

Abstract
Progress in experimental and theoretical biology is likely to provide us with the opportunity to assemble detailed predictive models of mammalian cells. Using a functional format to describe the organization of mammalian cells, we describe current approaches for developing qualitative and quantitative models using data from a variety of experimental sources. Recent developments and applications of graph theory to biological networks are reviewed. The use of these qualitative models to identify the topology of regulatory motifs and functional modules is discussed. Cellular homeostasis and plasticity are interpreted within the framework of balance between regulatory motifs and interactions between modules. From this analysis we identify the need for detailed quantitative models on the basis of the representation of the chemistry underlying the cellular process. The use of deterministic, stochastic, and hybrid models to represent cellular processes is reviewed, and an initial integrated approach for the development of large-scale predictive models of a mammalian cell is presented.
Keywords: cell signaling, protein-protein interactions, network modeling, systems biology
INTRODUCTION
Over the past four decades, the progress in biochemistry and molecular biology in assembling lists of cellular components and their interactions, and in cell biology and physiology in understanding the functions of various components of the cell, have allowed us to start thinking about how these components and interactions come together to form the basic unit of life, a cell. Understanding how a functioning cell is configured from its parts cannot be achieved by experiments alone, but also requires substantial theoretical analyses and tight integration between the two. This integration should lead to the development of predictive models of a cell, so that by specifying the initial conditions, we can predict responses to a variety of external and internal stimuli. The development of predictive models poses considerable challenges at both theoretical and experimental levels. Nevertheless, in the past five years there has been steady progress on various fronts. The sequencing and characterization of many genomes (2, 15, 64, 76, 82, 111, 118), the development of technologies that allow for large-scale profiling of cellular components (20, 37, 53, 94) and their interactions (49, 97, 108), the development of a variety of modeling approaches using both chemical kinetics (75, 110, 101, 112) and graph theory (4, 9, 79, 80), and the reduction in cost of large-scale computation have allowed us to begin developing integrated approaches for understanding the principles underlying the organization and function of a cell. Here we describe current approaches and discuss how they may be integrated to obtain predictive models of mammalian cells.
WHAT IS A MAMMALIAN CELL?
The cell as the basic unit of life has been long accepted as a central tenet of biology. The nature of the cell in different species has a core set of common characteristics, but there are also considerable differences. Bacterial cells, and unicellular eukaryotes such as yeast, share similarities with cells from higher organisms in a variety of functions and have served as useful model systems. Much of the early understanding of the cell cycle and its regulation has come from genetic manipulation of yeast (46), and many findings in yeast have been confirmed universally. Similarly, metabolism and chemotaxis in bacteria have served as useful model systems to understand these functions in cells of higher organisms. Nevertheless, mammalian cells have distinct attributes and functions that need to be dealt with directly. Most mammalian cells belong to tissues and organs and thus have a substantial level of internal spatial complexity. Intracellular components are distributed anisotropically, and cellular activity results in a continuous redistribution of intracellular components. This spatial feature is compounded by the increased complexity of the genome, in which the regulatory proteins have more isoforms in higher organisms. For many proteins that are a part of cellular signaling pathways, the human genome has more isoforms than the genome of the fly or yeast (111).
Isoforms raise several issues with regard to modeling. Isoforms are often distributed in different locations within the cell and have partially overlapping connectivity and hence often need to be dealt with as individual entities. For these reasons we think it is useful to focus directly on mammalian cells, as suggested by recent models of mammalian circadian rhythms (34). The physiology of mammalian cells, tissues, and organs are well studied and provide a wealth of information that can be used to constrain, train, and refine small- and medium-scale models and thus greatly facilitate the development of large-scale models. None of these reasons should be viewed as deterrents to studies on model organisms. Rather, studies on mammalian systems and model organisms are most often complementary and mutually beneficial.
The concept of a typical mammalian cell is a myth. In an adult organism, a neuron is quite different from a hematopoetic cell such as a T- or B-cell, which in turn is different from a pancreatic β-cell that secretes insulin. Such reasoning can be extended to other cell types such as muscle cells, fibroblasts, and epithelial cells. How then can we develop a general approach that would be applicable to building models of all these cell types? We have proposed that all cells have a central signaling network that can sense signals from both internal and external stimuli and process them through a network of interacting signaling pathways to regulate multiple cellular machines (57). Each cell type has its own specific receptors that define the types of input signals the cell can sense. Thus, neurons have neurotransmitter receptors that are both ionotropic and metabotropic. The ionotropic receptors are ion channels, and their activity is responsible for rapid synaptic signaling. The metabotropic receptors couple to G proteins and activate intracellular regulatory pathways. The T cell receptors interact with antigen-presenting cells to recognize specific antigens and then activate an intracellular signaling network to produce and secrete cytokines. The pancreatic β-cells have receptors to sense glucose and then, through the intracellular signaling network, regulate the secretion of insulin.
In each case, the extracellular signals are processed through a central regulatory network of interacting signaling pathways to regulate various cellular machines, i.e., the electrical response system, the secretion apparatus, transcriptional and translational machinery, and the motility machinery. Thus, all cells can be thought of as having a common functional configuration: a central signaling network that regulates a cell type–specific combination of cellular machines to evoke the observed phenotypic behaviors. This is similar to considering the different cells as chemical plants with a central regulatory system that controls a number of interconnected reactors to allow for the generation of specified products. This rationale is depicted in Figure 1.
Figure 1.
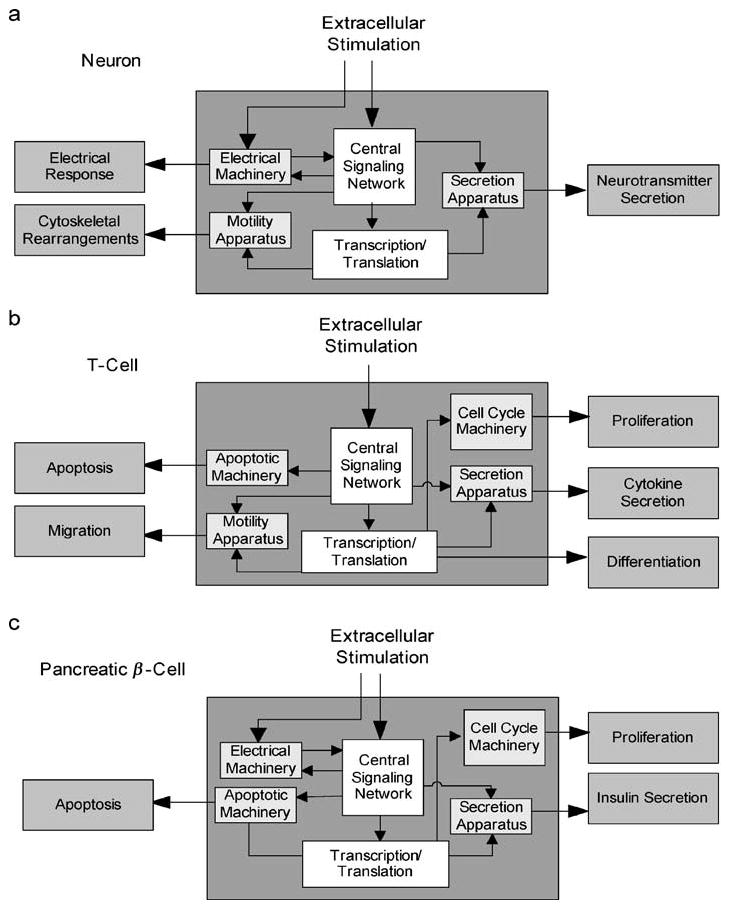
A common regulatory architecture for different mammalian cell types. Three mammalian cells, (a) neuron, (b) T-cell, and (c) pancreatic β-cell, are stimulated by different ligands and exhibit different phenotypic behavior, although the overall organization and mechanisms used for discriminating, processing, and responding to signals are conserved. The phenotypic responses involve differing sets of cellular machines for different cell types.
OBTAINING AN OVERVIEW
The sequencing of various genomes allowed us to begin developing estimates of the numbers of components in a cell. If the mammalian genome is thought to have approximately 30,000 genes, and if we assume that each gene may have on average at least two alternatively spliced transcripts, then there would be approximately 60,000 transcripts in total. If about half of these are expressed in any given cell type, then we would expect about 30,000 different proteins and mRNA species in one cell. If one assumes a similar number of other components such as lipids, sugars, ions, and nucleotides, an upper estimate of the different types of components that comprise a mammalian cell would be about 100,000. Each component interacts on average with 4 other components leading to a system of about 200,000 interactions. How can we understand such a system? Studying one component or an organized group of components such as a signaling pathway is unlikely to provide insight into the logic underlying the overall organization of a cell. Here an initial top-down view could be useful. Such a view could provide insight into how individual components are organized to form higher order units and what types of behaviors these units may be capable of accomplishing. The assembly of such units to form an integrated functional system could be the highest level of organization. To assemble such a system, the currently known components and their interactions (discovered experimentally through biochemical, genetic, or cell physiological experiments) can be combined to form an in silico network. Such a network can be visualized and computationally analyzed by a variety of mathematical approaches. The understanding that biological systems can be represented as networks has led to a surge of interest in applying graph theory approaches to analyze the topology of these networks. Before such analysis can be performed, the networks need to be assembled. This can be done experimentally by large-scale data gathering or by in silico assembly of interactions based on binary relationships between components and known pathways.
Assembling Networks
Several approaches have been used for building cellular signaling and regulatory networks in a format that can be computationally analyzed. The most common approach uses a top-down format, in which a large set of interactions are simultaneously determined on the basis of genomic specifications. At the mRNA level this approach uses data obtained from mRNA profiling experiments using microarrays (36, 58, 104). At the protein level, interactions are determined by large-scale, yeast two-hybrid screens (40, 51, 59, 68, 120) or by copurification experiments coupled to mass spectrometry (49). Such data yield large-scale networks, but the technologies used, although promising, currently provide limited information. These techniques do not specify key biological properties, including the directionality of the connections and the relationship between components, such as activation or inhibition. Thus, these datasets yield “undirected graphs” that provide limited representations of the system.
Another approach is the construction of networks based on the biochemistry, cell biology, and cell physiology literature. From the experimental studies of individual binary interactions and small groups of interactions, we can assemble larger networks. This can be done in several ways. The use of natural language processing (NLP), a subfield of artificial intelligence, has led to text-mining programs that search the published literature to extract data from published papers (35, 71, 84). Whether such efforts will yield error-free, large-scale maps is not known. However, such approaches are surely needed if we are to capitalize on the currently available, vast functional data that define components and their interactions in different mammalian cell types. The computer-based automatic search efforts are complemented by bottom-up approaches, in which interactions are identified from data manually extracted from primary publications (39, 88). Science’s STKE (42) and the Alliance for Cell Signaling (67) use this approach, which has been most fruitful to date. STKE has published more than 50 pathways for a variety of cell types and organisms. The networks in STKE are relatively small with 10 to 50 components. The bottom-up approach contains a lower incidence of false positives (113) but a higher incidence of false negatives. False positives are interactions that are not relevant in the biological context but exist within the datasets. False negatives are interactions that are relevant and exist in the biological systems but are unknown or otherwise not included in the datasets. Currently, there are no large-scale (1000 or more components) mammalian cellular signaling regulatory network maps ready for computational analysis. The construction of these datasets is still in its infancy, in contrast with metabolic networks (60). However, several databases do exist (Table 1).
TABLE 1.
List of cellular components interactions databases available on the internet
| Web-based dataset sources mining intracellular molecular interactions | |||
|---|---|---|---|
| Abbreviation | Name/description | URL | PMID |
| AfCS | Alliance for Cell Signaling | http://www.signaling-gateway.org/ | 12478304 |
| aMAZE | WorkBench for information on cellular processes | http://www.amaze.ulb.ac.be/introduction/ | 14681453 |
| BIND | The Biomolecular Interaction Network Database | http://bind.ca/ | 15608229 |
| BioCarta | Charting Pathways of Life | http://www.biocarta.com/genes/newPathways.asp | N/A |
| BioCyc | Knowledge Library of Pathway/Genome Databases | http://biocyc.org/ | 14751985 |
| BRITE | Database storing binary relations of genes and proteins | http://www.genome.jp/brite/brite.html | 11752249 |
| DIP | Database of Interacting Proteins | http://dip.doe-mbi.ucla.edu/ | 14681454 |
| DOQCS | Database of Quantitative Cellular Signaling | http://doqcs.ncbs.res.in/ | 12584128 |
| Drastic | Signal Transduction in Plant cells | http://www.drastic.org.uk/ | N/A |
| GeneNet | Gene network database | http://wwwmgs.bionet.nsc.ru/mgs/gnw/genenet/ | 15608230 |
| GenMAPP | Gene Microarray Data Profiler | http://www.genmapp.org/ | 11984561 |
| GRID | Genetic and physical interactions in fly and yeast | http://biodata.mshri.on.ca/grid/servlet/Index | 12620108 |
| HPRD | Human Protein Reference Database | http://www.hprd.org/ | 14681466 |
| IntAct | Open source protein interaction data | http://www.ebi.ac.uk/intact/index.jsp | 14681455 |
| KEGG | KEGG Pathway Database | http://www.genome.jp/kegg/regulation.html | 11752249 |
| KinaseDB | Kinase Pathway Database | http://kinasedb.ontology.ims.u-tokyo.ac.jp:8081/ | 12799355 |
| MICPS/MIPS | Munich Information Center for Protein Sequence | http://mips.gsf.de/proj/yeast/tables/interaction/ | 15531608 |
| MINT | Molecular interaction relational database | http://mint.bio.uniroma2.it/mint/index.php | 1191183 |
| PKR | Protein Kinase Resource | http://pkr.sdsc.edu/html/index.shtml | N/A |
| PPID | Protein-Protein Interactions Database | http://defiant.inf.ed.ac.uk:8000/ | 15114355 |
| RegulonDB | Transcriptional Regulation and Operon Organization | http://www.cifn.unam.mx/ComputationalGenomics/regulondb/ | 11125053 |
| SENTRA | Database of Sensory signal transduction proteins | http://www-wit.mcs.anl.gov/sentra/ | 11752334 |
| SigPath | Database annotating quantitative interactions data | http://icb.med.cornell.edu/crt/SigPath/index.xml | 15340175 |
| SPAD | Signaling Pathway Database | http://www.grt.kyushu-u.ac.jp/spad/ | N/A |
| STKE | Signal Transduction Knowledge Environment | http://stke.sciencemag.org/cm/ | 12438188 |
Cellular interactions may be represented graphically as networks. On the basis of their level of abstraction, four types of graphs may be drawn: undirected (Type I), directed (Type II), directed and weighted (Type III), and directed/weighted with spatial specifications (Type IV). For any set of components (Figure 2a, see color insert), most protein-protein interaction networks publicly available today are undirected graphs and describe only binding interactions (40, 51) (Figure 2b). To achieve biologically relevant insights from simulations of these networks, the representation needs to include the direction of the interactions. In Type II graphs the directionality of information flow, as well as the activating or inhibitory effect of a source component on its target component, is specified (Figure 2c). These are called directed graphs. For further accuracy, the different states of each component can be specified. Consequently, the connections can be state specific and weighted to indicate the strength of the connections (Figure 2d). If spatial information is available, the different components can be assigned to different compartments, thus exhibiting spatially restricted connectivity (Figure 2e). Such representations will yield an ensemble of networks for the same set of components.
Figure 2.
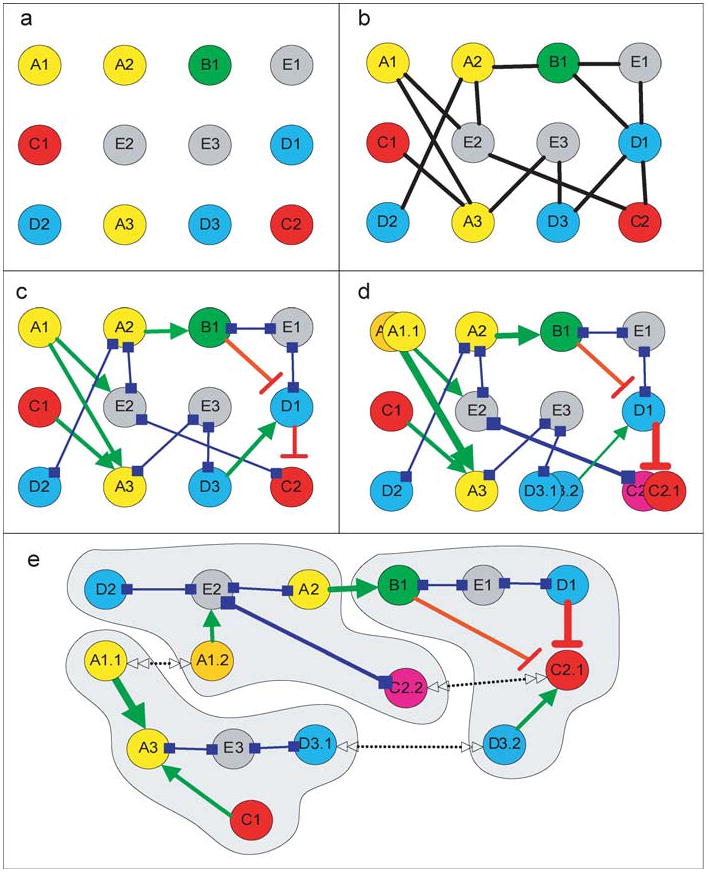
Qualitative representation of networks. (a) Components are identified by cloning of genes and/or purification of proteins and assigned function. Components with different functions are color coded and labeled alphabetically. Numbers identify individual components in a class. (b) Type I representation: The network is represented as an undirected graph. (c) Type II representation: The network is represented as a directed graph. The nature of the binary interactions is specified: activation in green, inhibition in red, and neutral in blue. Neutral interactions often indicate binding to scaffold and anchor proteins. (d) Type III representation: Both states of the nodes and weights of the connections are specified. The different states of the components are labeled by a second digit. Specifying the state of the nodes also leads to differential connectivity, and connectivity of differing strengths is specified by line thickness. (e) Type IV representation: Spatial information is included. The same components are in different compartments, where they have different connectivity. Double-sided arrows indicate the ability of the components to move between compartments.
Network Analysis by Graph Theory
Understanding the topology of the network is useful for determining the range of behaviors that can be produced. For this, the network can be probed by statistical approaches. Such analyses have been useful in understanding computer networks and social networks. Several properties of the networks can be identified, such as characteristic path length, clustering, and presence and organization of regulatory motifs. These properties are discussed in detail below. From these properties an overview of the capabilities of a complex system can be obtained.
These analyses generally assume the network connectivity to be static (66), i.e., the connectivity within the network is maintained at all times. Although this is true for some networks such as computer networks, such constant connectivity is not true in cellular regulatory networks. Here connections are made and broken depending on the state of the components; hence, the system is quite dynamic. The dynamics of regulatory networks may be simulated by Boolean models (63). A recent simulation of the gene regulatory network of segment polarity genes in Drosophila using a Boolean model (5) provides a reasonable description of the functioning of the network comparable to that obtained when the network was described by a system of differential equations (112). Another approach to understanding the dynamics of the regulatory topology is through artificial network models. Artificial networks can be created by applying network growth algorithms. These algorithms generate networks with properties similar to real biological networks (9, 62, 85, 92). This approach provides initial insight into the evolutionary processes that generated the biological networks. Several graph theory–related approaches applied to characterize biological cellular networks are described below.
Clustering Coefficient: Measuring Local Connectivity
Watts & Strogatz (115) defined two important network properties: clustering coefficient and characteristic path length. Clustering coefficient, a local property, measures the density of connectivity within the network. A high clustering coefficient of a network, compared with random networks with similar connectivity distribution, may be indicative of modularity within the network. As shown in Figure 3a, the clustering coefficient captures the abundance of triangles in the network. The nodes in this motif are often localized in physical proximity and thus clustering coefficient becomes a measure of colocalized components that interact with one another. Caldarelli et al. (24) have pointed out that clustering coefficient does not capture the abundance of rectangles, another elemental measure of interacting nodes (Figure 3b). They formulated a method to measure local interactions by considering rectangles in the calculation to obtain a “grid-coefficient.” Both clustering and grid coefficients are useful in defining modularity in cellular networks owing to the abundance of scaffolds and anchoring proteins in mammalian cells (87). Such proteins often bring together groups of cellular components that interact with one another in a spatially restricted manner to yield functional responses.
Figure 3.

Schematics of clustering and grid coefficients. (a) The central node has four neighbors. The neighbors are connected with two connections of possible six. The clustering coefficient for the central node is 2/6 = 0.33. The two triangles used in the calculation are highlighted. Computing the average clustering coefficient of all nodes in a network measures the network’s local connectivity and can define the existence of modularity within networks when compared with random networks (115). (b) The grid coefficient considers the rectangles as well as the triangles when calculating the level of clustered interactions (22). The two rectangles and two triangles used to compute the grid coefficient for the black node are highlighted.
Characteristic Path Length and Small-World Networks
Characteristic path length, also called the degree of separation, is a measure of how sparsely the nodes are connected within the network. This is a global property. The shortest path between any two pairs of nodes is calculated for all possible pairs of nodes. The concept originated in the field of human psychology. Stanley Milgram (77) found that any person in the United States was separated from any other person by only six links. This has recently been verified by using chains of emails (31). The characteristic path length indicates the average of the minimum number of steps required to reach any other component within the network when one starts at one component. The clustering coefficient and the characteristic path length often have a predictable relationship for real networks (115). A relatively high clustering and a similar path length compared with randomized or shuffled networks are used to characterize real networks as small world. Such a characterization implies the presence of many shortcuts for connectivity and information propagation within the network (115).
Scale-Free Networks
Barabasi and coworkers have found that real-world networks follow a connectivity distribution that follows a power law (56, 92). For such systems, when counts of nodes are plotted against their connectivity (also called degree) on a log-log plot, a straight line with a slope between -2 and -3 is obtained. They termed this topology scale free. Such a designation indicates a predictable relationship between the number of nodes and a certain number of links; it follows a defined power-law function irrespective of the size of the network. Barabasi and coworkers analyzed 43 metabolic networks inferred from genomic sequences for a number of organisms (56) and showed that they all have similar clustering coefficients, regardless of the network size, and that these networks are scale free. The metabolic networks are organized as modules within modules to form a hierarchical network (92). By building modular hierarchical artificial networks, the authors showed that these networks display the same properties as the metabolic networks that have both high clustering coefficients and highly connected hubs (92).
If biological networks are both small world and scale free, these networks may share some basic properties such as enhanced information propagation speed owing to the presence of shortcut links, synchronization ability, robustness or error tolerance (4), and computational power (6, 55, 106). Implicit in the definition of scale-free networks is the assumption that these networks grow through the “rich nodes get richer” hypothesis (9), which states that highly connected nodes are more likely to increase their connectivity than less connected nodes as the size of the network increases. A study that examined the connectivity of proteins through evolution by comparing connectivity of the same proteins in Escherichia coli and yeast found that “oldest” proteins in both E. coli and yeast had on average 6.2 links per protein, whereas “newest” proteins found only in yeast had 0.5 links per protein. These observations have been interpreted to support the “rich nodes get richer” hypothesis (33). Further analyses of more biological networks are needed to determine if this hypothesis is generally valid.
Scale-free networks commonly have a cutoff property as defined by Stanley and coworkers (6). In such networks, a scale-free connectivity distribution might be followed by a tail that has a Gaussian or exponential pattern, thereby limiting the number of highly connected nodes. This cutoff is thought to occur for two reasons: aging and limited physical growth capacity of nodes for additional links. Old nodes may stop acquiring new links after they reach a certain evolutionary age, indicating that each node has a time limit for active growth, during which it can acquire additional links. Alternatively, the physical capacity for connectivity for individual nodes can be limited, implying that once a node reaches its maximal possible physical connectivity capacity it stops growing. Understanding how the networks are configured with respect to their scalability should be useful in determining their regulatory capabilities.
Identifying Regulatory Motifs: Information Processing Within Networks
A different approach in characterizing networks is the identification of motifs. Motifs are sets of interactions involving 2 to 10 nodes. These organized sets of interactions are capable of higher order functions such as switching, information storage, and amplification, and hence represent the functional capabilities within the network. The configurations of links between sets of components constitute different types of network motifs. Milo and coworkers (79, 80, 99) defined combinations for interconnectivity of three- and four-component motifs and searched for their prevalence in biological-directed graphs such as worm and yeast gene regulatory networks. They identified a signature pattern of motifs that may characterize these networks. The motifs can be useful indicators of signal integration, sorting, and information processing within cellular networks. For instance, the prevalence of three-component triangular motifs and the clustering coefficient together indicate local modularity and information-processing capability within the modules.
The search for network motifs may also be used to predict unknown interactions or validate interactions. Albert & Albert (3) used a combination of motif search algorithms with the SUGGEST machine learning algorithm (121) to predict and provide an additional line of support for interactions in the yeast protein-protein interactions dataset (71). Algorithms such as SUGGEST are typically used to predict the products a customer is likely to buy on the basis of previous purchases and grouping of products. Motif classification can also be used to predict function. Bu et al. (23) used the yeast protein-protein interactions data and graph theory to predict the functions of proteins on the basis of motif characteristics and interactions with proteins of known function. The general validity and overall usefulness of these approaches remain to be determined.
Motif searches are potentially valuable because they provide initial insight into the regulatory capabilities of the networks. Although there is some debate about the validity of the null networks used as controls to identify the evolutionary-favored motifs (8, 78), the ability to define various types of motifs can be useful. For instance, both feedback and feed-forward motifs have substantial information processing capability. A positive feedback loop in the growth factor receptor-MAPK pathway is shown in Figure 4a. This motif can function as a bistable switch (12). A positive feed-forward motif from cAMP to its transcriptional regulator CREB is shown in Figure 4b. Both types of motif allow for persistence of signals within the intracellular network after the extracellular signal has dissipated. This is important for network function because both the amplitude and duration of activation of key intracellular components determine if biochemical signaling events are converted to physiological responses.
Figure 4.
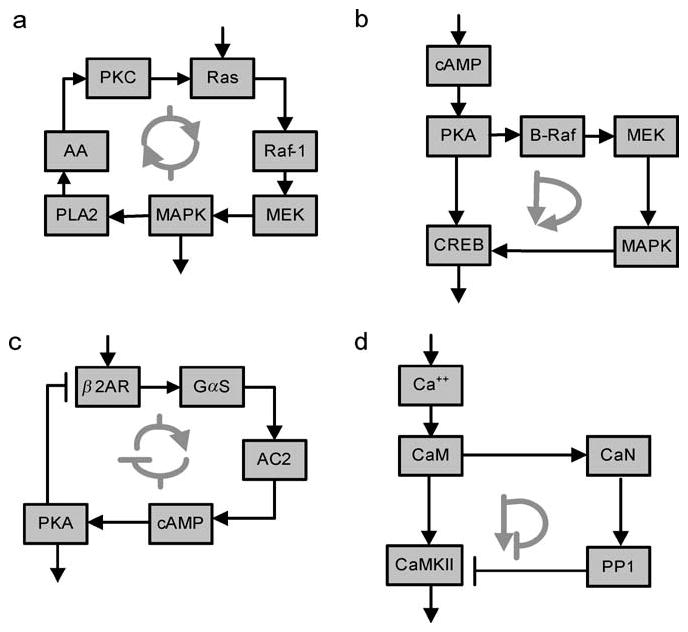
Regulatory motifs in signaling networks. (a) Positive feedback loop that functions as a bistable switch. (b) Positive feed-forward loop. (c) Negative feedback loop. (d) Negative feed-forward loop. See Reference 11 for details. Abbreviations: PKC, protein kinase C; MAPK, mitogen-activated protein kinase; cPLA2, phospholipase A2; MEK, mitogen-activated protein kinase kinase; AA, arachidonic acid; cAMP, cyclic 3,5 adenosine monophosphate; PKA, protein kinase A; CREB, cAMP response element binding protein; β2AR, β2-adrenergic receptor; Gαs, stimulatory G protein; AC2, adenylyl cyclase 2; CaM, calmodulin; CaN, calcenurin; PP1, protein phosphatase-1; CaMKII, Ca2+/calmodulin stimulated kinase II.
Negative feedback loop and negative feed-forward loop motifs also exist. Negative feedback loops are commonly used to desensitize signaling pathways to limit the deleterious effects of repeated signals within short time periods. A prototypic negative feedback loop in the β-adrenergic receptor pathway is shown in Figure 4c. Negative feed-forward loop motifs are also observed in signaling networks. A feed-forward loop from calmodulin that participates in the induction of long-term potentiation (LTP) in hippocampal neurons is shown in Figure 4d. We had referred to the negative feed-forward loop motif as a gate (17, 18).
ORGANIZING PRINCIPLES FOR FUNCTION
Statistical analyses of networks provide a bird’s eye view of the regulatory capabilities of the system. An understanding of the various motifs that are present and how they are juxtaposed with respect to one another describes the overall regulatory topology of the cellular network. Such representation may be used to describe the cell as a whole and its regulatory and functional capabilities.
Although initially we focused only on signal transduction pathways as connections maps or networks, all cellular entities, including cellular machines such as the actin-myosin-based motility machinery or the transcription and translation machineries, can be represented as networks of interacting components. Because the signaling pathways regulate the components of cellular machines to affect overall function, all the components of the cell can be considered a part of a large network of interacting components. Such a hypothesis raises important issues. If all the components are connected, does the activity of any one component affect all other components? How are the well-defined biological effects that are regulated by a single signaling pathway obtained? The answers to questions such as these may lie in understanding the intrinsic modularity that exists within cellular networks.
Modularity
Hartwell et al. (45) had suggested that cell biology is moving from molecular to modular biology. Modularity has been a recurring theme of regulatory biology. Modules are often made of the regulatory motifs and thus have information-processing capability. Broadly, a module can be defined as a group of components that interact in an ordered fashion to evoke an effect in response to a stimulus. Growth factor stimulation of MAPK to trigger the transcription of immediate early genes such as fos can be considered a functional module. But such definitions are both conditional and contextual. Soon after the receptor tyrosine kinase–Ras–MAPK pathway had been recognized as a distinct module involved in proliferation and growth (32), it was discovered that this pathway is regulated by another well-known pathway, the cAMP pathway (29, 119), resulting in regulation of Ras-induced oncogenic transformation (26). This and other interactions involving the cAMP pathway led to the notion of gates, whereby one pathway regulates information flow through another pathway (18, 52). Although such regulation implicitly described a modular arrangement, it was only until later, when we explicitly modeled a signaling network (12), that the functional basis for modularity became apparent. We were able to describe a small synaptic signaling network in neurons as a set of interconnected modules that we termed timer switch, coincidence-detector, gate, and output response units (13). These modules contain regulatory motifs such as positive feedback and negative feed-forward loops. Such organizational and functional descriptions serve as one of the criteria for the definitions of modules. There are other criteria for identifying modules such as spatial separation within the cell. Often spatial separation is achieved by localization of a component within an organelle. One well-studied example is the mitochondria. This organelle contains the enzymes for ATP synthesis and key components for triggering cell death or apoptosis. Similarly, the role of the plasma membrane in defining the electrical properties of the cell is well understood. Spatial and functional criteria for modularity are not mutually exclusive and often each contributes to certain features of the overall modularity. Signaling pathway modules can be defined in both spatial and functional terms. The initial signal is received at the cell membrane and transduced to a downstream component that is either a small molecule or a protein kinase. The signal then travels to the appropriate cellular organelle to obtain functional effects. IP3 stimulation of Ca2+ release from the endoplasmic reticulum internal stores and MAPK1,2 stimulation of early gene expression in the nucleus have the same design logic. Here, integration of function occurs across organelles, so if each pathway is thought of as a module, the definition is functional. The boundaries between functional modules are also not fixed. They can vary depending on the regulatory interactions and on the basis of the functional outcome. How the boundaries between modules are defined is crucial for our understanding of the dynamics of various modules, as well as the system as a whole. Functional modules have also been recognized in metabolic networks in which their boundaries are better defined by the identity of the metabolite involved. Mathematical approaches (22) have been proposed to reduce the complexity in defining interactions between functional modules.
Balance Between Modules: Plasticity and Homeostasis
If the concept that cellular networks consist of an ensemble of interconnected modules is accepted, then the ability of the cell to function as an integrated unit may arise from the balance of activities between the modules. This may be a core design principle for mammalian cells. Such balance between modules allows the cell to exhibit the seemingly opposing capabilities of homeostasis and plasticity while exhibiting overall robustness. To understand the basis for such balance between modules and the definitions of the contours defining a module, it is necessary to develop quantitative descriptions of interactions in terms of the concentrations of the components, and the kinetic rates or the probabilities of their interactions. Before we delve into quantitative chemistry-based approaches, it is useful to analyze the reasons why understanding the balance between modules (and implicitly motifs) requires quantitative reasoning.
When a cell receives few external stimuli, many of the nodes in the network are unconnected and most of the modules are isolated, exhibiting only local connectivity. In contrast, when there is a multitude of external stimuli, such that most of the connections are operational, the network is highly connected to yield a small-world topology. A series of intermediate states are possible depending on the number, intensity, and duration of the extracellular signals. In response to extracellular stimulation of sufficient intensity and duration, connections are made or eliminated in a transient manner. This dynamic nature of the components and their interactions would result in a series of configurations engaging several modules. Such dynamic behavior is prominent in neurons, for which the changing nature of synaptic communication is well documented.
LTP is a use-dependent increase in synaptic efficiency (16) that can last for hours in vitro and for months in the intact animal (1). LTP is considered a family of phenomena because different types of synaptic stimulation activate different intracellular components to induce multiple phases of potentiation (83). The different forms of LTP can be viewed as physiological outcomes of interactions between different network modules. Less connected states with greater numbers of autonomous modules might yield relatively transient physiological changes. Stimuli that produce early LTP, which persists for 1 to 3 h and reflects posttranslational mechanisms, typically involve Ca2+ entry through postsynaptic NMDA-type glutamate receptors and the activation of calcium/calmodulin-dependent kinase II (CaMKII), leading to the potentiation of synaptic currents carried by AMPA-type glutamate receptor channels. This is achieved through phosphorylation of the channels in the membrane (93, 98) and by the insertion of new channels into the synapse (48, 65). This process can be considered the less connected network, in which few signaling pathways and a single cellular machine, the postsynaptic electrical response machine, are involved. When only a few modules are functional, the overall change to the cell is both spatially restricted and short-lived. On the other hand, when multiple stimuli such as NMDA receptor-mediated Ca2+ entry are coupled to the activation of the β-adrenergic receptors coupled to cAMP production (18, 41), much of the central regulatory network is engaged to activate most of the key protein kinases in the neuron: protein kinase A (PKA), protein kinase C (PKC), MAPK1,2, and CaMKII. These kinases become active for an extended period and engage the translational, transcriptional, and postsynaptic electrical response machines, and perhaps the motility machineries, to produce stable changes in the postsynaptic electrical response machinery. This is predicted to be the highly connected small-world state in which many modules are involved and interact with one another. The maintenance of LTP depends on the transcription of plasticity-related genes, which is accomplished by PKA and MAPK1,2 phosphorylation and activation of transcription factors such as CREB, thus transferring the extracellular information to change the cell’s phenotype. A similar format involving the sequential induction of transcription factors is thought to participate in lineage commitment in B-cells (43). Depending on the type of cell, the results are persistent physiological or structural changes and often new phenotypes. Thus, regulated modular interactions can result in plasticity.
The balance between plasticity and homeostasis may be achieved by reconfiguring components and connections within a selected module in response to appropriate extrinsic stimulation. Because the network that is operative at a given time constitutes only a subset of the possible connections, the dynamics of module reconfiguration protect the cell against global changes in connectivity by restricting altered activity within specified modules. The dynamic reconfigurations that result in changing the contours of the modules can play a compensatory role, resulting in overall homeostasis. Such protection can occur even as long-term modifications, involving altered gene expression, are happening. In the neuron and other differentiated cells that are capable of long-term functional changes, the signaling network enables plasticity while protecting the phenotype of the cell by restricting persistent changes to selected modules, which prevents the perturbation of other modules. Although the signals for LTP change the activity of both transcriptional and translational machineries, as well as the cell motility apparatus, the overall long-term changes are restricted to the functional structure and composition of dendritic spines and the neurotransmitter secretion and synaptic electrical response machineries.
Plasticity-related signaling components often represent highly connected nodes within the cellular network and thus are considered important interfaces between modules. However, these nodes serve other functions independent of plasticity. For example, some forms of LTP in hippocampal neurons require PKA, which has many direct effectors. To avoid disturbing diverse PKA-regulated functions (including metabolic processes) during the establishment of LTP, the activation of PKA is restricted to modules within the network that support plasticity. This type of local state change makes it necessary to consider the cellular network a system in which nodes have multiple states and the different states may allow different connectivities (Figure 2d,e). Two strategies enforce this functional segregation, the posttranslational modification of components, often through phosphorylation/dephosphorylation reactions, and the spatial translocation of components. These mechanisms are not mutually exclusive, as translocation often occurs after changes in phosphorylation states of components. Translocation can be local, acting at short distances such as stimulus-dependent binding of protein kinase to scaffold proteins leading to local reconfigurations (28), or they may be long-range, such as the movement of MAPK1,2 to the nucleus (86).
The boundaries between modules, and consequently the balance between interacting modules, are determined by basic biochemical mechanisms. The basis for regulated connectivity between modules is a change in concentration of the components and/or their activity within the module. Thus, the essential definition for connection and balance between modules is quantitative. Local reconfiguration can produce an increase in concentration and/or activity, resulting in increased connectivity in a restricted region. The result could be the formation of a modular highly connected local network. Such a highly internally connected module can trap a signal until it can be transferred to the next module. Extensive internal connectivity can bring together separate pathways that have been stimulated to subthreshold levels to form coincident detectors (positive feed-forward loops), enabling appropriate signals to propagate through the system while reducing the likelihood of false extracellular cues. Such dramatic increases in connectivity within modules are expected to be transient owing to the activation of negative feedback loops by phosphorylation/dephosphorylation reactions, sequestration, or the regulated degradation of components. This mechanism can also support another function of local highly connected modules: to hold the signal until it is safely dissipated locally, thus protecting against the inappropriate engagement of cellular machines. Increasing MAPK phosphatase activity allows MAPK1,2 activity to be local and transient. MAPK1,2, in this case, stimulates the synthesis of autocrine factors through phospholipase A2 at the membrane but does not trigger proliferation (14). Modularity ensures that signals within the cells are sufficiently dispersed, enabling autonomous behavior by the various modules. This feature is also used to route extracellular stimuli to regulate individual cellular machines and thus achieve the observed physiological effects in which one pathway evokes one physiological response while the rest of the cell is unperturbed.
Network Consciousness
When evoking long-term changes, the activity of each component within a module is constrained by the limits of its own intrinsic activity. This includes the coupled deactivation reactions, the limits of activity of the immediate upstream components, the activity of other components within the module, and the activity of components that connect modules. Such intra- and intermodule balance results from changes in the effective concentration of components owing to the regulated synthesis and degradation or changes in activity of the components. Thus, the biochemical basis for homeostasis is likely to be in the balance in coupling between components during information transfer. Within each module there are self-monitoring reactions that define the basal state and the limits of perturbation that the module can tolerate. This collection of coupled self-monitoring reactions defines the capability of modules to maintain their individual identity while interacting with other modules. This concept can give rise to a property we call network consciousness. While acknowledging the semantic implications of this term, we suggest that it expresses the capability of a cellular network as a whole “to know” the existence of the various parts of the network, as well as to recognize external stimuli, by virtue of interactions between modules. Network consciousness allows external stimuli to be routed appropriately to different parts of the network to evoke the appropriate physiological responses. A common analogy would be the constant communication between mobile telephones and the central system, such that the network knows how to route calls even when the locations of phones change.
In the neuronal intracellular network the interactions between the cAMP pathway and the CaMKII pathway by the gating mechanism (17) provide a biochemical example for understanding network consciousness. Here the cAMP pathway and the CaMKII pathways may be considered different modules that interact with each other owing to the substrate selectivity of the protein kinases and phosphatases. Because the extent and duration of the CaMKII signal are regulated by the protein phosphatase 1 (PP1), which in turn is inhibited by cAMP levels through PKA, the identity of adenylyl cyclases that produce cAMP determines whether the CaMKII pathway is conscious of the presence of a gate. At low fractional occupancy of the β-adrenergic receptors, if the functional adenylyl cyclases are the isoforms (AC5, AC6) with low basal activities (27, 89, 102), then the effect of inhibiting PP1 might be minimal. As a result, the high PP1 activity limits the duration and extent of the CaMKII activity and holds the gate shut. In contrast, in the presence of adenylyl cyclases with high basal activity (AC1, AC2, or AC8), PP1 activity is low, basal CaMKII activity is high, and hence even low fractional occupancy of the β-adrenergic receptor might allow subthreshold activation of CaMKII to induce LTP. The balance of basal activities between the cAMP and CaMKII pathways can determine connectivity and the probability of a physiological response. The ability of the various modules to define their intrinsic activity, or self-identity, arises from the identity of the isoforms of the components present in the module. In the example given above, the differences in basal activity of the different adenylyl cyclases give rise to different biochemical capabilities for the same module and thus can change its ability to interact with the second module containing CaMKII when signals from external stimulus, the β-adrenergic receptor, are received.
Signaling gates (i.e., negative feed-forward motifs) provide balance between modules. These gates are operative not only at the level of upstream signaling components but also at the level of gene expression, in which both cAMP and MAPK1,2 (86) pathways are involved in gating that regulates neuronal plasticity. In neurons it appears that open-gate configuration favors plasticity. In contrast, in proliferating cells the closed-gate configuration is essential for regulating cell proliferation and homeostasis. Here, cAMP-dependent inhibition of signaling from Ras to Raf (119) regulates signal flow to MAPK1,2 and, as a result, the proliferation response. In addition, controls at the level of transcription regulation, through key inhibitors such as p53 or the cAMP-regulated p27, reduce network connectivity and are essential for maintaining homeostasis. Thus, the ability of the network to be conscious of coupled or uncoupled modules is likely an essential feature for the overall integrity and function of the cell. The coupling between modules is defined by the regulatory motifs within the modules as well as motifs that may bridge modules. Hence, to define the modules and understand how they can function in a coordinated manner, it is essential to understand the topology of regulatory motifs within the network.
Substantial perturbations in network consciousness may cause cell death or disease. The signals that trigger cell death are integrated into the mitochondria, leading to a tight coupling between cellular energy production and control of cell death. The origins of cancer reside in the lack of control of signaling networks, leading to uncontrolled proliferation. The homeostatic mechanisms that allow the cell to adjust to increased proliferation perpetuate this perturbation, leading to a pathophysiological state. The ability to define the coupled reactions that give rise to cellular network consciousness may help us understand how multicellular systems such as tissues and organs are assembled and maintained, or explain the origins of many cellular diseases. Currently, it is not possible to estimate how many of these reactions and relationships between modules need to be defined to specify where the limits of cellular homeostasis and plasticity reside. However, it is certain that qualitative analysis is not likely to produce complete answers to these questions.
QUANTITATIVE REPRESENTATION OF CELLULAR NETWORKS
Quantitative representation of cellular networks involves the description of cellular processes in chemical or physiochemical terms. Indeed, all cellular interactions can be described in terms of chemical reactions. If we are to build detailed predictive models of mammalian cells, we need to understand how the large-scale qualitative networks used for statistical analysis can be represented in quantitative biochemical terms. All the chemical reactions in a cell can be described as either noncovalent binding or enzymatic reactions. These two classes of reactions represent the basic chemistry underlying all cellular functions including electrical responses and force generation. At a formal level, cellular networks will have to be understood on the basis of thermodynamic considerations. Biological systems including cellular networks are never at a steady state but rather on a trajectory approaching steady state (90), and formal mathematical treatments for such systems were developed in the 1960s (61). Here we do not discuss these systems in such a formal way but rather focus on utilitarian approaches that facilitate the matching of appropriate mathematical representations to the specified set of biochemical reactions underlying the process of interest and indicate the type of experimental data that need to be obtained.
Chemical Representation: Deterministic, Stochastic, and Hybrid Systems
Cellular reactions can be characterized as either deterministic or stochastic. For deterministic reactions we consider bulk concentrations, not individual molecules. Because a large proportion of these reactions occur in an aqueous milieu, the most common representation of chemical reactions uses standard solution chemistry, in which the trajectory of a reaction is defined by the initial concentrations of the reactants and the reaction rates. The underlying assumption is that the system is a well-stirred reactor in which all the reactants have equal access to each other. The reactions are governed by the laws of mass action. This is the most common type of representation used to model cell signaling pathways (14, 47, 72, 95) as well as metabolic pathways (73, 96). This assumption is often applied to many processes that occur in cellular networks and can be used to simplify numerical simulations. Deterministic reactions can be spatially specified by considering multiple compartments with specified fluxes between compartments. Such models have been developed most notably for Ca2+-regulated signaling processes (114) and for the movement of the small GTPase Ran between the cytoplasm and the nucleus (103).
However, other reactions occur between reactants that are few in number and/or present in a restricted location. These reactions are not governed by the laws of mass action. Rather, each molecule behaves in a nondeterministic fashion on the basis of its location and thermal motion, and thus the reactants interact with one another in a probabilistic fashion. Such reactions are termed stochastic reactions. Many important biological processes are governed by stochastic reactions. The regulation of gene expression, in which transcriptional regulators bind to specific sites at or near the initiation site for transcription, is a stochastic phenomenon. Such systems have been successfully modeled using a stochastic kinetic approach (7, 74, 117). Recently, a detailed analysis of signaling networks under small-volume conditions by Bhalla (10, 11) showed that some properties observed in deterministic models may not be observed in stochastic modules when few molecules in small spaces are considered. Such analyses are just beginning and substantial research is required to define the conditions under which purely deterministic or stochastic processes operate.
It is simplistic to categorize cellular components as participating exclusively in either deterministic or stochastic reactions. More likely, components may behave deterministically under some conditions and stochastically in other situations. Thus, hybrid models that consider a continuum of deterministic and stochastic reactions are likely to yield biologically realistic representations. Few hybrid models exist. Greenstein & Winslow (44) have developed an integrative model of local control of Ca2+ release on the basis of the balance of the activities of L-type Ca2+ channels and ryanodine receptors in the sarcoplasmic reticulum in myocytes. They analyzed the system within the context of global properties using both stochastic and deterministic reactions. This approach yielded a scalable model that could account for both local and global behaviors of the system.
Mathematical Representation and Numerical Computation
Deterministic reactions are commonly represented by ordinary differential equations. If flux of molecules and multiple compartments are involved, a system of partial differential equations may be used. These equations are solved to obtain concentrations of reactants or products as a function of time. Although analytical solutions can sometimes be obtained, typically systems of ordinary differential equations that describe regulatory modules such as feedback or feed-forward loops are sufficiently nonlinear, making analytical solutions difficult, if not impossible, to obtain. Most often the equations are solved numerically, and the values obtained for sets of reactions can be matched to input-output relationships that have been experimentally observed (12). Several freely available programs such as Kinetikit/Genesis (http://www.ncbs.res.in/~bhalla/kkit/download.html), Jarnac(http://www.cds.caltech.edu/~hsauro/Jarnac.htm), or JSim(http://nsr.bioeng.washington.edu/PLN/Software) can be used for the numerical computation of systems of ordinary differential equations. When flux of cellular components and spatial representations are involved, it becomes necessary to use partial differential equations to represent biological processes. Such systems of equations are more difficult to assemble and solve. Currently, Virtual Cell (69, 101), a software suite that enables researchers to develop spatially explicit models, is one of the few programs available for biologists attempting to model systems that require the use of partial differential equations.
For stochastic processes, Monte Carlo–style algorithms are typically used. The most common approach to model stochastic reactions uses the algorithm originally described by Gillespie (38). Several solvers for stochastic processes are available. M-Cell is a stochastic simulator used for spatially realistic simulations. It has been used to describe the binding of the neurotransmitter acetylcholine to its nicotinic receptor channel and the activation states of the receptor at the synapse (105). Another stochastic simulator that is freely available is Stochsim (100). Recently, Kinetikit/Genesis, a program suitable for the development of large-scale deterministic models, has been adapted for numerical computation of hybrid models (109). Although each of these programs represents a valuable contribution, the development of stochastic programs is still in its infancy. Substantial development is needed to make these programs user-friendly for the biologist.
To go from qualitative network representations suitable for statistical analysis to detailed numerical models based on chemical reactions and the appropriate mathematical representations, it would be advantageous to reduce the complexity of the system in terms of numbers of components and interactions. One practical approach may be to compute smaller sets of reactions representing regulatory motifs of the types described in the qualitative network analysis and determine the input-output relationships for these motifs. Subsequently, the individually computed motifs may be assembled as modules to construct a large-scale network. Assembling modules containing regulatory motifs will lead to a new level of nonlinear relationships that will have to be solved.
Spatial Specifications
A unique feature of all eukaryotic cells, including mammalian cells, is the utilization of spatial separation of components and interactions to achieve selective regulation of function. For proteins, the molecular basis of such spatial separation resides in the signal sequence that specifies the appropriate location within the cell (19). However, for many cellular components the localization is not fixed. It changes in an activity-dependent manner. The translocation results in changes in connectivity and, in some cases, kinetics. To build realistic models of a cell from its parts, we must account for both of these properties. If we are to use the qualitative descriptions of networks and quantitative analysis in an integrated manner, we need to develop representations that include spatial specifications at both levels. A sustained collaborative effort among a community of scientists is needed to develop a generally acceptable format of representation that includes spatial specifications. Such representation must capture the dynamics of the systems so that various forms of transport of components, from thermal diffusion to ATP-utilizing mechanisms involving specific molecular motors, are represented. One approach for such representation is to account for diffusion coefficients and then specify modification factors to this constant on the basis of the mechanism involved in the movement of the component of interest. This requires experimental measurements of movement rates. Few such measurements currently exist. However, the rapid development of fluorescent imaging techniques in live cells indicates that gathering this type of data is now technologically feasible.
Current Datasets: Experimental Approaches and Quantitative Databases
Much of the functional data that currently exist can be found in the biochemistry, cell biology, and cell physiology literature. These sources can be best thought of as data that deal with the components and interactions that make up the various signaling pathway modules and cellular machine modules. The advantages in considering data from these sources include the directionality of the interactions and, in many cases for which quantitative measurements have been performed, the kinetic data specifying input-output relationships that are useful in constraining quantitative models. The major disadvantage in obtaining data from these sources is that the data typically represent the minimal number of components required for a certain effect or function. Such minimal definitions do not indicate how many other components are associated with the module being studied. Hence, it is necessary that these function-based biochemical and cell physiological studies be coupled with proteomic approaches that provide a broad overview of the components that may be involved in the function of a module. Even well-studied functional entities in the cell, such as the mitochondria or the nuclear pore complex, when subjected to proteomic analysis, have shown the presence of yet unrecognized components (30, 81). A recent study on the well-characterized tumor necrosis factor alpha signaling pathway identified 10 new modulators that had not been discovered by the biochemical and cell physiological studies (21). It would be best if the emerging high-throughput genome-wide approaches were coupled with smaller scale functional studies to yield precise definitions of components and interactions.
At a qualitative level, to define large networks the two approaches used most often are the identification of interactions by yeast two-hybrid screens (40, 51, 59, 68, 120) and the isolation of complexes followed by mass spectrometric identification of components. The latter approach is also capable of identifying posttranslational modifications and hence can provide additional information about the regulatory status of components. A major limitation of both approaches is that they provide no information about directionality of interactions, and such information must be obtained from functional studies. Another popular approach for large-scale data gathering is the use of microarrays to profile mRNA levels under varying conditions (54). Although such datasets do provide useful signatures for disease states (91) and the treatment outcomes (70), it appears unlikely that microarray data can be useful in developing quantitative models of cell function. The major problem lies in the lack of information regarding how levels of mRNA correlate with protein concentration. A recent study explicitly analyzing such correlation found that only about 40% of the mRNA and corresponding protein levels correlated well (107). It may be necessary to obtain reliable measurements of the various levels of proteins individually albeit in high throughput.
To develop quantitative models, we need several types of parameters. These include concentrations of components, both global as well as local, so that concentrations are always correlated with spatial localization. Global concentrations may be obtained by biochemical approaches such as quantitative immunoblotting, whereas local concentrations can be obtained only by microscopy, ideally by live-cell imaging. We also need to obtain reaction rates for various reactions and the overall enzymatic rates. This must be done one reaction at a time, but if we are to gather such kinetic parameters for thousands of interactions, technology development is needed to obtain these measurements in a high throughput. Currently, most of the kinetic parameters come from biochemical characterization experiments, which have been conducted under a variety of conditions. It will be useful to achieve some level of standardization so that we can obtain kinetic parameters under similar conditions.
The requirement for both qualitative and quantitative data for mammalian cellular networks is matched by the requirement for appropriate databases to store these parameters and interactions and to allow for the assembly of reactions and models for both qualitative and quantitative analysis. Several potentially useful projects to develop databases for computation of models are currently underway. A list of freely accessible databases for both mammalian and nonmammalian systems is provided in Table 1. Campagne et al. (25) have begun to develop a quantitative database in which reactions of modules can be assembled and exported to simulation software for numerical computation. Such quantitative databases would have to be fully integrated with genomic and proteomic databases. Eventually, the development of a database that has the best features of many of the current databases would be ideal. Such a database should link all the known genomic, structural, and other qualitative information of each component to its quantitative characteristics in specified cell types. Interactions should be described in terms of directionality, location, and context for regulatory behavior. To achieve these types of characterization, we need to develop a common vocabulary for database entries. The SBML project (50) is an initial step toward the goal of developing a common vocabulary and exchangeable models.
DEVELOPING PREDICTIVE MODELS
Ultimately our goal is to obtain detailed models of a mammalian cell such that we can understand and predict its behavior as it encounters a variety of external stimuli and undergoes internal changes. Such predictions at a high level would not only describe the various functions that result in the cellular phenotype, but also explain how variants of the same cell with somewhat different genotypes resulting from single-nucleotide polymorphisms can yield differing responses to the same stimuli. In predicting the trajectory of a cell through its life, we should be able to predict the limits of its tolerance to perturbation and the mechanisms by which catastrophic failures can occur. Such models would be similar to detailed engineering models of passenger jets that are now designed and initially analyzed for performance solely on computers. However, models of cells are likely to be more complicated owing to the turnover of components on a regular basis (116). This situation is akin to the passenger jet having the rivets on the wings replaced at timed intervals during flight. Does such complexity make the development of detailed models unrealistic? Current progress suggests that development of such models is indeed feasible. Much of the noise and fluctuations seen at the most detailed level of organization are often smoothed out as higher orders of organization arise, producing overall robustness of behavior. For this reason it is useful to start with a top-down approach and identify the motifs and modules in the cellular network. This should yield the broad overview without the details of the functional characteristics of each module. This overview description can then be used to drill-down to develop and analyze differential equation-based models to define the input-output (I/O) relationships. These modules can then be reassembled to obtain a large-scale model that describes the cell as a whole. The proposed approach is schematically summarized in Figure 5 and is undoubtedly simplistic. For example, the term experiment covers a vast area. There are many technical challenges we need to overcome in gathering the data needed to construct qualitative interaction maps for whole cells, as well as to constrain and validate the quantitative models. There are considerable challenges at the level of modeling as well. As we assemble modules that have been modeled in detail, the higher order models should capture not only the balance of activity between modules, but also the reach-through capability of key components within modules. This type of analysis, although challenging, should be rewarding, because it is through the assembly of the modules that we will identify the core design principles of a cell. Such a design may include a definition of intrinsic uncertainty whereby there are limits in our ability to predict cellular behavior, even when we know all the parts and interactions. These limits can be discovered only through building and testing models.
Figure 5.
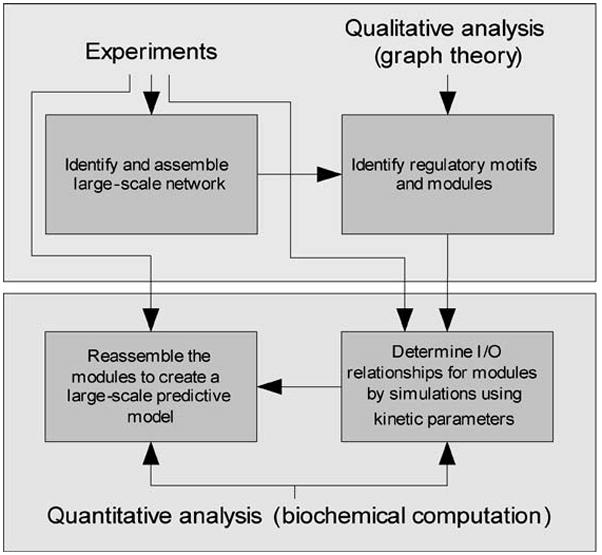
An integrative approach for the development of predictive models.
Acknowledgments
A.M. is supported by the predoctoral training grant: Integrated Training Program in Pharmacological Sciences, GM-62754. R.I. thanks John Doyle (Caltech) for suggesting the relevant analogy of mobile phones and the central network. Research in the Iyengar laboratory is supported by GM-54508 and CA-81050.
Contributor Information
Avi Ma’ayan, Email: avi.maayan@mssm.edu.
Robert D. Blitzer, Email: robert.blitzer@mssm.edu.
Ravi Iyengar, Email: ravi.iyengar@mssm.edu.
LITERATURE CITED
- 1.Abraham WC, Logan B, Greenwood JM, Dragunow M. Induction and experience-dependent consolidation of stable long-term potentiation lasting months in the hippocampus. J Neurosci. 2002;22:9626–34. doi: 10.1523/JNEUROSCI.22-21-09626.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, et al. The genome sequence of Drosophila melanogaster. Science. 2000;287:2185–95. doi: 10.1126/science.287.5461.2185. [DOI] [PubMed] [Google Scholar]
- 3.Albert I, Albert R. Conserved network motifs allow protein-protein interaction prediction. Bioinformatics. 2004;20:3346–52. doi: 10.1093/bioinformatics/bth402. [DOI] [PubMed] [Google Scholar]
- 4.Albert R, Jeong H, Barabasi AL. Error and attack tolerance of complex networks. Nature. 2000;406:378–82. doi: 10.1038/35019019. [DOI] [PubMed] [Google Scholar]
- 5.Albert R, Othmer HG. The topology of the regulatory interactions predicts the expression pattern of the segment polarity genes in Drosophila melanogaster. J Theor Biol. 2003;223:1–18. doi: 10.1016/s0022-5193(03)00035-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amaral LA, Scala A, Barthelemy M, Stanley HE. Classes of small-world networks. Proc Natl Acad Sci USA. 2000;97:11149–52. doi: 10.1073/pnas.200327197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arkin A, Ross J, McAdams HH. Stochastic kinetic analysis of developmental pathway bifurcation in phage lambda-infected Escherichia coli cells. Genetics. 1998;149:1633–48. doi: 10.1093/genetics/149.4.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Artzy-Randrup Y, Fleishman SJ, Ben-Tal N, Stone L. Comment on “Network motifs: simple building blocks of complex networks” and “Superfamilies of evolved and designed networks. Science. 2004;305:1107. doi: 10.1126/science.1099334. [DOI] [PubMed] [Google Scholar]
- 9.Barabasi AL, Albert R. Emergence of scaling in random networks. Science. 1999;286:509–12. doi: 10.1126/science.286.5439.509. [DOI] [PubMed] [Google Scholar]
- 10.Bhalla US. Signaling in small sub-cellular volumes. I. Stochastic and diffusion effects on individual pathways. Biophys J. 2004;87:733–44. doi: 10.1529/biophysj.104.040469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bhalla US. Signaling in small sub-cellular volumes. II. Stochastic and diffusion effects on synaptic network properties. Biophys J. 2004;87:745–53. doi: 10.1529/biophysj.104.040501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bhalla US, Iyengar R. Emergent properties of networks of biological signaling pathways. Science. 1999;283:381–87. doi: 10.1126/science.283.5400.381. [DOI] [PubMed] [Google Scholar]
- 13.Bhalla US, Iyengar R. Functional modules in biological signalling networks. Novartis Found Symp. 2001;239:4–13. doi: 10.1002/0470846674.ch2. [DOI] [PubMed] [Google Scholar]
- 14.Bhalla US, Ram PT, Iyengar R. MAP kinase phosphatase as a locus of flexibility in a mitogen-activated protein kinase signaling network. Science. 2002;297:1018–23. doi: 10.1126/science.1068873. [DOI] [PubMed] [Google Scholar]
- 15.Blattner FR, Plunkett G, 3rd, Bloch CA, Perna NT, Burland V, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–74. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 16.Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 17.Blitzer RD, Connor JH, Brown GP, Wong T, Shenolikar S, et al. Gating of CaMKII by cAMP-regulated protein phosphatase activity during LTP. Science. 1998;280:1940–42. doi: 10.1126/science.280.5371.1940. [DOI] [PubMed] [Google Scholar]
- 18.Blitzer RD, Wong T, Nouranifar R, Iyengar R, Landau EM. Postsynaptic cAMP pathway gates early LTP in hippocampal CA1 region. Neuron. 1995;15:1403–14. doi: 10.1016/0896-6273(95)90018-7. [DOI] [PubMed] [Google Scholar]
- 19.Blobel G. Protein targeting. Chembiochem. 2000;18:86–102. doi: 10.1002/1439-7633(20000818)1:2<86::AID-CBIC86>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 20.Boutros M, Kiger AA, Armknecht S, Kerr K, Hild M, et al. Genome-wide RNAi analysis of growth and viability in Drosophila cells. Science. 2004;303:832–35. doi: 10.1126/science.1091266. [DOI] [PubMed] [Google Scholar]
- 21.Bouwmeester T, Bauch A, Ruffner H, Angrand PO, Bergamini G, et al. A physical and functional map of the human TNF-alpha/NF-kappa B signal transduction pathway. Nat Cell Biol. 2004;6:97–105. doi: 10.1038/ncb1086. [DOI] [PubMed] [Google Scholar]
- 22.Bruggeman FJ, Westerhoff HV, Hoek JB, Kholodenko BN. Modular response analysis of cellular regulatory networks. J Theor Biol. 2002;218:507–20. [PubMed] [Google Scholar]
- 23.Bu D, Zhao Y, Cai L, Xue H, Zhu X, et al. Topological structure analysis of the protein-protein interaction network in budding yeast. Nucleic Acids Res. 2003;31:2443–50. doi: 10.1093/nar/gkg340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Caldarelli G, Pastor-Satorras R, Vespignani A. Cycles structure and local ordering in complex networks. Eur Phys J B. 2004;38:183–86. [Google Scholar]
- 25.Campagne F, Neves S, Chang CW, Skrabanek L, Ram PT, et al. Quantitative information management for the biochemical computation of cellular networks. Sci STKE. 2004;248:PL11. doi: 10.1126/stke.2482004pl11. [DOI] [PubMed] [Google Scholar]
- 26.Chen J, Iyengar R. Suppression of Ras-induced transformation of NIH 3T3 cells by activated G alphas. Science. 1994;263:1278–81. doi: 10.1126/science.8122111. [DOI] [PubMed] [Google Scholar]
- 27.Chen Y, Harry A, Li J, Smit MJ, Bai X, et al. Adenylyl cyclase 6 is selectively regulated by protein kinase A phosphorylation in a region involved in G-alpha stimulation. Proc Natl Acad Sci USA. 1998;94:14100–4. doi: 10.1073/pnas.94.25.14100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Colledge M, Dean RA, Scott GK, Langeberg LK, Huganir RL, et al. Targeting of PKA to glutamate receptors through a MAGUK-AKAP complex. Neuron. 2000;27:107–19. doi: 10.1016/s0896-6273(00)00013-1. [DOI] [PubMed] [Google Scholar]
- 29.Cook SJ, McCormick F. Inhibition by cAMP of Ras-dependent activation of Raf. Science. 1993;262:1069–72. doi: 10.1126/science.7694367. [DOI] [PubMed] [Google Scholar]
- 30.Cronshaw JM, Krutchinsky AN, Zhang W, Chait BT, Matunis MJ. Proteomic analysis of the mammalian nuclear pore complex. J Cell Biol. 2002;158:915–27. doi: 10.1083/jcb.200206106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dodds PS, Muhamad R, Watts DJ. An experimental study of search in global social networks. Science. 2003;301:827–29. doi: 10.1126/science.1081058. [DOI] [PubMed] [Google Scholar]
- 32.Egan SE, Weinberg RA. The pathway to signal achievement. Nature. 1993;365:781–83. doi: 10.1038/365781a0. [DOI] [PubMed] [Google Scholar]
- 33.Eisenberg E, Levanon EY. Preferential attachment in the protein network evolution. Phys Rev Lett. 2003;91:138701. doi: 10.1103/PhysRevLett.91.138701. [DOI] [PubMed] [Google Scholar]
- 34.Forger DB, Peskin CS. A detailed predictive model of the mammalian circadian clock. Proc Natl Acad Sci USA. 2003;100:14806–11. doi: 10.1073/pnas.2036281100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Friedman C, Kra P, Yu H, Krauthammer M, Rzhetsky A. GENIES: a natural-language processing system for the extraction of molecular pathways from journal articles. Bioinformatics. 2001;17:S74–S82. doi: 10.1093/bioinformatics/17.suppl_1.s74. [DOI] [PubMed] [Google Scholar]
- 36.Gardner TS, di Bernardo D, Lorenz D, Collins JJ. Inferring genetic networks and identifying compound mode of action via expression profiling. Science. 2003;301:102–5. doi: 10.1126/science.1081900. [DOI] [PubMed] [Google Scholar]
- 37.Giaever G, Chu AM, Ni L, Connelly C, Riles L, et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 2002;418:387–91. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- 38.Gillespie DT. A general method for numerically simulating the stochastic time evolution of coupled chemical reactions. J Comput Phys. 1976;22:403–34. [Google Scholar]
- 39.Gilman AG, Simon MI, Bourne HR, Harris BA, Long R, et al. Overview of the Alliance for Cellular Signaling. Nature. 2002;420:703–6. doi: 10.1038/nature01304. [DOI] [PubMed] [Google Scholar]
- 40.Giot L, Bader JS, Brouwer C, Chaudhuri A, Kuang B, et al. A protein interaction map of Drosophila melanogaster. Science. 2003;302:1727–36. doi: 10.1126/science.1090289. [DOI] [PubMed] [Google Scholar]
- 41.Giovannini MG, Blitzer RD, Wong T, Asoma K, Tsokas P, et al. Mitogen-activated protein kinase regulates early phosphorylation and delayed expression of calcium/calmodulin-dependent protein kinase II in long-term potentiation. J Neurosci. 2001;21:7053–62. doi: 10.1523/JNEUROSCI.21-18-07053.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gough NR. Science’s signal transduction knowledge environment: the connections maps database. Ann NY Acad Sci. 2002;971:585–87. doi: 10.1111/j.1749-6632.2002.tb04532.x. [DOI] [PubMed] [Google Scholar]
- 43.Graf T. Differentiation plasticity of hematopoietic cells. Blood. 2002;99:3089–101. doi: 10.1182/blood.v99.9.3089. [DOI] [PubMed] [Google Scholar]
- 44.Greenstein JL, Winslow RL. An integrative model of the cardiac ventricular myocyte incorporating local control of Ca2+ release. Biophys J. 2002;83:2918–45. doi: 10.1016/S0006-3495(02)75301-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hartwell LH, Hopfield JJ, Leibler S, Murray AW. From molecular to modular cell biology. Nature. 1999;402:C47–C52. doi: 10.1038/35011540. [DOI] [PubMed] [Google Scholar]
- 46.Hartwell LH, Kastan MB. Cell cycle control and cancer. Science. 1994;266:1821–28. doi: 10.1126/science.7997877. [DOI] [PubMed] [Google Scholar]
- 47.Haugh JM, Wells A, Lauffenburger DA. Mathematical modeling of epidermal growth factor receptor signaling through the phospholipase C pathway: mechanistic insights and predictions for molecular interventions. Biotechnol Bioeng. 2000;70:225–38. [PubMed] [Google Scholar]
- 48.Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, et al. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287:2262–67. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- 49.Ho Y, Gruhler A, Heilbut A, Bader GD, Moore L, et al. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature. 2002;415:180–83. doi: 10.1038/415180a. [DOI] [PubMed] [Google Scholar]
- 50.Hucka M, Finney A, Sauro HM, Bolouri H, Doyle JC, et al. The systems biology markup language (SBML): a medium for representation and exchange of biochemical network models. Bioinformatics. 2003;19:524–31. doi: 10.1093/bioinformatics/btg015. [DOI] [PubMed] [Google Scholar]
- 51.Ito T, Chiba T, Ozawa R, Yoshida M, Hattori M, et al. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc Natl Acad Sci USA. 2001;98:4569–74. doi: 10.1073/pnas.061034498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Iyengar R. Gating by cyclic AMP: expanded role for an old signaling pathway. Science. 1996;271:461–63. doi: 10.1126/science.271.5248.461. [DOI] [PubMed] [Google Scholar]
- 53.Janes KA, Albeck JG, Peng LX, Sorger PK, Lauffenburger DA, et al. A high-throughput quantitative multiplex kinase assay for monitoring information flow in signaling networks: application to sepsis-apoptosis. Mol Cell Proteomics. 2003;2:463–73. doi: 10.1074/mcp.M300045-MCP200. [DOI] [PubMed] [Google Scholar]
- 54.Jeffrey SS, Fero MJ, Borresen-Dale AL, Botstein D. Expression array technology in the diagnosis and treatment of breast cancer. Mol Interv. 2004;2:101–9. doi: 10.1124/mi.2.2.101. [DOI] [PubMed] [Google Scholar]
- 55.Jeong H, Mason SP, Barabasi AL, Oltvai ZN. Lethality and centrality in protein networks. Nature. 2001;411:41–42. doi: 10.1038/35075138. [DOI] [PubMed] [Google Scholar]
- 56.Jeong H, Tombor B, Albert R, Oltvai ZN, Barabasi AL. The large-scale organization of metabolic networks. Nature. 2000;407:651–54. doi: 10.1038/35036627. [DOI] [PubMed] [Google Scholar]
- 57.Jordan JD, Landau EM, Iyengar R. Signaling networks: the origins of cellular multitasking. Cell. 2000;103:193–200. doi: 10.1016/s0092-8674(00)00112-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kamme F, Salunga R, Yu J, Tran DT, Zhu J, et al. Single-cell microarray analysis in hippocampus CA1: demonstration and validation of cellular heterogeneity. J Neurosci. 2003;23:3607–15. doi: 10.1523/JNEUROSCI.23-09-03607.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kanehisa M, Goto S, Kawashima S, Nakaya A. The KEGG databases at GenomeNet. Nucleic Acids Res. 2001;30:42–46. doi: 10.1093/nar/30.1.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Karp PD. Metabolic databases. Trends Biochem Sci. 1998;23:114–16. doi: 10.1016/s0968-0004(98)01184-0. [DOI] [PubMed] [Google Scholar]
- 61.Katchalsky A, Curran PF. Nonequilibrium Thermodynamics in Biophysics. Cambridge, MA: Harvard Univ. Press; 1967. [Google Scholar]
- 62.Kim JW, Hunt B, Ott E. Evolving networks with multispecies nodes and spread in the number of initial links. Phys Rev E Stat Nonlin Soft Matter Phys. 2002;66:046115. doi: 10.1103/PhysRevE.66.046115. [DOI] [PubMed] [Google Scholar]
- 63.Kuaffman SA. The Origins of Order. Oxford, UK: Oxford Univ. Press; 1993. [Google Scholar]
- 64.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 65.Lee HK, Barbarosie M, Kameyama K, Bear MF, Huganir RL. Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature. 2000;405:955–59. doi: 10.1038/35016089. [DOI] [PubMed] [Google Scholar]
- 66.Levchenko A. Dynamical and integrative cell signaling: challenges for the new biology. Biotechnol Bioeng. 2004;84:773–82. doi: 10.1002/bit.10854. [DOI] [PubMed] [Google Scholar]
- 67.Li J, Ning Y, Hedley W, Saunders B, Chen Y, et al. The Molecule Pages database. Nature. 2002;420:716–17. doi: 10.1038/nature01307. [DOI] [PubMed] [Google Scholar]
- 68.Li S, Armstrong CM, Bertin N, Ge H, Milstein S, et al. A map of the interactome network of the metazoan C. elegans. Science. 2004;303:540–43. doi: 10.1126/science.1091403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Loew LM, Schaff JC. The Virtual Cell: a software environment for computational cell biology. Trends Biotechnol. 2001;19:401–6. doi: 10.1016/S0167-7799(01)01740-1. [DOI] [PubMed] [Google Scholar]
- 70.Lossos IS, Czerwinski DK, Alizadeh AA, Wechser MA, Tibshirani R, et al. Prediction of survival in diffuse large-B-cell lymphoma based on the expression of six genes. N Engl J Med. 2004;350:1828–37. doi: 10.1056/NEJMoa032520. [DOI] [PubMed] [Google Scholar]
- 71.Marcotte EM, Xenarios I, Eisenberg D. Mining literature for protein-protein interactions. Bioinformatics. 2001;17:359–63. doi: 10.1093/bioinformatics/17.4.359. [DOI] [PubMed] [Google Scholar]
- 72.Markevich NI, Hoek JB, Kholodenko BN. Signaling switches and bistability arising from multisite phosphorylation in protein kinase cascades. J Cell Biol. 2004;164:353–59. doi: 10.1083/jcb.200308060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Martiel JL, Goldbeter A. Metabolic oscillations in biochemical systems controlled by covalent enzyme modification. Biochimie. 1981;63:119–24. doi: 10.1016/s0300-9084(81)80175-7. [DOI] [PubMed] [Google Scholar]
- 74.McAdams HH, Arkin A. Stochastic mechanisms in gene expression. Proc Natl Acad Sci USA. 1997;94:814–19. doi: 10.1073/pnas.94.3.814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mendes P. GEPASI: a software package for modelling the dynamics, steady states and control of biochemical and other systems. Comput Appl Biosci. 1993;9:563–71. doi: 10.1093/bioinformatics/9.5.563. [DOI] [PubMed] [Google Scholar]
- 76.Mewes HW, Albermann K, Bahr M, Frishman D, Gleissner A, et al. Overview of the yeast genome. Nature. 1997;387:7–65. doi: 10.1038/42755. [DOI] [PubMed] [Google Scholar]
- 77.Milgram S. The small world problem. Psychol Today. 1967;2:60–67. [Google Scholar]
- 78.Milo R, Itzkovitz S, Kashtan N, Levitt R, Alon U. Response to “Comment on ‘Network motifs: simple building blocks of complex networks’ and ‘Superfamilies of evolved and designed networks’”. Science. 2004;305:1107D. [Google Scholar]
- 79.Milo R, Itzkovitz S, Kashtan N, Levitt R, Shen-Orr S, et al. Superfamilies of evolved and designed networks. Science. 2004;303:1538–42. doi: 10.1126/science.1089167. [DOI] [PubMed] [Google Scholar]
- 80.Milo R, Shen-Orr S, Itzkovitz S, Kashtan N, Chklovskii D, et al. Network motifs: simple building blocks of complex networks. Science. 2002;298:824–27. doi: 10.1126/science.298.5594.824. [DOI] [PubMed] [Google Scholar]
- 81.Mootha VK, Bunkenborg J, Olsen JV, Hjerrild M, Wisniewski JR, et al. Integrated analysis of protein composition, tissue diversity, and gene regulation in mouse mitochondria. Cell. 2003;115:629–40. doi: 10.1016/s0092-8674(03)00926-7. [DOI] [PubMed] [Google Scholar]
- 82.Mural RJ, Adams MD, Myers EW, Smith HO, Miklos GL, et al. A comparison of whole-genome shotgun-derived mouse chromosome 16 and the human genome. Science. 2002;296:1661–71. doi: 10.1126/science.1069193. [DOI] [PubMed] [Google Scholar]
- 83.Nguyen PV, Abel T, Kandel ER. Requirement of a critical period of transcription for induction of a late phase of LTP. Science. 1994;265:1104–7. doi: 10.1126/science.8066450. [DOI] [PubMed] [Google Scholar]
- 84.Nikitin A, Egorov S, Daraselia N, Mazo I. Pathway studio: the analysis and navigation of molecular networks. Bioinformatics. 2003;19:2155–57. doi: 10.1093/bioinformatics/btg290. [DOI] [PubMed] [Google Scholar]
- 85.Pastor-Satorras R, Smith E, Sole RV. Evolving protein interaction networks through gene duplication. J Theor Biol. 2003;222:199–210. doi: 10.1016/s0022-5193(03)00028-6. [DOI] [PubMed] [Google Scholar]
- 86.Patterson SL, Pittenger C, Morozov A, Martin KC, Scanlin H, et al. Some forms of cAMP-mediated long-lasting potentiation are associated with release of BDNF and nuclear translocation of phospho-MAP kinase. Neuron. 2001;32:123–40. doi: 10.1016/s0896-6273(01)00443-3. [DOI] [PubMed] [Google Scholar]
- 87.Pawson T, Scott JD. Signaling through scaffold, anchoring, and adaptor proteins. Science. 1998;278:2075–80. doi: 10.1126/science.278.5346.2075. [DOI] [PubMed] [Google Scholar]
- 88.Peri S, Navarro JD, Amanchy R, Kristiansen TZ, Jonnalagadda CK, et al. Development of human protein reference database as an initial platform for approaching systems biology in humans. Genome Res. 2003;13:2363–71. doi: 10.1101/gr.1680803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pieroni JP, Harry A, Chen J, Jacobowitz O, Magnusson RP, et al. Distinct characteristics of the basal activities of adenylyl cyclases 2 and 6. J Biol Chem. 1995;270:21368–73. doi: 10.1074/jbc.270.36.21368. [DOI] [PubMed] [Google Scholar]
- 90.Prigogine I, Nicolis G, Babloyantz A. Nonequilibrium problems in biological phenomena. Ann NY Acad Sci. 1974;231:99–105. doi: 10.1111/j.1749-6632.1974.tb20557.x. [DOI] [PubMed] [Google Scholar]
- 91.Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nat Genet. 2002;33:49–54. doi: 10.1038/ng1060. [DOI] [PubMed] [Google Scholar]
- 92.Ravasz E, Somera AL, Mongru DA, Oltvai ZN, Barabasi AL. Hierarchical organization of modularity in metabolic networks. Science. 2002;297:1551–55. doi: 10.1126/science.1073374. [DOI] [PubMed] [Google Scholar]
- 93.Raymond LA, Blackstone CD, Huganir RL. Phosphorylation and modulation of recombinant GluR6 glutamate receptors by cAMP-dependent protein kinase. Nature. 1993;361:637–41. doi: 10.1038/361637a0. [DOI] [PubMed] [Google Scholar]
- 94.Remy I, Michnick SW. Visualization of biochemical networks in living cells. Proc Natl Acad Sci USA. 2001;98:7678–83. doi: 10.1073/pnas.131216098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Riccobene TA, Omann GM, Linderman JJ. Modeling activation and desensitization of G-protein coupled receptors provides insight into ligand efficacy. J Theor Biol. 1999;200:207–22. doi: 10.1006/jtbi.1999.0988. [DOI] [PubMed] [Google Scholar]
- 96.Rohwer JM, Meadow ND, Roseman S, Westerhoff HV, Postma PW. Understanding glucose transport by the bacterial phosphoenolpyruvate:glycose phosphotransferase system on the basis of kinetic measurements in vitro. J Biol Chem. 2000;275:34909–21. doi: 10.1074/jbc.M002461200. [DOI] [PubMed] [Google Scholar]
- 97.Schwikowski B, Uetz P, Fields S. A network of protein-protein interactions in yeast. Nat Biotechnol. 2000;18:1257–61. doi: 10.1038/82360. [DOI] [PubMed] [Google Scholar]
- 98.Seidenman KJ, Steinberg JP, Huganir R, Malinow R. Glutamate receptor subunit 2 serine 880 phosphorylation modulates synaptic transmission and mediates plasticity in CA1 pyramidal cells. J Neurosci. 2003;23:9220–28. doi: 10.1523/JNEUROSCI.23-27-09220.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Shen-Orr SS, Milo R, Mangan S, Alon U. Network motifs in the transcriptional regulation network of Escherichia coli. Nat Genet. 2002;31:64–68. doi: 10.1038/ng881. [DOI] [PubMed] [Google Scholar]
- 100.Shimizu TS, Bray D. Modelling the bacterial chemotaxis receptor complex. Novartis Found Symp. 2003;247:162–77. [PubMed] [Google Scholar]
- 101.Slepchenko BM, Schaff JC, Macara I, Loew LM. Quantitative cell biology with the Virtual Cell. Trends Cell Biol. 2003;13:570–76. doi: 10.1016/j.tcb.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 102.Smit MJ, Verzijl D, Iyengar R. Identity of adenylyl cyclase isoform determines the rate of cell cycle progression in NIH 3T3 cells. Proc Natl Acad Sci USA. 1998;95:15084–89. doi: 10.1073/pnas.95.25.15084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Smith AE, Slepchenko BM, Schaff JC, Loew LM, Macara IG. Systems analysis of Ran transport. Science. 2002;295:488–91. doi: 10.1126/science.1064732. [DOI] [PubMed] [Google Scholar]
- 104.Steffen M, Petti A, Aach J, D’haeseleer P, Church G. Automated modelling of signal transduction networks. BMC Bioinformatics. 2002;3:34. doi: 10.1186/1471-2105-3-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Stiles JR, Bartol TM, Jr, Salpeter EE, Salpeter MM. In: Computational Neuroscience. Bower JM, editor. New York: Plenum; 1998. pp. 279–84. [Google Scholar]
- 106.Strogatz SH. Exploring complex networks. Nature. 2001;410:268–76. doi: 10.1038/35065725. [DOI] [PubMed] [Google Scholar]
- 107.Tian Q, Stepaniants SB, Mao M, Weng L, Feetham MC, et al. Integrated genomic and proteomic analyses of gene expression in mammalian cells. Mol Cell Proteomics. 2004;3:960–69. doi: 10.1074/mcp.M400055-MCP200. [DOI] [PubMed] [Google Scholar]
- 108.Tong AH, Lesage G, Bader GD, Ding H, Xu H, et al. Global mapping of the yeast genetic interaction network. Science. 2004;303:808–13. doi: 10.1126/science.1091317. [DOI] [PubMed] [Google Scholar]
- 109.Vasudeva K, Bhalla US. Adaptive stochastic-deterministic chemical kinetic simulations. Bioinformatics. 2003;20:78–84. doi: 10.1093/bioinformatics/btg376. [DOI] [PubMed] [Google Scholar]
- 110.Vayttaden SJ, Bhalla US. Developing complex signaling models using GENESIS/Kinetikit. Sci STKE. 2004;2004:PL4. doi: 10.1126/stke.2192004pl4. [DOI] [PubMed] [Google Scholar]
- 111.Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, et al. The sequence of the human genome. Science. 2001;291:1304–51. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 112.von Dassow G, Meir E, Munro EM, Odell GM. The segment polarity network is a robust developmental module. Nature. 2000;406:188–92. doi: 10.1038/35018085. [DOI] [PubMed] [Google Scholar]
- 113.von Mering C, Krause R, Snel B, Cornell M, Oliver SG, et al. Comparative assessment of large-scale data sets of protein-protein interactions. Nature. 2002;417:399–403. doi: 10.1038/nature750. [DOI] [PubMed] [Google Scholar]
- 114.Wagner J, Fall CP, Hong F, Sims CE, Allbritton NL, et al. Wave of IP3 production accompanies the fertilization Ca2+ wave in the egg of the frog, Xenopus laevis: theoretical and experimental support. Cell. 2004;35:433–47. doi: 10.1016/j.ceca.2003.10.009. [DOI] [PubMed] [Google Scholar]
- 115.Watts DJ, Strogatz SH. Collective dynamics of ‘small-world’ networks. Nature. 1998;393:440–42. doi: 10.1038/30918. [DOI] [PubMed] [Google Scholar]
- 116.Weng G, Bhalla US, Iyengar R. Complexity in biological signaling systems. Science. 1999;284:92–96. doi: 10.1126/science.284.5411.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wolf DM, Arkin AP. Motifs, modules and games in bacteria. Curr Opin Microbiol. 2003;6:125–34. doi: 10.1016/s1369-5274(03)00033-x. [DOI] [PubMed] [Google Scholar]
- 118.Wood V, Gwilliam R, Rajandream MA, Lyne M, Lyne R, et al. The genome sequence of Schizosaccharomyces pombe. Nature. 2002;415:871–80. doi: 10.1038/nature724. [DOI] [PubMed] [Google Scholar]
- 119.Wu J, Dent P, Jelinek T, Wolfman A, Weber MJ, et al. Inhibition of the EGF-activated MAP kinase signaling pathway by adenosine 3′,5′-monophosphate. Science. 1993;262:1065–69. doi: 10.1126/science.7694366. [DOI] [PubMed] [Google Scholar]
- 120.Xenarios I, Fernandez E, Salwinski L, Duan XJ, Thompson MJ, et al. DIP: the database of interacting proteins: 2001 update. Nucleic Acids Res. 2000;29:239–41. doi: 10.1093/nar/29.1.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhu CP, Xiong SJ, Tian YJ, Li N, Jiang KS. Scaling of directed dynamical small-world networks with random responses. Phys Rev Lett. 2004;92:218702. doi: 10.1103/PhysRevLett.92.218702. [DOI] [PubMed] [Google Scholar]