

Abstract
Neuronal intermediate filament inclusion disease (NIFID), a rare form of frontotemporal lobar degeneration (FTLD), is characterized neuropathologically by focal atrophy of the frontal and temporal lobes, neuronal loss, gliosis, and neuronal cytoplasmic inclusions (NCI) containing epitopes of ubiquitin and neuronal intermediate filament proteins. Recently, the ‘fused in sarcoma’ (FUS) protein (encoded by the FUS gene) has been shown to be a component of the inclusions of familial amyotrophic lateral sclerosis with FUS mutation, NIFID, basophilic inclusion body disease, and atypical FTLD with ubiquitin-immunoreactive inclusions (aFTLD-U). To further characterize FUS proteinopathy in NIFID, and to determine whether the pathology revealed by FUS immunohistochemistry (IHC) is more extensive than α-internexin, we have undertaken a quantitative assessment of ten clinically and neuropathologically well-characterized cases using FUS IHC. The densities of NCI were greatest in the dentate gyrus (DG) and in sectors CA1/2 of the hippocampus. Anti-FUS antibodies also labeled glial inclusions (GI), neuronal intranuclear inclusions (NII), and dystrophic neurites (DN). Vacuolation was extensive across upper and lower cortical layers. Significantly greater densities of abnormally enlarged neurons and glial cell nuclei were present in the lower compared with the upper cortical laminae. FUS IHC revealed significantly greater numbers of NCI in all brain regions especially the DG. Our data suggest: (1) significant densities of FUS-immunoreactive NCI in NIFID especially in the DG and CA1/2; (2) infrequent FUS-immunoreactive GI, NII, and DN; (3) widely distributed vacuolation across the cortex, and (4) significantly more NCI revealed by FUS than α-internexin IHC.
Keywords: Neurofilament intermediate filament inclusion disease (NIFID), ‘Fused in sarcoma’ (FUS), Neuronal cytoplasmic inclusions (NCI), Density, Neuronal intranuclear inclusions (NII)
Introduction
Neuronal intermediate filament inclusion disease (NIFID) is a rare form of frontotemporal lobar degeneration (FTLD) characterized clinically by an early-onset and a variable phenotype that includes frontotemporal dementia (FTD), pyramidal, and extrapyramidal signs [13, 16, 24]. Neuropathologically, there is degeneration of the cerebral cortex, striatum, and brain stem with neuronal loss in the frontal, parietal, and temporal cortex [13, 16, 17]. Abnormal neuronal intermediate filament (IF) aggregates in the form of neuronal cytoplasmic inclusions (NCI) have been regarded as the pathological hallmark of NIFID [18]. In addition, abnormally enlarged neurons (EN) and gliosis are often observed in affected areas [9, 16].
The densities of pathological changes in areas of the frontal and temporal lobe have been quantified previously in NIFID using antibodies that recognize either a phosphorylated epitope of the heavy neurofilament subunit (NF-H) [15, 17] or α-internexin [2–4, 9] one of the four proteins that comprise the type IV neuronal intermediate filament (IF) proteins [20, 21]. Not all inclusions in NIFID, however, are immunolabelled by anti-NF-H or anti-neuronal IF antibodies, and therefore, the primary molecular defect is uncertain [30]. Recently, ‘fused in sarcoma’ (FUS) protein (encoded by the FUS gene) has been shown to be a component of the inclusions of familial amyotrophic lateral sclerosis (FALS) with FUS mutation [26, 34, 35], and three FTLD entities, viz., NIFID [30, 37], basophilic inclusion body disease (BIBD) [29, 37], and atypical FTLD with ubiquitin-immunoreactive inclusions (aFTLD-U) [31]. In addition, the pathology of NIFID may include glial inclusions (GI), neuronal intranuclear inclusions (NII), and dystrophic neurites (DN) [30] and these lesions have not been observed previously in NIFID using α-internexin immunohistochemistry (IHC). Hence, there are currently four entities included in a possible molecular group of FTLD-FUS proteinopathies. Previously, Neumann et al. [30] carried out a semi-quantitative analysis of the FUS pathology in five cases of NIFID. Hence, the rationale of this study was to quantify the FUS pathology in ten cases of NIFID in more detail to better categorize these cases compared with the other FTLD-FUS proteinopathies and to better understand the role of FUS in NIFID. Consequently, we measured the densities of NCI, GI, NII, DN together with vacuolation, number of surviving neurons, EN, and glial cell nuclei in FUS immunolabelled sections of the frontal and temporal lobes of ten cases of NIFID.
Materials and methods
Cases
Ten cases of NIFID (see Table 1) obtained from Canada, Norway, Spain, Japan (one case from each), and from France, the UK, and the USA (two cases from each) were studied [19]. Patients had no family history of psychiatric or neurological disorders. Presenting symptoms included personality change, apathy, blunted affect, and disinhibition in seven patients and memory loss and cognitive impairments in five patients. Motor weakness was evident at presentation in three patients and extrapyramidal features in eight patients. All cases displayed the previously defined histological hallmarks of NIFID, viz., α-internexin-immunoreactive NCI, together with EN and gliosis in the cerebral cortex and striatum [19]. None of the cases had pathological features immunoreactive for phosphorylated tau, cytoskeletal proteins specific for astrocytes (GFAP) [17, 19] or microglia (CD68) [17, 19], prion protein, α-synuclein [16], or TDP-43 [11] thus eliminating a diagnosis of a tauopathy, synucleinopathy, prion disease, or TDP-43 proteinopathy. The degree of brain atrophy varied between cases and was tentatively related to the four-stage scheme for severity of atrophy in FTLD [14]. In this scheme, case G had the least (stage 1) and cases I and J the most severe atrophy (stages 3/4). The remaining cases had intermediate levels of atrophy (stages 2/3) with cases A and B having a greater degree of atrophy compared with C, D, E, F, and H. One case (B) was also diagnosed as having associated motor neuron disease [19].
Table 1.
Demographic features and gross brain weight of the neuronal intermediate filament inclusion disease (NIFID) cases studied
| Case | Sex | Age at onset (years) | Age at death (years) | Duration (years) | Brain weight (g) | Atrophy |
|---|---|---|---|---|---|---|
| A | F | 23 | 28 | 5 | 860 | Fr, T, P, C |
| B | F | 25 | 29 | 4 | 710 | Fr, T, P, C |
| C | M | 32 | 35 | 3 | NA | Fr, T, C |
| D | F | 38 | 41 | 3 | 904 | Fr, T, C |
| E | M | 39 | 42.5 | 3.5 | 950 | Fr, T, C |
| F | M | 47 | 50 | 3 | 1,200 | Fr, T, C |
| G | M | 48 | 52 | 4 | 1,310 | Fr |
| H | M | 48 | 61 | 13 | 850 | Fr, T, C |
| I | F | 52 | 54.7 | 2.7 | 813 | Fr, T, P, C, A |
| J | M | 56 | 60 | 4 | 1,250 | Fr, T, P, C, A |
M male, F female, NA data not available, Fr frontal cortex, T temporal cortex, P parietal cortex, C caudate/putamen, A amygdala
Histological methods
After death, the consent of the next of kin was obtained for brain removal, following local Institute Review Board (Human Studies Committee, Washington University School of Medicine) and the 1995 Declaration of Helsinki (as modified Edinburgh, 2000). Brain tissue was preserved in buffered 10% formalin or 4% paraformaldehyde. Tissue blocks were taken from the frontal lobe at the level of the genu of the corpus callosum to study the superior frontal cortex (SFC), and temporal lobe at the level of the lateral geniculate body. Frontal and temporal lobe blocks were available for seven of the cases while three cases had frontal lobe blocks only. Within the temporal lobe, the inferior temporal gyrus (ITG), parahippocampal gyrus (PHG), CA sectors of the hippocampus (HC) (CA1/CA2), and the dentate gyrus (DG) were studied. Tissue was fixed in 10% phosphate buffered formal–saline and embedded in paraffin wax. Following microwave pretreatment, IHC was performed on 6–8 μm sections with a commercial anti-FUS rabbit polyclonal antibody (1:1,500; Sigma–Aldrich, St. Louis, MO). Sections were counterstained with haematoxylin and were taken adjacent to those previously immunolabelled with α-internexin [9].
Morphometric methods
In each neocortical region, NCI, GI, NII, DN, vacuoles, surviving neurons, EN, and glial cell nuclei were quantified along a strip of tissue (3,200–6,400 μm in length) parallel to the pia mater, using 250 × 50 μm sample fields arranged contiguously. The sample fields were located both in the upper (approximating to laminae II/III) and lower (approximating to laminae V/VI) cortex, the short edge of the sample field being orientated parallel with the pia mater and aligned with guidelines marked on the slide. In the HC, the histological features were counted from sector CA1 to CA2, the short dimension of the contiguous sample field being aligned with the alveus. Pathological changes were also observed in the DG granule cells [19] and to quantify these lesions, the sample field was aligned with the upper edge of the granule cell layer. Surviving neurons were identified as cells containing at least some stained cytoplasm in combination with larger shape and non-spherical outline [1]. By contrast, small spherical or asymmetrical nuclei without cytoplasm, but with the presence of a thicker nuclear membrane and more heterogeneous chromatin, were identified as glial cells. EN had enlarged perikarya, lacked NCI, had a shrunken nucleus displaced to the periphery of the cell, and a maximum cell diameter of at least three times the diameter of the nucleus [1]. The number of discrete vacuoles greater than 5 μm in diameter was also recorded in each sample field [8, 10].
Data analysis
The data were analyzed by analysis of variance (ANOVA) of incomplete randomized blocks with subsequent comparisons between the groups made by Fisher’s ‘protected least significant difference’ (PLSD) as a post hoc procedure (STATISTICA software, Statsoft Inc., 2300 East 14th St, Tulsa, Ok, 74104, USA). First, the densities of each histological feature in the upper cortical laminae of the neocortical regions studied were compared with those in sectors CA1/2 and the DG using an one-way ANOVA. A similar analysis was then carried out but substituting densities in the lower cortical laminae. Second, densities of each histological feature were compared between the upper and lower neocortical laminae using two-factor, split-plot ANOVA with brain region as a main-plot factor and cortical lamina as the sub-plot factor. Third, densities of the NCI revealed by FUS and by α-internexin IHC [9] were compared using a two-factor, split-plot ANOVA. Fourth, to investigate the effect of confounding variables, the correlations between the densities of the FUS-immunoreactive inclusions and disease onset, age at death, disease duration, brain weight, and brain atrophy were tested using Pearson’s correlation coefficient (r).
Results
The NCI (Fig. 1a, b) were commonly round, oval, or cup-shaped but other morphologies were present including crescent and tangle-shaped inclusions [30]. The NII (Fig. 1a) were variable but most were circular or filamentous in shape [32] while the DN (Fig. 1c) were characteristically long and contorted. The GI (Fig. 1d) occurred within astrocytes in the white matter (Fig. 1d) and in oligodendrocytes, and the latter morphologically resembled analogous inclusions reported in various tauopathies such as corticobasal degeneration (CBD) [6, 28], progressive supranuclear palsy (PSP) [23, 25, 36], and argyrophilic grain disease (AGD) [33].
Fig. 1.
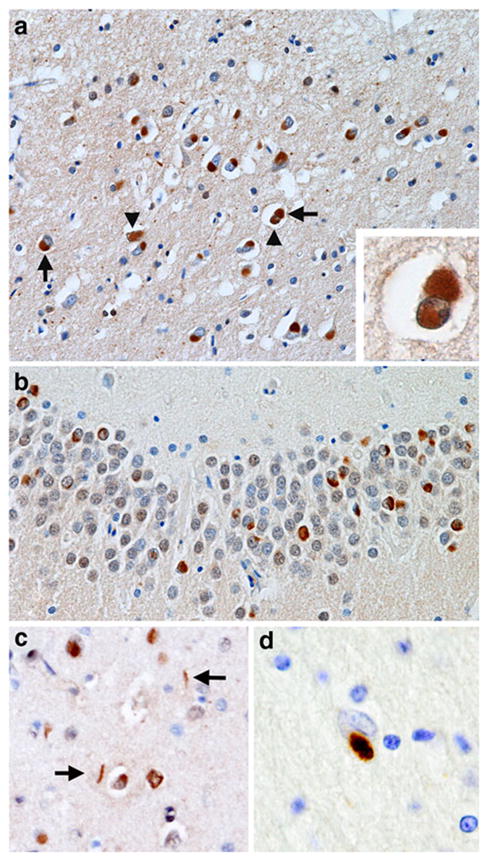
Spectrum of ‘fused in sarcoma’ (FUS) immunoreactive inclusions in neuronal intermediate filament inclusion disease (NIFID). a A neuronal cytoplasmic inclusion (NCI) (arrow) and neuronal intranuclear inclusion (NII) (arrowhead) in the superficial laminae of the frontal lobe (×200); inset shows a neuron containing both an NCI and NII; b NCI in the dentate fascia (×400); c NCI and dystrophic neurites (DN) in the frontal lobe (×200); d a glial cytoplasmic inclusion (GI) in an astrocyte in the white matter of the frontal lobe (×400); a–d FUS immunohistochemistry
The densities of the FUS-immunoreactive pathological features in each region of each case are shown in Table 2 and the group means are shown in Fig. 2. There were significant differences in the densities of NCI between brain regions both in the upper (F = 4.72, P<0.01) and lower cortex (F = 5.33, P<0.01). A major difference between regions is the high density of NCI in the DG which was especially evident in four of the seven cases in which temporal lobe blocks were available. Densities of NCI were also significantly higher in sectors CA1/2 of the hippocampus compared with regions of the neocortex in four of the cases. The densities of the GI (range 0–0.16 per 50 × 250 μm field), NII (range 0.02–0.12 per field), and DN (range 0.01–0.03 per field) were highly variable, significantly lower than those of the NCI, and exhibited no differences between brain regions. In addition, there were no significant differences in density of the FUS-immunoreactive pathological features between the upper and lower cortex. There were no essential differences in FUS pathology between the case with associated MND (B) compared with the other cases.
Table 2.
Densities (per 50 × 250 μm sample field) of ‘fused in sarcoma’ (FUS)-immunoreactive neuronal cytoplasmic inclusions (NCI), glial inclusions (GI), neuronal intranuclear inclusions (NII), and dystrophic neurites (DN) in various brain regions in each of the ten cases of neuronal intermediate filament inclusion disease (NIFID)
| Brain region | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Case | Lesion | SFC (U) | SFC (L) | ITG (U) | ITG (L) | PHG (U) | PHG (L) | CA1/2 | DG |
| A | NCI | 0.56 | 0.73 | 1.56 | 1.35 | 0.5 | 0.59 | 0.68 | 3.68 |
| GI | 0.16 | 0.15 | 0.09 | 0.10 | 0 | 0.13 | 0 | 0 | |
| NII | 0.09 | 0.15 | 0.16 | 0.45 | 0.16 | 0.22 | 0.32 | 0.06 | |
| DN | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| B | NCI | 2.13 | 1.38 | 0.68 | 0.78 | 2.48 | 2.06 | 2.06 | 4.25 |
| GI | 0.06 | 0.09 | 0.03 | 0.06 | 0.19 | 0.06 | 0.09 | 0 | |
| NII | 0.13 | 0.09 | 0.06 | 0.06 | 0.14 | 0.19 | 0.03 | 0 | |
| DN | 0 | 0 | 0 | 0 | 0.05 | 0 | 0 | 0 | |
| C | NCI | 1.59 | 1.78 | – | – | – | – | – | – |
| GI | 0.16 | 0.19 | – | – | – | – | – | – | |
| NII | 0.06 | 0.19 | – | – | – | – | – | – | |
| DN | 0 | 0 | – | – | – | – | – | – | |
| D | NCI | 1.00 | 0.59 | 1.84 | 2.63 | 1.78 | 1.16 | 2.31 | 4.31 |
| GI | 0.06 | 0.06 | 0.13 | 0.31 | 0.06 | 0.03 | 0 | 0 | |
| NII | 0.09 | 0.09 | 0.09 | 0.06 | 0.03 | 0.09 | 0.09 | 0.03 | |
| DN | 0 | 0 | 0.13 | 0.06 | 0.03 | 0.06 | 0.16 | 0.16 | |
| E | NCI | 1.34 | 1.00 | – | – | – | – | – | – |
| GI | 0.03 | 0.03 | – | – | – | – | – | – | |
| NII | 0.06 | 0.09 | – | – | – | – | – | – | |
| DN | 0 | 0 | – | – | – | – | – | – | |
| F | NCI | 2.20 | 0.14 | 1.50 | 2.62 | 0.13 | 1.81 | 1.78 | 1.16 |
| GI | 0.60 | 0 | 0.28 | 0.31 | 0.06 | 0.10 | 0.16 | 0 | |
| NII | 0.16 | 0 | 0.19 | 0.25 | 0.13 | 0.09 | 0.03 | 0.06 | |
| DN | 0.04 | 0 | 0.06 | 0 | 0 | 0 | 0 | 0 | |
| G | NCI | 2.59 | 1.00 | 0 | 0.31 | 0.19 | 0.38 | 1.34 | 1.03 |
| GI | 0.25 | 0.09 | 0 | 0 | 0 | 0 | 0.16 | 0 | |
| NII | 0.16 | 0.06 | 0 | 0 | 0 | 0.06 | 0.13 | 0 | |
| DN | 0 | 0.03 | 0 | 0 | 0 | 0 | 0 | 0 | |
| H | NCI | 0.31 | 0.47 | 0.06 | 0.06 | 0.19 | 0.25 | 0.59 | 1.19 |
| GI | 0 | 0.09 | 0.06 | 0 | 0 | 0.19 | 0.03 | 0 | |
| NII | 0.13 | 0.03 | 0 | 0 | 0 | 0 | 0.03 | 0 | |
| DN | 0.06 | 0 | 0 | 0 | 0.06 | 0 | 0 | 0 | |
| I | NCI | 1.22 | 0.59 | – | – | – | – | – | |
| GI | 0.13 | 0.06 | – | – | – | – | – | ||
| NII | 0.16 | 0 | – | – | – | – | – | ||
| DN | 0 | 0.03 | – | – | – | – | – | ||
| J | NCI | 1.71 | 1.97 | 0.75 | 1.44 | 1.06 | 0.86 | 1.94 | 6.06 |
| GI | 0.16 | 0.16 | 0.13 | 0.06 | 0.10 | 0.05 | 0.19 | 0 | |
| NII | 0.06 | 0.06 | 0.09 | 0 | 0.03 | 0.09 | 0.06 | 0 | |
| DN | 0 | 0.13 | 0 | 0.06 | 0 | 0.05 | 0.03 | 0 | |
SFC superior frontal cortex, ITG inferior temporal gyrus, PHG parahippocampal gyrus, CA1/2 pyramidal cell layer sectors of the hippocampus, DG dentate gyrus, U upper cortex, L lower cortex, – tissue section not available
Fig. 2.

Densities of ‘fused in sarcoma’ (FUS)-immunoreactive neuronal cytoplasmic inclusions (NCI), glial inclusions (GI), neuronal intranuclear inclusions (NII), and dystrophic neurites (DN) in various brain regions (SFC superior frontal cortex, ITG inferior temporal gyrus, PHG parahippocampal gyrus, CA1/2 pyramidal cell layer sectors of the hippocampus, DG dentate gyrus, U upper cortex, L lower cortex) in ten cases of neuronal intermediate filament inclusion disease (NIFID). Analysis of variance (ANOVA) One-way (upper cortical laminae, CA1/2, DG): between brain regions NCI F = 4.72 (P<0.01); one-way (lower cortical laminae, CA1/2, DG): between brain regions NCI F = 5.33 (P<0.01); GI upper laminae F = 1.11 (P>0.05), lower laminae F = 0.07 (P>0.05); NII upper laminae F = 2.07 (P>0.05), lower laminae F = 1.07 (P>0.05); DN F = 0.21 (P>0.05), lower laminae F = 0.07 (P>0.05); comparison between upper and lower laminae (ANOVA two-factor, split-plot) NCI laminae F = 0.29 (P>0.05), interaction with brain region F = 1.99 (P>0.05); GI laminae F = 1.14 (P>0.05), interaction with brain region F = 1.27 (P>0.05); NII laminae F = 0.44 (P>0.05), interaction with brain region F = 1.55 (P>0.05); DN laminae F = 0.03 (P>0.05), interaction with brain region F = 0.35 (P>0.05)
The density of the vacuoles, surviving neurons, EN, and glial cell nuclei are shown in Fig. 3. There were significant differences in the densities of vacuoles between brain regions both in the upper (F = 3.16, P<0.05) and lower cortex (F = 2.94, P<0.05) with lower densities in the DG compared with sectors CA1/2 and the neocortex. The densities of the surviving neurons (in neocortex only), EN, and glial cell nuclei (in all regions) were similar between brain regions. However, there were significantly greater densities of EN (F = 7.93, P<0.01) and glial cell nuclei (F = 35.32, P<0.001) in the lower compared with the upper cortex.
Fig. 3.

Densities of vacuoles, abnormally enlarged neurons (EN), surviving neurons, and glial cell nuclei in the upper and lower laminae in various brain regions (SFC superior frontal cortex, ITG inferior temporal gyrus, PHG parahippocampal gyrus, CA1/2 pyramidal cell layer sectors of the hippocampus, DG dentate gyrus, U = upper cortex, L lower cortex) in ten cases of neuronal intermediate filament inclusion disease (NIFID). ANOVA one-way (upper cortical laminae, CA1/2, DG): between brain regions vacuoles F = 3.16 (P<0.05); one-way (lower cortical laminae, CA1/2, DG): between brain regions vacuoles F = 2.94 (P<0.05); upper laminae F = 0.53 (P>0.05), lower laminae F = 0.59 (P>0.05); surviving neurons upper laminae F = 0.74 (P>0.05), lower laminae F = 0.59 (P>0.05); glial cell nuclei F = 6.19 (P<0.001), lower laminae F = 6.05 (P<0.001); comparison between upper and lower laminae (ANOVA two-factor, split-plot) vacuoles laminae F = 2.87 (P>0.05), interaction with brain region F = 0.09 (P>0.05); EN laminae F = 7.93 (P<0.01), interaction with brain region F = 0.81 (P>0.05); surviving neurons laminae F = 0.79 (P>0.05), interaction with brain region F = 0.02 (P>0.05); glial cell nuclei laminae F = 35.32 (P<0.001), interaction with brain region F = 0.06 (P>0.05)
A comparison of the densities of the NCI as revealed by FUS and α-internexin IHC is shown in Fig. 4 and the densities of α-internexin-immunoreactive NCI in each case in Table 3. In both the upper (F = 4.62, P<0.01) and lower cortex (F = 6.12, P<0.01) greater numbers of NCI were revealed by FUS compared with α-internexin IHC. A significant brain region × antibody interaction also suggests that the differences in density between FUS and α-internexin varied between brain regions; the greatest difference being observed in the DG.
Fig. 4.
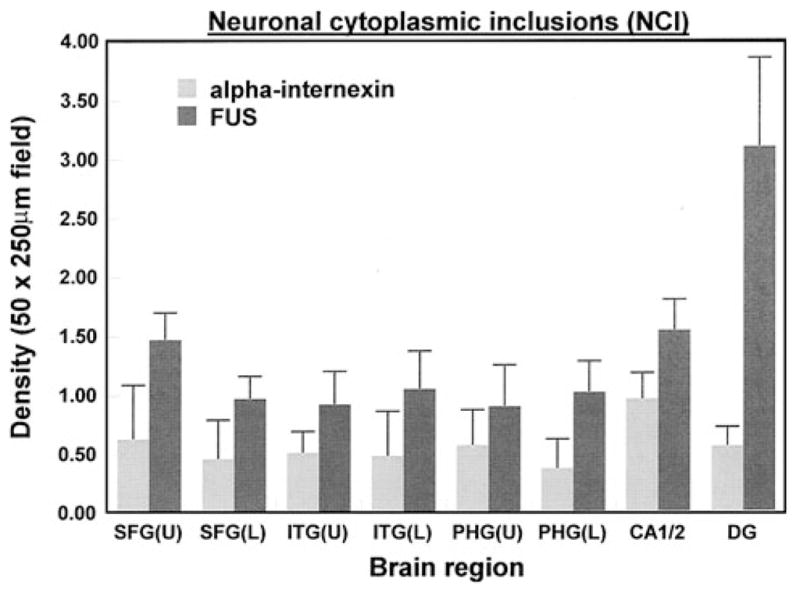
A comparison of the densities of the NCI as revealed by ‘fused in sarcoma’ (FUS) and α-internexin ANOVA (two-factor split plot) upper laminae region F = 4.62 (P<0.01), antibody F = 20.89 (P<0.001), interaction F = 3.81 (P<0.05); lower laminae region F = 6.12 (P<0.01), Antibody F = 17.08 (P<0.001), interaction F = 3.83 (P<0.05)
Table 3.
Densities (per 50 × 250 μm sample field) of α-internexin-immunoreactive neuronal cytoplasmic inclusions (NCI) in various brain regions in each of the ten cases of neuronal intermediate filament inclusion disease (NIFID)
| Brain region | ||||||||
|---|---|---|---|---|---|---|---|---|
| Case | SFC (U) | SFC (L) | ITG (U) | ITG (L) | PHG (U) | PHG (L) | CA1/2 | DG |
| A | 0.23 | 0 | 0.23 | 0.17 | 0.10 | 0 | 0.20 | 0.12 |
| B | 1.31 | 0.57 | 0.69 | 0.21 | 0 | 0 | 0 | 0 |
| C | 1.03 | 0.25 | – | – | – | – | – | – |
| D | 0.40 | 0.59 | 0.49 | 0.51 | 0.70 | 0.42 | 0.63 | 0.49 |
| E | 0.49 | 0.70 | – | – | – | – | – | – |
| F | 0.35 | 0.30 | 0.58 | 1.12 | 0.32 | 0.26 | 1.13 | 1.34 |
| G | 1.99 | 0.96 | 0.33 | 0.37 | 0.86 | 0.63 | 0.84 | 0.38 |
| H | 0.60 | 0.58 | 0.52 | 0.32 | 0.90 | 0.44 | 1.07 | 0.39 |
| I | 0.37 | 0.83 | – | – | – | – | – | – |
| K | 0.07 | 0.19 | 0.49 | 0.16 | 0.42 | 0.15 | 0.78 | 0.25 |
Data from Armstrong et al. [9]
SFC superior frontal cortex, ITG inferior temporal gyrus, PHG parahippocampal gyrus, CA1/2 pyramidal cell layer sectors of the hippocampus, DG dentate gyrus, U upper cortex, L lower cortex, – tissue section not available
There were no significant correlations between the densities of NCI and disease onset, age at death, disease duration, brain weight or atrophy. However, the densities of NII in several regions were inversely correlated with disease onset in the lower laminae of the SFC (r = −0.75, P<0.05) and in the upper (r = −0.78, P<0.01) and lower layers (r = −0.83, P<0.01) of the PHG. In addition, the densities of the GI (r = 0.79, P<0.01) and DN (r = 0.77, P<0.05) were positively correlated with disease duration in the PHG and SFC, respectively (Table 3).
Discussion
The first objective was to quantify the FUS-immunoreactive pathological changes in NIFID. These data suggest that NIFID is characterized by: (1) FUS-immunoreactive NCI in frontal and temporal lobe gyri with similar densities in the upper and lower cortex, (2) relatively numerous NCI in the DG granule cells and the large pyramidal neurons of sectors CA1/2 of the hippocampus, (3) relatively infrequent FUS-immunoreactive GI, NII, and DN compared with NCI, (4) similar densities of vacuoles and surviving neurons in the upper and lower cortical layers, and (5) low densities of EN, and (6) a significant gliosis in affected areas, with a greater density of glial cells in the lower laminae.
There was evidence that in some brain regions NII may be more frequent in early-onset cases, and GI and DN in longer duration cases. The abundance of NII will be affected by the number of surviving neurons and there is some evidence that neuronal survival was maintained, at least in the early-onset cases. The greater densities of GI and DN in longer duration cases may be a reflection of the increasing neuronal loss with duration of disease. However, correlations were only present in a small number of areas and the number of cases of NIFID studied quantitatively to date is small and hence such conclusions are tentative.
The abundant and extensive FUS proteinopathy in NIFID reported here and elsewhere [30] suggests that FUS may be central to the pathogenesis of NI-FID. FUS binds to DNA, is normally at higher concentration in the nucleus [26], and has diverse cellular functions including DNA repair, homologous recombination, transcriptional regulation, and pre-mRNA processing [12]. In affected pyramidal and granular cells of the hippocampus, however, FUS immunoreactivity is not present in the nucleus but in the cytosol and somatodendritic particles at which sites it may co-localize with Sam 68, a nuclear RNA binding protein [12]. FUS may therefore contribute to steering, anchoring, or regulating mRNAs at synaptic sites within the hippocampus. Hence, the fact that in hippocampal neurons, FUS is present in the cytoplasm rather than the nucleus, may explain the high densities of FUS-immunoreactive NCI in these regions.
There were no significant differences in the densities of the vacuoles, surviving neurons or FUS-immunoreactive lesions between upper and lower cortex. In association areas of control brain, the density of the surviving neurons in laminae II/III is usually greater than in laminae V/VI [7]. Hence, the absence of a larger density peak in the upper cortex suggests a specific neuronal loss affecting laminae II/III in NIFID [3]. However, EN and glial cell nuclei occurred at greater density in the lower cortex. The density of EN was low in NIFID compared with the tauopathies corticobasal degeneration (CBD) [6] and Pick’s disease (PiD) [5] and, therefore, these lesions are unlikely to play a significant role in the pathology. Hence, the distribution of the glial cell nuclei together with the vacuoles suggests degeneration across the cortical laminae in NIFID and especially affecting the lower laminae [9].
The second objective was to compare the quantitative estimates of density as revealed by FUS IHC with those previously reported using anti-α-internexin antibodies. Consistent with the results of the semi-quantitative study of Neumann et al. [30], significantly greater densities of NCI were revealed by FUS than α-internexin IHC especially in the DG and sectors CA1/2. In addition, FUS also revealed the presence of GI, NII, and DN which were not previously observed in NIFID using α-internexin IHC [9]. In addition, Neumann et al. [30] using double labeling studies, reported that many cells had only FUS-immunoreactive inclusions while all cells with IF-immunoreactive inclusions were also labeled by FUS. Hence, despite the fact that no mutations in the FUS gene have been found in NIFID to date [30], FUS together with neuronal IF proteins is likely to be involved in the pathogenesis of NIFID. In addition, four cases were examined quantitatively using anti-ubiquitin IHC, and although these cases revealed greater numbers of ubiquitin-immunoreactive NCI than by α-internexin IHC, ubiquitin-immunoreactive NCI densities were either similar to or less than revealed by FUS in different regions suggesting that FUS aggregation may precede that of ubiquitin. Thus, FUS proteinopathy links NIFID with three other entities having FUS-immunoreactive NCI: familial cases of ALS with FUS mutation [22, 26, 34, 35], BIBD which exhibits intense FUS labeling of nuclei in unaffected areas together with FUS-immunoreactive NCI in affected regions [29], and aFTLD-U [31].
In conclusion, NIFID is an uncommon neurodegenerative disease and one of several disorders that constitute FTLD-FUS [27]. Neuropathologically, NIFID is characterized by widespread degeneration of the frontal and temporal lobes with significant densities of FUS-immunoreactive NCI in these regions, especially in the DG. FUS-immunoreactive GI, NII, and DN are present but infrequent. Vacuolation is extensive across both upper and lower cortex while significant gliosis can be observed in the lower cortex. Significantly more NCI are demonstrated with FUS than with than neuronal IF protein IHC. To distinguish NIFID from other closely related disorders will require both FUS and anti-neuronal IF IHC, the former to separate NIFID from other FTLDs and the latter to distinguish NIFID from other FTLD-FUS entities. The role FUS plays in the pathogenesis of NIFID and other FTLD-FUS entities remains to be determined.
Acknowledgments
We thank Deborah Carter and Toral Patel of the Betty Martz Laboratory for Neurodegenerative Research for expert technical assistance and we thank the families of patients whose generosity made this research possible. Support for this work was provided by grants from the National Institute on Aging of the National Institutes of Health (P50-AG05681, P01-AG03991), the Hope Center for Neurological Disorders, the Buchanan Fund, the Charles F. and Joanne Knight Alzheimer’s Disease Research Centre, the McDonnell Center for Molecular and Cellular Neurobiology, and the Barnes-Jewish Foundation.
Footnotes
Conflict of interest The authors declare that they have no conflict of interest.
Contributor Information
Richard A. Armstrong, Email: R.A.Armstrong@aston.ac.uk, Vision Sciences, Aston University, Birmingham B4 7ET, UK
Marla Gearing, Department of Pathology and Laboratory Medicine, Centre for Neurodegenerative Disease, Emory University School of Medicine, Atlanta, GA, USA.
Eileen H. Bigio, Department of Pathology, Northwestern University Medical School, Chicago, IL, USA
Felix F. Cruz-Sanchez, Institute of Neurological and Gerontological Sciences, International University of Catalonia, Barcelona, Spain
Charles Duyckaerts, Laboratoire de Neuropathologie, Hôpital de la Salpêtrière, AP-HP, 75651 Paris, France.
Ian R. A. Mackenzie, Department of Pathology and Laboratory Medicine, Vancouver General Hospital, Vancouver, BC, Canada
Robert H. Perry, Department of Neuropathology, Newcastle General Hospital, Newcastle-upon-Tyne NE4 6BE, UK
Kari Skullerud, Department of Pathology, Rikshospitalet, 0027 Oslo, Norway.
Hedeaki Yokoo, Department of Pathology, Gunma University School of Medicine, Maebashi, Japan.
Nigel J. Cairns, Charles F. and Joanne Knight Alzheimer’s Disease Research Center, Washington University School of Medicine, St Louis, MO, USA. Department of Pathology and Immunology, Washington University School of Medicine, St Louis, MO, USA. Department of Neurology, Washington University School of Medicine, St Louis, MO, USA
References
- 1.Armstrong RA. Correlations between the morphology of diffuse and primitive β-amyloid (Aβ) deposits and the frequency of associated cells in Down’s syndrome. Neuropathol Appl Neurobiol. 1996;22:527–530. doi: 10.1111/j.1365-2990.1996.tb01131.x. [DOI] [PubMed] [Google Scholar]
- 2.Armstrong RA, Cairns NJ. Topography of α-internexin-positive neuronal aggregates in 10 patients with neuronal intermediate filament inclusion disease. Eur J Neurol. 2006;13:528–532. doi: 10.1111/j.1468-1331.2006.01284.x. [DOI] [PubMed] [Google Scholar]
- 3.Armstrong RA, Cairns NJ. Laminar degeneration of the frontal and temporal cortex in neuronal intermediate filament inclusion disease (NIFID). A study using α-internexin immunohistochenistry. Clin Neuropathol. 2006;25:209–215. [PubMed] [Google Scholar]
- 4.Armstrong RA, Cairns NJ. Spatial patterns of the pathological changes in neuronal intermediate filament inclusion disease (NIFID), an α-internexin immunohistochemical study. J Neural Transm. 2007;114:451–456. doi: 10.1007/s00702-006-0595-2. [DOI] [PubMed] [Google Scholar]
- 5.Armstrong RA, Cairns NJ, Lantos PL. Quantification of pathological lesions in the frontal and temporal lobe of ten patients diagnosed with Pick’s disease. Acta Neuropathol. 1999;97:456–462. doi: 10.1007/s004010051014. [DOI] [PubMed] [Google Scholar]
- 6.Armstrong RA, Cairns NJ, Lantos PL. A quantitative study of the pathological lesions in the neocortex and hippocampus of 12 patients with corticobasal degeneration. Exp Neurol. 2000;163:348–356. doi: 10.1006/exnr.2000.7392. [DOI] [PubMed] [Google Scholar]
- 7.Armstrong RA, Lantos PL, Cairns NJ. Laminar distribution of ballooned neurons and tau positive neurons with inclusions in patients with corticobasal degeneration. Neurosci Res Commun. 2000;27:85–93. [Google Scholar]
- 8.Armstrong RA, Lantos PL, Cairns NJ. Spatial correlations between the vacuolation, prion protein deposits, and surviving neurons in the cerebral cortex in sporadic Creutzfeldt–Jakob disease. Neuropathology. 2001;21:266–271. doi: 10.1046/j.1440-1789.2001.00406.x. [DOI] [PubMed] [Google Scholar]
- 9.Armstrong RA, Kerty E, Skullerud K, Cairns NJ. Neuropathological changes in ten cases of neuronal intermediate filament inclusion disease (NIFID): a study using α-internexin immunohistochemistry and principal components analysis (PCA) J Neural Transm. 2006;113:1207–1215. doi: 10.1007/s00702-005-0387-0. [DOI] [PubMed] [Google Scholar]
- 10.Armstrong RA, Ironside J, Lantos PL, Cairns NJ. A quantitative study of the pathological changes in the cerebellum of 15 cases of variant Creutzfeldt–Jakob disease. Neuropathol Appl Neurobiol. 2009;35:36–45. doi: 10.1111/j.1365-2990.2008.00979.x. [DOI] [PubMed] [Google Scholar]
- 11.Armstrong RA, Ellis W, Hamilton RL, et al. Neuropathological heterogeneity in frontotemporal lobar degeneration with TDP-43 proteinopathy: a quantitative study of 94 cases using principal components analysis. J Neural Transm. 2010;117:227–239. doi: 10.1007/s00702-009-0350-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Belly A, Moreau-Gachelin F, Sadoue R, Goldberg Y. Delocalization of the multifunctional RNA splicing factor TLS/FUS in hippocampal neurons: exclusion from the nucleus and accumulation in dendritic granules and spine heads. Neurosci Lett. 2005;379:152–157. doi: 10.1016/j.neulet.2004.12.071. [DOI] [PubMed] [Google Scholar]
- 13.Bigio EH, Lipton AM, White CL, Dickson DW, Hirano A. Frontotemporal dementia and motor neurone degeneration with neurofilament inclusion bodies: additional evidence for overlap between FTD and ALS. Neuropathol Appl Neurobiol. 2003;29:239–253. doi: 10.1046/j.1365-2990.2003.00466.x. [DOI] [PubMed] [Google Scholar]
- 14.Broe M, Hodges JR, Schofield E, Shepherd CE, Kril JJ, Halliday GM. Staging disease severity in pathologically confirmed cases of frontotemporal dementia. Neurology. 2003;60:1005–1011. doi: 10.1212/01.wnl.0000052685.09194.39. [DOI] [PubMed] [Google Scholar]
- 15.Cairns NJ, Armstrong RA. Clustering of neuronal inclusions in “dementia with neurofilament inclusions”. Acta Neuropathol. 2003;106:125–128. doi: 10.1007/s00401-003-0710-5. [DOI] [PubMed] [Google Scholar]
- 16.Cairns NJ, Perry RH, Jaros E, et al. Patients with a novel neurofilamentopathy: dementia with neurofilament inclusions. Neurosci Lett. 2003;341:177–180. doi: 10.1016/s0304-3940(03)00100-9. [DOI] [PubMed] [Google Scholar]
- 17.Cairns NJ, Zhukareva V, Uryu K, et al. α-Internexin is present in the pathological inclusions of neuronal intermediate filament inclusion disease. Am J Pathol. 2004;164:2153–2161. doi: 10.1016/s0002-9440(10)63773-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cairns NJ, Jaros E, Perry RH, Armstrong RA. Temporal lobe pathology of human patients with neurofilament inclusion disease. Neurosci Lett. 2004;354:245–247. doi: 10.1016/j.neulet.2003.10.020. [DOI] [PubMed] [Google Scholar]
- 19.Cairns NJ, Grossman M, Arnold SE, et al. Clinical and neuropathologic variation in neuronal intermediate filament inclusion disease (NIFID) Neurology. 2004;63:1376–1384. doi: 10.1212/01.wnl.0000139809.16817.dd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ching GY, Liein RKH. Roles of head and tail domains in alpha-internexin’s self-assembly and coassembly with the neurofilament triplet proteins. J Cell Sci. 1998;111:321–333. doi: 10.1242/jcs.111.3.321. [DOI] [PubMed] [Google Scholar]
- 21.Ching GY, Chien CL, Flores R, Liein RKH. Overexpression of alpha-internexin causes abnormal neurofilamentous accumulations and motor coordination deficits in transgenic mice. J Neurosci. 1999;19:2974–2986. doi: 10.1523/JNEUROSCI.19-08-02974.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chio A, Restagno G, Brunetti M, et al. Two Italian kindreds with familial amyotrophic lateral sclerosis due to FUS mutation. Neurobiol Aging. 2009;30:1272–1275. doi: 10.1016/j.neurobiolaging.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ikeda K, Akiyama H, Kondo H, et al. Thorn-shaped astrocytes: possibly secondarily induced tau-positive glial fibrillary tangles. Acta Neuropathol. 1995;90:620–625. doi: 10.1007/BF00318575. [DOI] [PubMed] [Google Scholar]
- 24.Josephs KA, Holton JL, Rossor MN, et al. Neurofilament inclusion body disease: a new proteinopathy? Brain. 2003;126:2291–2303. doi: 10.1093/brain/awg231. [DOI] [PubMed] [Google Scholar]
- 25.Komori T. Tau positive glial inclusions in progressive supranuclear palsy, corticobasal degeneration and Pick’s disease. Brain Pathol. 1999;9:663–679. doi: 10.1111/j.1750-3639.1999.tb00549.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kwiatkowski TJ, Bosco DA, LeClerc AL, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- 27.Mackenzie IR, Neumann M, Bigio EH, et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol. 2010;119:1–4. doi: 10.1007/s00401-009-0612-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matsumoto S, Udaka F, Kameyama M, Kusaka H, Itoh H, Imai T. Subcortical neurofibrillary tangles, neuropil threads and argentophilic glial inclusions in corticobasal degeneration. Clin Neuropath. 1996;15:209–214. [PubMed] [Google Scholar]
- 29.Munoz DG, Neumann M, Kusaka H, Yokata O, Ishihara K, Terada S, Kuroda S, Mackenzie IR. FUS pathology in basophilic inclusion body disease. Acta Neuropathol. 2009;118:617–627. doi: 10.1007/s00401-009-0598-9. [DOI] [PubMed] [Google Scholar]
- 30.Neumann M, Roeher S, Kretzschmar HA, Rademakers R, Baker M, Mackenzie IRA. Abundant FUS-immunoreactive pathology in neuronal intermediate filament inclusion disease (NIFID) Acta Neuropathol. 2009;118:605–616. doi: 10.1007/s00401-009-0581-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Neumann M, Eademakers R, Roeher S, Baker M, Kretzschmar HA, Mackenzie IRA. A new type of frontotemporal lobar degeneration with FUS pathology. Brain. 2009;132:2922–2931. doi: 10.1093/brain/awp214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pirici D, Vandenberghe R, Rademakers R, et al. Characterization of ubiquitinated intraneuronal inclusions in a novel Belgian frontotemporal lobar degeneration family. J Neuropathol Exp Neurol. 2006;65:289–301. doi: 10.1097/01.jnen.0000205147.39210.c7. [DOI] [PubMed] [Google Scholar]
- 33.Probst A, Tolnay M. Argyrophilic grain disease, a frequent and largely underestimated cause of dementia in old patients. Rev Neurol. 2002;158:155–165. [PubMed] [Google Scholar]
- 34.Valdmanis PN, Daoud H, Dion PA, Rouleau GA. Recent advances in the genetics of amyotrophic lateral sclerosis. Curr Neurol Neurosci Rep. 2009;9:198–205. doi: 10.1007/s11910-009-0030-9. [DOI] [PubMed] [Google Scholar]
- 35.Vance C, Rogelj B, Hortobagyi T, et al. Mutations in FUS an RNA processing protein cause familial amyotrophic lateral sclerosis Type 6. Science. 2009;323:1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yamada T, McGeer PL, McGeer EG. Appearance of paired nucleated tau-positive glia in patients with progressive supranuclear palsy brain tissue. Neurosci Lett. 1992;135:99–102. doi: 10.1016/0304-3940(92)90145-w. [DOI] [PubMed] [Google Scholar]
- 37.Yokota O, Tsuchiya K, Terada S, et al. Basophilic inclusion body disease and neuronal intermediate filament inclusion disease: a comparative clinicopathological study. Acta Neuropathol. 2008;115:561–575. doi: 10.1007/s00401-007-0329-z. [DOI] [PubMed] [Google Scholar]