP1 contains a strongly negatively charged domain lent some plausibility to this hypothesis, since LpL and several apolipoproteins within chylomicrons contain positively charged domains (and since those positively charged domains are known to mediate both protein–protein and protein-heparan sulfate interactions) (2, 50). To explore this possibility, we constructed a full-length mouse Gpihbp1 cDNA expression vector (39). When we transfected LDL receptor–deficient CHO cells with the Gpihbp1 expression vector, we observed high levels of GPIHBP1 expression at the cell surface, as judged by immunocytochemistry. Importantly, we found that this cell-surface GPIHBP1 could be released with PIPLC (39). To assess the ability of GPIHBP1 to bind LpL, we examined the binding of avian LpL to a mutant CHO cell line (pgsA-745) that cannot synthesize HSPGs (51). The GPIHBP1-expressing pgsA-745 cells bound 10–20-fold more LpL than cells transfected with empty vector; this binding was saturable, and nonspecific binding was minimal (Figure 21). All of the LpL binding could be released with PIPLC (39). Recently, we also found that GPIHBP1 also binds to human LpL, and that this LpL can be released by treatment with PIPLC or heparin (39).
Figure 21.

Testing the ability of cell lines expressing GPIHBP1 to bind LpL. (A) Binding of LpL to pgsA-745 CHO cells stably transfected with a cDNA encoding mouse Gpihbp1 or empty vector. Cells were incubated for 2 h with increasing amounts of avian LpL. Bound LpL was analyzed by ELISA. The binding of avian LpL to pgsA-745 CHO cells could be eliminated by treating the cells with a phosphatidylinositol-specific phospholipase C. Reproduced, with permission from Elsevier, from the article by Beigneux and coworkers (39).
We also tested the binding of fluorescently-labeled chylomicrons to GPIHBP1 on LDL receptor–deficient CHO cells. We found that cells transfected with an empty vector did not bind chylomicrons, whereas the cells transfected with the Gpihbp1 expression vector bound chylomicrons quite avidly (Figure 22) (39). Chylomicron binding was dramatically reduced by treating the transfected cells with PIPLC (39). The ability of GPIHBP1 to bind both to LpL and to chylomicrons was consistent with our hypothesis that GPIHBP1 plays a critical role in lipolysis, perhaps by drawing chylomicrons and LpL into close proximity along the luminal surface of capillaries (39).
Figure 22.

Binding of DiI-labeled chylomicrons (red) to nonpermeabilized CHO-ldlA7 cells that had been transiently transfected with a mouse Gpihbp1 cDNA. Binding was measured at 4° C. GPIHBP1 expression was detected with rabbit anti-GPIHBP1 antiserum and FITC-labeled anti-rabbit IgG (green). Reproduced, with permission from Elsevier, from the article by Beigneux and coworkers (39).
Chylomicrons contain several apolipoproteins (e.g., apo-B48, apo-E, and apo-AV) with positively charged domains; these apolipoproteins are known to bind, via electrostatic interactions, to negatively charged molecules such as heparin sulfate or HSPGs (52–54). It is easy to imagine that one or more of these apolipoproteins on chylomicron particles could mediate binding to the strongly negatively charged amino-terminal domain of GPIHBP1. We imagined that apo-AV could be a good candidate as a ligand for GPIHBP1, since Apoav deficiency in mice causes hypertriglyceridemia associated with decreased LpL-mediated lipolysis (55–57). Indeed, our recent experiments have shown, quite unequivocally, that apo-AV–phospholipid disks bind strongly to Gpihbp1-transfected cells (39). In the future, it will be interesting to assess the binding of apo-E–phospholipid disks to GPIHBP1.
We have also examined the binding of LpL and chylomicrons to Gpihbp1-transfected and nontransfected wild-type CHO-K1 cells (which synthesize HSPGs normally). For LpL, we observed a highly significant 70% increase of binding to the transfected cells compared to non-transfected cells. When comparing CHO-K1 cells and CHO-745 cells both transfected with Gpihbp1, CHO-K1 cells bound 60% more LPL. Total binding to the transfected cells reflects binding to Gpihbp1 and probably also to heparan sulfate chains. For chylomicrons, increased chylomicron binding to the transfected cells was easily detectable. For apo-AV, the levels of binding to the nontransfected cells were very high—so high that differences between Gpihbp1-transfected and nontransfected cells were obscured (Dr. Peter Gin, unpublished observations).
It would obviously be desirable to assess binding of all of these ligands to endothelial cells rather than transfected cells. However, preliminary studies suggest that GPIHBP1 is not expressed in aortic endothelial cells or late-passage microvascular endothelial cells from the heart (Dr. Brandon Davies, unpublished observations).
Although GPIHBP1 on the surface of CHO cells clearly has the capability of binding to LpL and chylomicrons in vitro, we emphasize that the precise role of GPIHBP1 in lipolysis is incompletely understood. While GPIHBP1 likely acts as a platform for lipolysis, the details are sketchy. One possibility is that GPIHBP1 binds LpL and chylomicrons, and that both of these roles are essential. Another possibility is that the main role of GPIHBP1 is in binding chylomicrons, and that LpL binds secondarily to the lipoproteins, perhaps being recruited from surrounding HSPGs. Still another possibility is that the principal role of GPIHBP1 is the binding of LpL, and that LpL, rather than GPIHBP1 itself, plays a predominant role in binding chylomicrons in the bloodstream.
Might Apo-B48 Play a Role in the Binding of Chylomicrons to GPIHBP1?
Another interesting hypothesis is that GPIHBP1 could act as a receptor for apo-B48, the key structural protein of chylomicrons. Abundant evidence indicates that apo-B48–containing remnants are taken up by liver receptors via interactions with apo-E on these particles (28). Thus far, however, there has been little convincing evidence that apo-B48 itself specifically mediates binding to any cell-surface receptor. However, if apo-B48 were to bind directly to GPIHBP1 on the surface of endothelial cells within capillaries, it would make some physiologic sense, as it would provide a mechanism to unload the cargo of apo-B48–containing intestinal lipoproteins (i.e., triglycerides) to physiologically relevant tissues (i.e., muscle and adipose tissue—tissues where GPIHBP1 is expressed).
Both apo-B48 and chylomicrons (defined as intestinal lipoproteins that are initially secreted into the lymph) are found only in mammals (58). Similarly, GPIHBP1 is found in all mammals, including platypus (an aquatic egg-laying mammal), but is absent from the genomes of lower organisms. We suspect that apo-B48 and GPIHBP1 are both mammalian adaptations to optimize the delivery of triglycerides to vital tissues. In the future, we believe that it will be interesting to determine if apo-B48 binds specifically to GPIHBP1. It will also be interesting to determine if the processing of apo-B100–containing lipoproteins is equally dependent on GPIHBP1 and to determine if Gpihbp1−/−Apob100/100 mice would manifest equally severe hypertriglyceridemia.
Spontaneous Atherosclerotic Lesions in Chow-fed Gpihbp1−/− Mice
We had predicted that the Gpihbp1−/− mice on a chow diet would not manifest spontaneous atherosclerotic lesions, simply because the lipoproteins in the plasma of these mice were so immense. However, preliminary observations suggest that Gpihbp1−/− mice do indeed develop atherosclerosis (Mr. Michael Weinstein, Drs. Liya Yan, Loren Fong, unpublished observations). If this finding holds up, it would raise several issues. Can large triglyceride-rich lipoproteins somehow drive the formation of atherosclerotic lesions? Is the atherosclerosis in these mice largely dependent on the level of cholesterol in the triglyceride-rich lipoproteins? If so, would the atherosclerosis in these mice be eliminated with ezetimibe? Given the striking delay in the clearance of chylomicrons, would dietary oxidized lipids exhibit a markedly prolonged circulation time? Would oxidized lipids in the diet increase the susceptibility of Gpihbp1−/− mice to atherosclerosis? In the future, it would be interesting to address each of these questions.
Acknowledgments
The authors thank many dedicated postdoctoral fellows, students, and research associates for their help with the studies described in this chapter. Supported by grants from the NIH (R01 HL087228 and R01 HL76839) and by a beginning Grant-in-Aid from the American Heart Association, Western States Affiliate.
References
- 1.Grundy SM. Cholesterol and coronary heart disease. A new era. J Am Med Assoc. 1986;256:2849–2858. [PubMed] [Google Scholar]
- 2.Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science. 1986;232:34–47. doi: 10.1126/science.3513311. [DOI] [PubMed] [Google Scholar]
- 3.NIH Consensus Development Panel. Triglyceride, high-density lipoprotein, and coronary heart disease. J Am Med Assoc. 1993;269:505–510. [PubMed] [Google Scholar]
- 4.Feussner G, Wagner A, Kohl B, Ziegler R. Clinical features of type III hyperlipoproteinemia: Analysis of 64 patients. Clin Investig. 1993;71:362–366. doi: 10.1007/BF00186624. [DOI] [PubMed] [Google Scholar]
- 5.Fazio S, Lee Y-L, Ji Z-S, Rall SC., Jr Type III hyperlipoproteinemic phenotype in transgenic mice expressing dysfunctional apolipoprotein E. J Clin Invest. 1993;92:1497–1503. doi: 10.1172/JCI116728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brunzell JD, Bierman EL. Chylomicronemia syndrome. Interaction of genetic and acquired hypertriglyceridemia. Med Clin North Am. 1982;66:455–468. doi: 10.1016/s0025-7125(16)31430-4. [DOI] [PubMed] [Google Scholar]
- 7.Chait A, Brunzell JD. Chylomicronemia syndrome. Adv Intern Med. 1991;37:249–273. [PubMed] [Google Scholar]
- 8.Benlian P, De Gennes JL, Foubert L, Zhang H, Gagne SE, Hayden M. Premature atherosclerosis in patients with familial chylomicronemia caused by mutations in the lipoprotein lipase gene. N Engl J Med. 1996;335:848–854. doi: 10.1056/NEJM199609193351203. [DOI] [PubMed] [Google Scholar]
- 9.Duff GL, McMillan GC. The effect of alloxan diabetes on experimental cholesterol atherosclerosis in the rabbit. I. The inhibition of experimental cholesterol atherosclerosis in alloxan diabetes. II. The effect of alloxan diabetes on the retrogression of experimental cholesterol atherosclerosis. J Exp Med. 1949;89:611–630. doi: 10.1084/jem.89.6.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nordestgaard BG, Zilversmit DB. Large lipoproteins are excluded from the arterial wall in diabetic cholesterol-fed rabbits. J Lipid Res. 1988;29:1491–1500. [PubMed] [Google Scholar]
- 11.Nordestgaard BG, Zilversmit DB. Comparison of arterial intimal clearances of LDL from diabetic and nondiabetic cholesterol-fed rabbits. Differences in intimal clearance explained by size differences. Arteriosclerosis. 1989;9:176–183. doi: 10.1161/01.atv.9.2.176. [DOI] [PubMed] [Google Scholar]
- 12.Nordestgaard BG, Stender S, Kjeldsen K. Reduced atherogenesis in cholesterol-fed diabetic rabbits. Giant lipoproteins do not enter the arterial wall. Arteriosclerosis. 1988;8:421–428. doi: 10.1161/01.atv.8.4.421. [DOI] [PubMed] [Google Scholar]
- 13.Paigen B, Morrow A, Brandon C, Mitchell D, Holmes P. Variation in susceptibility to atherosclerosis among inbred strains of mice. Atherosclerosis. 1985;57:65–73. doi: 10.1016/0021-9150(85)90138-8. [DOI] [PubMed] [Google Scholar]
- 14.Ishida BY, Paigen B. Atherosclerosis in the mouse. In: Lusis AJ, Sparkes SR, editors. Genetic Factors in Atherosclerosis: Approaches and Model Systems. Basel: Karger; 1989. pp. 189–222. [Google Scholar]
- 15.Paigen B, Morrow A, Holmes PA, Mitchell D, Williams RA. Quantitative assessment of atherosclerotic lesions in mice. Atherosclerosis. 1987;68:231–240. doi: 10.1016/0021-9150(87)90202-4. [DOI] [PubMed] [Google Scholar]
- 16.Rubin EM, Krauss RM, Spangler EA, Verstuyft JG, Clift SM. Inhibition of early atherogenesis in transgenic mice by human apolipoprotein AI. Nature. 1991;353:265–267. doi: 10.1038/353265a0. [DOI] [PubMed] [Google Scholar]
- 17.Ishibashi S, Brown MS, Goldstein JL, Gerard RD, Hammer RE, Herz J. Hypercholesterolemia in low density lipoprotein receptor knockout mice and its reversal by adenovirus-mediated gene delivery. J Clin Invest. 1993;92:883–893. doi: 10.1172/JCI116663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ishibashi S, Goldstein JL, Brown MS, Herz J, Burns DK. Massive xanthomatosis and atherosclerosis in cholesterol-fed low density lipoprotein receptor–negative mice. J Clin Invest. 1994;93:1885–1893. doi: 10.1172/JCI117179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Piedrahita JA, Zhang SH, Hagaman JR, Oliver PM, Maeda N. Generation of mice carrying a mutant apolipoprotein E gene inactivated by gene targeting in embryonic stem cells. Proc Natl Acad Sci USA. 1992;89:4471–4475. doi: 10.1073/pnas.89.10.4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang SH, Reddick RL, Piedrahita JA, Maeda N. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science. 1992;258:468–471. doi: 10.1126/science.1411543. [DOI] [PubMed] [Google Scholar]
- 21.Plump AS, Smith JD, Hayek T, Aalto-Setälä K, Walsh A, Verstuyft JG, Rubin EM, Breslow JL. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E–deficient mice created by homologous recombination in ES cells. Cell. 1992;71:343–353. doi: 10.1016/0092-8674(92)90362-g. [DOI] [PubMed] [Google Scholar]
- 22.Nakashima Y, Plump AS, Raines EW, Breslow JL, Ross R. ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler Thromb. 1994;14:133–140. doi: 10.1161/01.atv.14.1.133. [DOI] [PubMed] [Google Scholar]
- 23.Véniant MM, Kim E, McCormick S, Borén J, Nielsen LB, Raabe M, Young SG. Insights into apolipoprotein B biology from transgenic and gene-targeted mice. J Nutr. 1999;129:451S–455S. doi: 10.1093/jn/129.2.451S. [DOI] [PubMed] [Google Scholar]
- 24.Véniant MM, Pierotti V, Newland D, Cham CM, Sanan DA, Walzem RL, Young SG. Susceptibility to atherosclerosis in mice expressing exclusively apolipoprotein B48 or apolipoprotein B100. J Clin Invest. 1997;100:180–188. doi: 10.1172/JCI119511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Véniant MM, Sullivan MA, Kim SK, Ambroziak P, Chu A, Wilson MD, Hellerstein MK, Rudel LL, Walzem RL, Young SG. Defining the atherogenicity of large and small lipoproteins containing apolipoprotein B100. J Clin Invest. 2000;106:1501–1510. doi: 10.1172/JCI10695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Véniant MM, Withycombe S, Young SG. Lipoprotein size and atherosclerosis susceptibility in Apoe−/− and Ldlr−/− mice. Arterioscler Thromb Vasc Biol. 2001;21:1567–1570. doi: 10.1161/hq1001.097780. [DOI] [PubMed] [Google Scholar]
- 27.Véniant MM, Zlot CH, Walzem RL, Pierotti V, Driscoll R, Dichek D, Herz J, Young SG. Lipoprotein clearance mechanisms in LDL receptor–deficient “apo-B48-only” and “apo-B100-only” mice. J Clin Invest. 1998;102:1559–1568. doi: 10.1172/JCI4164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ishibashi S, Herz J, Maeda N, Goldstein JL, Brown MS. The two-receptor model of lipoprotein clearance: Tests of the hypothesis in “knockout” mice lacking the low density lipoprotein receptor, apolipoprotein E, or both proteins. Proc Natl Acad Sci USA. 1994;91:4431–4435. doi: 10.1073/pnas.91.10.4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Farese RV, Jr, Véniant MM, Cham CM, Flynn LM, Pierotti V, Loring JF, Traber M, Ruland S, Stokowski RS, Huszar D, Young SG. Phenotypic analysis of mice expressing exclusively apolipoprotein B48 or apolipoprotein B100. Proc Natl Acad Sci USA. 1996;93:6393–6398. doi: 10.1073/pnas.93.13.6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Teng B, Burant CF, Davidson NO. Molecular cloning of an apolipoprotein B messenger RNA editing protein. Science. 1993;260:1816–1819. doi: 10.1126/science.8511591. [DOI] [PubMed] [Google Scholar]
- 31.Linton MF, Farese RV, Jr, Chiesa G, Grass DS, Chin P, Hammer RE, Hobbs HH, Young SG. Transgenic mice expressing high plasma concentrations of human apolipoprotein B100 and lipoprotein(a) J Clin Invest. 1993;92:3029–3037. doi: 10.1172/JCI116927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Purcell-Huynh DA, Farese RV, Jr, Johnson DF, Flynn LM, Pierotti V, Newland DL, Linton MF, Sanan DA, Young SG. Transgenic mice expressing high levels of human apolipoprotein B develop severe atherosclerotic lesions in response to a high-fat diet. J Clin Invest. 1995;95:2246–2257. doi: 10.1172/JCI117915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Powell-Braxton L, Véniant M, Latvala RD, Hirano K-I, Won WB, Ross J, Dybdal N, Zlot CH, Young SG, Davidson NO. A mouse model of human familial hypercholesterolemia: Markedly elevated low density lipoprotein cholesterol levels and severe atherosclerosis on a low-fat chow diet. Nat Med. 1998;4:934–938. doi: 10.1038/nm0898-934. [DOI] [PubMed] [Google Scholar]
- 34.Neaton JD, Wentworth D. Serum cholesterol, blood pressure, cigarette smoking, and death from coronary heart disease. Overall findings and differences by age for 316 099 white men. Arch Intern Med. 1992;152:56–64. [PubMed] [Google Scholar]
- 35.Linton MF, Atkinson JB, Fazio S. Prevention of atherosclerosis in apolipoprotein E–deficient mice by bone marrow transplantation. Science. 1995;267:1034–1037. doi: 10.1126/science.7863332. [DOI] [PubMed] [Google Scholar]
- 36.Yang R, Powell-Braxton L, Ogaoawara AK, Dybdal N, Bunting S, Ohneda O, Jin H. Hypertension and endothelial dysfunction in apolipoprotein E knockout mice. Arterioscler Thromb Vasc Biol. 1999;19:2762–2768. doi: 10.1161/01.atv.19.11.2762. [DOI] [PubMed] [Google Scholar]
- 37.Verreth W, De Keyzer D, Pelat M, Verhamme P, Ganame J, Bielicki JK, Mertens A, Quarck R, Benhabiles N, Marguerie G, Mackness B, Mackness M, Ninio E, Herregods MC, Balligand JL, Holvoet P. Weight-loss-associated induction of peroxisome proliferator-activated receptor-alpha and peroxisome proliferator-activated receptor-gamma correlate with reduced atherosclerosis and improved cardiovascular function in obese insulin-resistant mice. Circulation. 2004;110:3259–3269. doi: 10.1161/01.CIR.0000147614.85888.7A. [DOI] [PubMed] [Google Scholar]
- 38.Camus MC, Chapman MJ, Forgez P, Laplaud PM. Distribution and characterization of the serum lipoproteins and apoproteins in the mouse, Mus musculus. J Lipid Res. 1983;24:1210–1228. [PubMed] [Google Scholar]
- 39.Beigneux A, Davies B, Gin P, Weinstein M, Farber E, Qiao X, Peale F, Bunting S, Walzem R, Wong J, Blaner W, Ding Z, Melford K, Wongsiriroj N, Sauvage Fd, Fong L, Bensadoun A, Young S. Glycosylphosphatidylinositol-anchored high density lipoprotein–binding protein 1 plays a critical role in the lipolytic processing of chylomicrons. Cell Metabolism. 2007;5:279–291. doi: 10.1016/j.cmet.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ioka RX, Kang M-J, Kamiyama S, Kim D-H, Magoori K, Kamataki A, Ito Y, Takei YA, Sasaki M, Suzuki T, Sasano H, Takahashi S, Sakai J, Fujino T, Yamamoto TT. Expression Cloning and Characterization of a Novel Glycosylphosphatidylinositol-anchored High Density Lipoprotein-binding Protein, GPI-HBP1. J Biol Chem. 2003;278:7344–7349. doi: 10.1074/jbc.M211932200. [DOI] [PubMed] [Google Scholar]
- 41.Acton SL, Scherer PE, Lodish HF, Krieger M. Expression cloning of SR-BI, a CD36-related class B scavenger receptor. J Biol Chem. 1994;269:21003–21009. [PubMed] [Google Scholar]
- 42.Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science. 1996;271:518–520. doi: 10.1126/science.271.5248.518. [DOI] [PubMed] [Google Scholar]
- 43.Brunzell JD, Deeb SS. Familial lipoprotein lipase deficiency, apo C-II deficiency, and hepatic lipase deficiency. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 2001. [Google Scholar]
- 44.Strauss JG, Frank S, Kratky D, Hammerle G, Hrzenjak A, Knipping G, von Eckardstein A, Kostner GM, Zechner R. Adenovirus-mediated rescue of lipoprotein lipase-deficient mice. Lipolysis of triglyceride-rich lipoproteins is essential for high density lipoprotein maturation in mice. J Biol Chem. 2001;276:36083–36090. doi: 10.1074/jbc.M104430200. [DOI] [PubMed] [Google Scholar]
- 45.Weinstock PH, Bisgaier CL, Aalto-Setälä K, Radner H, Ramakrishnan R, Levak-Frank S, Essenburg AD, Zechner R, Breslow JL. Severe hypertriglyceridemia, reduced high density lipoprotein, and neonatal death in lipoprotein lipase knockout mice. Mild hypertriglyceridemia with impaired low density lipoprotein clearance in heterozygotes. J Clin Invest. 1995;96:2555–2568. doi: 10.1172/JCI118319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Coleman T, Seip RL, Gimble JM, Lee D, Maeda N, Semenkovich CF. COOH-terminal disruption of lipoprotein lipase in mice is lethal in homozygotes, but heterozygotes have elevated triglycerides and impaired enzyme activity. J Biol Chem. 1995;270:12518–12525. doi: 10.1074/jbc.270.21.12518. [DOI] [PubMed] [Google Scholar]
- 47.Nguyen AT, Braschi S, Geoffrion M, Fong LG, Crooke RM, Graham MJ, Young SG, Milne R. A mouse monoclonal antibody specific for mouse apoB48 and apoB100 produced by immunizing “apoB39-only” mice with mouse apoB48. Biochim Biophys Acta. 2006;1761:182–185. doi: 10.1016/j.bbalip.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 48.Langner CA, Birkenmeier EH, Ben-Zeev O, Schotz MC, Sweet HO, Davisson MT, Gordon JI. The fatty liver dystrophy (fld) mutation. A new mutant mouse with a developmental abnormality in triglyceride metabolism and associated tissue-specific defects in lipoprotein lipase and hepatic lipase activities. J Biol Chem. 1989;264:7994–8003. [PubMed] [Google Scholar]
- 49.Greenwalt DE, Scheck SH, Rhinehart-Jones T. Heart CD36 expression is increased in murine models of diabetes and in mice fed a high fat diet. J Clin Invest. 1995;96:1382–1388. doi: 10.1172/JCI118173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cardin AD, Jackson RL, Sparrow DA, Sparrow JT. Interaction of glycosaminoglycans with lipoproteins. Ann NY Acad Sci. 1989;556:186–193. doi: 10.1111/j.1749-6632.1989.tb22503.x. [DOI] [PubMed] [Google Scholar]
- 51.Lugemwa FN, Esko JD. Estradiol beta-D-xyloside, an efficient primer for heparan sulfate biosynthesis. J Biol Chem. 1991;266:6674–6677. [PubMed] [Google Scholar]
- 52.Cardin AD, Hirose N, Blankenship DT, Jackson RL, Harmony JAK. Binding of a high reactive heparin to human apolipoprotein E: Identification of two heparin-binding domains. Biochem Biophys Res Commun. 1986;134:783–789. doi: 10.1016/s0006-291x(86)80489-2. [DOI] [PubMed] [Google Scholar]
- 53.Cardin AD, Barnhart RL, Witt KR, Jackson RL. Reactivity of heparin with the human plasma heparin-binding proteins thrombin, antithrombin III, and apolipoproteins E and B-100. Thromb Res. 1984;34:541–550. doi: 10.1016/0049-3848(84)90258-5. [DOI] [PubMed] [Google Scholar]
- 54.Lookene A, Beckstead JA, Nilsson S, Olivecrona G, Ryan RO. Apolipoprotein A-V-heparin interactions: implications for plasma lipoprotein metabolism. J Biol Chem. 2005;280:25383–25387. doi: 10.1074/jbc.M501589200. [DOI] [PubMed] [Google Scholar]
- 55.Pennacchio LA, Olivier M, Hubacek JA, Cohen JC, Cox DR, Fruchart JC, Krauss RM, Rubin EM. An apolipoprotein influencing triglycerides in humans and mice revealed by comparative sequencing. Science. 2001;294:169–173. doi: 10.1126/science.1064852. [DOI] [PubMed] [Google Scholar]
- 56.Calandra S, Oliva CP, Tarugi P, Bertolini S. APOA5 and triglyceride metabolism, lesson from human APOA5 deficiency. Curr Opin Lipidol. 2006;17:122–127. doi: 10.1097/01.mol.0000217892.00618.54. [DOI] [PubMed] [Google Scholar]
- 57.Grosskopf I, Baroukh N, Lee SJ, Kamari Y, Harats D, Rubin EM, Pennacchio LA, Cooper AD. Apolipoprotein A-V deficiency results in marked hypertriglyceridemia attributable to decreased lipolysis of triglyceride-rich lipoproteins and removal of their remnants. Arterioscler Thromb Vasc Biol. 2005;25:2573–2579. doi: 10.1161/01.ATV.0000186189.26141.12. [DOI] [PubMed] [Google Scholar]
- 58.Teng B, Davidson NO. Evolution of intestinal apolipoprotein B mRNA editing. Chicken apolipoprotein B mRNA is not edited, but chicken enterocytes contain in vitro editing enhancement factor(s) J Biol Chem. 1992;267:21265–21272. [PubMed] [Google Scholar]