

Summary
The bacterial primosome comprises the replicative homo-hexameric ring helicase DnaB and the primase DnaG. It is an integral component of the replisome as it unwinds the parental DNA duplex to allow progression of the replication fork, synthesizes the initiation primers at the replication origin, oriC, and the primers required for Okazaki fragment synthesis during lagging strand replication. The interaction between the two component proteins is mediated by a distinct C-terminal domain (p16) of the primase. Both proteins mutually regulate each other’s activities and a putative network of conserved residues has been proposed to mediate these effects. We have targeted 10 residues from this network. To investigate the functional contributions of these residues to the primase, ATPase and helicase activities of the primosome, we have used site-directed mutagenesis and in vitro functional assays. Five of these residues (E464, H494, R495, Y548 and R555) exhibited some functional significance while the remaining five (E483, R484, E506, D512 and E530) exhibited no effects. E464 participates in functional modulation of the primase activity, whereas H494, R495 and R555 participate in allosteric functional modulation of the ATPase and/or helicase activities. Y548 contributes directly to the structural interaction with DnaB.
Introduction
The bacterial primosome (helicase DnaB–primase DnaG complex) is an essential component of the replisome. It co-ordinates primer synthesis and replication fork progression (Johnson and O’Donnell, 2005). Initiation of DNA replication is intimately linked with primosomal assembly (Mott and Berger, 2007). Assembly of the primosome is mediated by specialized loading mechanisms that tether the replicative ring helicase, DnaB, around ssDNA, followed by recruitment of the primase, DnaG, via a direct interaction with DnaB. This interaction signals the end of the initiation stage and the onset of the elongation stage. The primosome directs the initiation of leading strand synthesis and repeatedly regulates the cyclic synthesis of Okazaki fragments during lagging strand synthesis (Tougu and Marians, 1996a; Lee et al., 2006). The DnaB and DnaG proteins mutually co-regulate each other’s functions when in complex. For instance, DnaB alters the activity of DnaG by modulating the initiation specificity, stimulation of primer synthesis and reduction of primer size (Lu et al., 1996; Bhattacharyya and Griep, 2000; Mitkova et al., 2003; Johnson et al., 2000; Koepsell et al., 2006), whereas DnaG stimulates the ATPase and helicase activities of DnaB (Bird et al., 2000; Thirlway et al., 2004). The structural details of this interaction and the mechanism of mutual regulation of their activities have been the focus of long-term interest and are only just starting to be unravelled.
The observed optimal stoichiometry of the primosome is three molecules of DnaG bound to one hexamer of DnaB (Bird et al., 2000; Mitkova et al., 2003; Bailey et al., 2007), but a few complexes with two or one DnaG molecules bound to a DnaB hexamer have also been detected in vitro (Thirlway et al., 2004). This stoichiometric variability could be of functional significance in vivo, particularly as cross-talk between primase subunits in the DnaB–DnaG complex, which have been shown to allow the primase to select initiation sites on template DNA and therefore to regulate primer synthesis in trans (Corn et al.,2005). One could envisage that the simplest way to modulate this trans-activation is by varying the number of DnaG molecules in the primosome.
The hexameric DnaB ring exists in different structural states: a symmetrical hexamer with sixfold symmetry and a trimer of dimers with threefold symmetry, referred to as C6 and C3 conformations (Bujalowski et al., 1994; Yu et al., 1996; Patel and Picha, 2000). An intermediate conformation known as C3C6 has also been observed (Bailey et al., 2007). The biological significance of these conformations remains unclear. Atomic force microscopy imaging demonstrated that the binding of primase to the helicase results in an apparent conversion of the hexameric DnaB from a mixture of C6 and C3 rings to predominantly a complex with a C3 morphology (Thirlway et al., 2004). During the preparation of this manuscript, the crystal structure of the Bacillus stearothermophilus DnaB complexed to the p16 helicase-binding domain (HBD) of the primase DnaG was published (Bailey et al., 2007). It confirmed the C3 conformation as a consequence of stabilizing a threefold conformation of the NTDs (N-terminal domains), which form a characteristic ‘trimer of dimers’ collar seated on top of a more loosely packed ring of the CTDs (C-terminal domains), rather than the result of characteristic interactions of three of the NTDs (one from each dimer) with the CTD of neighbouring monomers within the three dimeric units, as was suggested before for the Escherichia coli DnaB (Yang et al., 2002). The functional significance of this conformational C6 to C3 shift is not clear at present but may represent the stimulated more processive DnaB–DnaG complex in vivo.
The p16-HBD of the primase mediates its interaction with the helicase (Tougu and Marians, 1996b; Bird et al., 2000; Bailey et al., 2007). It consists of two separate helical subdomains, a six-helix bundle (subdomain, C1), essential for stimulation of DnaB activity, and a helical hairpin (subdomain, C2) that binds to DnaB (Oakley et al., 2005; Syson et al., 2005; Su et al., 2006). A three-helical fragment (3HF) comprising the C2 subdomain and the last helix of the C1 subdomain of the B. stearothermophilus p16 binds to DnaB (Syson et al., 2005), while the single-amino-acid mutations Q576A and K580A in the last nine C-terminal residues of E. coli DnaG result in defective DnaB binding (Tougu and Marians, 1996b). However, the 3HF does not modulate the activity of DnaB, suggesting that the C1 subdomain of p16 is also required for modulation of helicase activity in the primosome (Syson et al., 2005). The crystal structure of the DnaB–p16 complex revealed that although both the C1 and C2 subdomains interact with the NTDs in the inner and outer faces of the ring respectively, the C2 subdomains are packed more tightly and together with the last helix of the C1 subdomains make up most of the interaction interface with the helicase, thus explaining the formation of a stable complex of 3HF with DnaB (Bailey et al., 2007).
The crystal and nuclear magnetic resonance (NMR) structures of the E. coli (Oakley et al., 2005; Su et al., 2006) and B. stearothermophilus (Syson et al., 2005) p16 domain of the primase revealed a unique and surprising structural homology with the p17 domain of the helicase (Fass et al., 1999; Weigelt et al., 1999), despite their poor overall sequence similarity. Although p17 appears to be essential for helicase activity, because the p33 domain of B. stearothermophilus DnaB is helicase-defective despite its good ATPase activity (Bird et al., 2000), a p16–p33 chimeric protein restored the helicase activity of p33. Therefore, p16 can effectively replace p17, thus directly supporting the notion of functional homology between these two domains (Chintakayala et al., 2007).
A spatially conserved and functionally equivalent network of surface residues in the C1 subdomain of p16 and in the p17 domain of DnaB could be significant for the mutual functional modulation of the DnaB and DnaG proteins in the primosome (Soultanas, 2005). Using a combination of site-directed mutagenesis and biochemical analysis, we interrogated the roles of 10 amino acid residues from this conserved network in DnaG and studied the effects of these mutations on the primase activity and on its ability to modulate the ATPase and helicase activities of DnaB. We found that the Y548A and R555A mutations result in defects in its ability to fully stimulate the ATPase and helicase activity of DnaB. The Y548 appears to participate directly in the interaction with DnaB, whereas R555 appears to be acting allosterically. The E464G and H494A mutations have reduced ability to stimulate the DNA-independent ATPase activity of DnaB. H494A also failed to fully stimulate DNA-dependent ATPase activity, whereas E464G was able to stimulate the DNA-dependent activity of DnaB. Surprisingly, despite the fact that the ability of H494A to stimulate the ATPase activity of DnaB was compromised, it still stimulated the helicase activity. The R495A mutation compromised its ability to stimulate the helicase activity but not the ATPase activity of DnaB. With the exception of E464G, all the other mutants retained the ability to synthesize primers, in a manner comparable to native DnaG, in the presence and absence of DnaB. The E464G mutation enhanced primase activity in the presence or absence of DnaB. The E483A, R484A, E506A, D512A and E530A mutations had no effect on the ability of DnaG to stimulate the activities of DnaB or to synthesize primers in the presence and absence of DnaB. Overall, our data provide evidence that some residues from the proposed conserved network (Y548, R555, E464, H494 and R495) are indeed involved in a complex modulation of the primosome function. Their structure/function contributions are discussed based upon the recent crystal structure of the DnaB–p16 complex (Bailey et al., 2007).
Results
The E464G mutation enhances the primase activity of DnaG
Several residues are spatially conserved between the p16-HBD of DnaG and the structurally homologous N-terminal p17 domain of DnaB (Soultanas, 2005). These residues could potentially form a putative functional network that may modulate the activity of the primosome. In order to investigate this notion, we have targeted 10 of these residues by site-directed mutagenesis and studied the in vitro effects of these mutations on the primase, ATPase and helicase activities of the primosome. We first optimized the conditions for the primase activity in the presence and absence of the helicase, using an in vitro primer-synthesis spectrophotometric assay (Koepsell et al., 2004). The B. stearothermophilus DnaG exhibited optimal primase activity at 50–56°C, synthesizing well-defined primers in the absence of DnaB (Fig. 1A). In the presence of DnaB, the size distribution of the primers was altered but not the amount of primers synthesized (Fig. 1B). A range of smaller primers were synthesized consistent with previous data, showing that DnaB modulates the size of primers synthesized by DnaG, and that mutations on DnaB can compromise this modulatory effect (Thirlway and Soultanas, 2006). Modulation of primer length by DnaB appears to be a general feature in a variety of bacterial primases (M.A. Larson, unpubl. data).
Fig. 1.

The primase activity of B. stearothermophilus DnaG.
A. The effect of temperature on the primase activity of DnaG. Reactions were carried out with 1.5 μM DnaG and the CTA-containing ssDNA template (2 μM), as described in Experimental procedures. Primers eluted at 8.8 min and ssDNA templates eluted at 13 min. The optimum temperature for the primase activity is 50–56°C.
B. The effect of DnaB on the primase activity of DnaG. Reactions were carried with 360 nM DnaG and as the concentration of DnaB increased 20 nM (hexamer), a range of smaller primers were synthesized by DnaG, compared with the larger well-defined primers synthesized in the absence of DnaB.
With the exception of E464G, all the mutations had no effect on the primase activity and on the ability of DnaG to be modulated by the helicase, DnaB. More specifically, in the presence and absence of DnaB, the amount and size of primers synthesized by these mutant DnaG proteins were similar to primer synthesis by native DnaG (Fig. 2). The E464G mutation resulted in a protein with increased primase activity both in the presence or absence of DnaB. The E464G protein was also able to synthesize primers larger than the ones synthesized by native DnaG. These data show that despite the fact that E464 is in the C1 subdomain of p16, which is distinct from the polymerization domain p37 (Bird et al., 2000; Chintakayala et al., 2007), it can still affect primase activity when mutated. Therefore, the p16 domain does not only act as the helicase interaction element but may also have an ‘auto-regulatory’ effect on the polymerization activity of the p37 domain in the native protein. Interestingly, the E464A mutation resulted in an insoluble DnaG protein, suggesting that the nature of the side-chain at this position affects the overall conformation of the protein (data not shown).
Fig. 2.
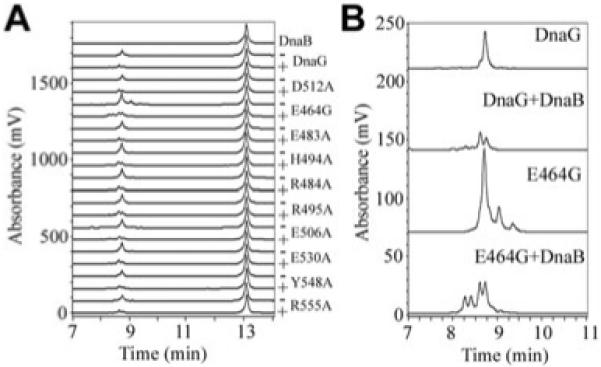
The effect of mutations on primase activity.
A. The primase activities of mutant DnaG proteins were assessed in the presence (plus) or absence (minus) of DnaB. All reactions were carried out with 540 nM DnaG in the presence or absence of 30 nM DnaB (hexamer), as described in Experimental procedures. A control chromatograph in the presence of DnaB without DnaG is shown at the top and the ssDNA templates used in the reactions are shown eluting at 13 min. With the exception of E464G, all the other mutant primases exhibited primase activities equivalent to the wild-type DnaG in the presence or absence of DnaB.
B. Enlarged chromatographs for the E464G primase activity in the presence or absence of DnaB, as indicated. Control chromatographs for the primase activity of the wild-type DnaG, in the presence or absence of DnaB are also shown for comparison. E464G exhibited a distinct stimulation of primase activity either in the presence or absence of DnaB, whereas its activity was modulated by DnaB in a manner analogous to that of the wild-type DnaG.
Primase mutations that affect its ability to stimulate the ATPase and helicase activities of DnaB
The effects of DnaG mutations on the DNA-independent and DNA-dependent ATPase activity of DnaB were examined by steady-state comparative in vitro spectrophotometric ATPase assays, in the presence or absence of native or mutant DnaG proteins and in the presence or absence of 15 molar excess of a single-stranded 42mer oligonucleotide. The DnaB ATPase profiles, turnover rate versus ATP concentration (0–1.20 mM), were compared. Over this range of ATP concentrations, the steady-state ATPase activity of DnaB exhibits Michaelis–Menten behaviour, whereas higher ATP concentrations do not display Michaelis-Menten kinetics, because of negative cooperativity (Bird et al., 2000; Soultanas and Wigley, 2002). In agreement with previous studies (Bird et al., 2000; Soultanas and Wigley, 2002), native DnaG stimulated the DNA-independent and DNA-dependent ATPase activity of DnaB (Fig. 3A and Fig. S1A). Two mutations (Y548A and R555A) resulted in mutant proteins that, despite their intact primase activities (Fig. 2), were unable to stimulate the ATPase activity of DnaB (Fig. 3A).
Fig. 3.

Primase mutations that affect its stimulatory effect on the helicase. The effects of primase mutations on its ability to modulate the helicase ATPase and helicase activities were examined with ATPase and helicase assays, as indicated. The Y548A and R555A mutant proteins exhibited defective ability to stimulate the ATPase (A) and helicase activities of DnaB (B), compared with native DnaG. The R495A mutant protein fully stimulated the ATPase activity (C) but exhibited defective ability to stimulate the helicase activity (D) of DnaB, compared with native DnaG. ATPase reactions were carried out with 30 nM hexamers of DnaB in the presence or absence of 90 nM R495A, while helicase reactions were carried out with 37.5 nM hexamers of DnaB in the presence or absence of 675 nM R495A, as described in Experimental procedures.
The ability to stimulate the helicase activity of DnaB was also examined by in vitro helicase assays designed to determine the displacement of a small oligonucleotide annealed to ssM13mp18 DNA. Native DnaG has been shown before to stimulate the helicase activity of DnaB (Bird et al., 2000; Soultanas and Wigley, 2002). Both mutant primases exhibited significantly compromised, but not abolished, ability to stimulate the DnaB helicase compared with native DnaG (Fig. 3B). These data suggest that both Y548 and R555 are involved in the modulation of primosome activity.
A primase mutation that affects its ability to stimulate the helicase but not the ATPase activity of DnaB
The R495A mutant fully stimulated the DNA-independent and DNA-dependent ATPase activity of DnaB (Fig. 3C and Fig. S1C) but was somewhat defective in stimulating the helicase activity (Fig. 3D). As we show below, the R495A mutation has not affected the affinity for binding to DnaB, and thus has instead uncoupled the ATPase and helicase activities. Therefore, the reduction of the stimulation of the helicase activity must be mediated via an allosteric mechanism that is uncoupled from the ATPase activity of DnaB.
Conserved residues that are not part of the structural/functional interaction network of the primosome
By comparison with the mutations described above, the E483A, R484A, E506A, D512A and E530A mutations did not affect the ability of the primase to stimulate the DNA-independent or DNA-dependent ATPase and helicase activities of DnaB (Fig. 4 and Fig. S1D), suggesting that these residues are not part of the functional network of residues that modulate the activity of the primosome. A mutant is considered to exhibit significant reduction in its stimulatory effect when this reduction is at least 20% less than the stimulatory activity of the native protein.
Fig. 4.

Primase mutations that do not affect its ability to stimulate the ATPase and helicase activities of DnaB. The effects of the primase E483A, R484A, E506A, D512A and E530A mutant proteins on the ATPase (A) and helicase (B) activities of DnaB were examined with ATPase and helicase assays, as indicated. All mutations did not affect the ability of the primase to stimulate the ATPase and helicase activities of DnaB. ATPase reactions were carried out with 30 nM hexamers of DnaB in the presence or absence of 90 nM mutant DnaG proteins, while helicase reactions were carried out with 37.5 nM hexamers of DnaB in the presence or absence of 675 nM mutant DnaG proteins, as described in Experimental procedures.
Binding of the DnaG mutants to DnaB
Both Y548A and R555A mutant proteins exhibited clear defects in their ability to stimulate the helicase and ATPase activities of DnaB. These functional defects could be the direct result of a defective interaction with DnaB or alternatively because of an allosteric effect via the interaction network of residues that mediate the functional stimulation of DnaB by DnaG. The fact that the B. stearothermophilus DnaB–DnaG complex is stable in vitro at room temperature allows us to test binding defects directly by analytical gel filtration. By mixing DnaB with excess DnaG and resolving the mixture through an appropriate gel filtration column, a stable DnaB–DnaG complex can be separated from the excess DnaG (Bird et al., 2000; Thirlway et al., 2004). Using this assay, the interaction of native DnaG, Y548A and R555A proteins with DnaB was examined with a Superdex S200 10/30 gel filtration column (Fig. 5).
Fig. 5.
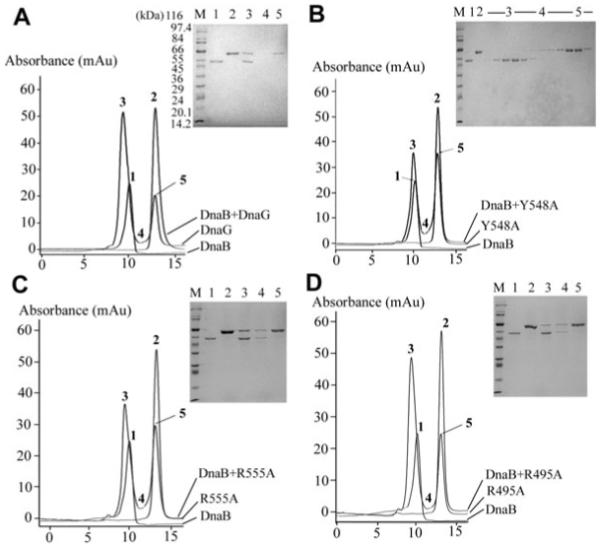
Examination of the primase–helicase interactions in vitro by analytical gel filtration. A mixture of DnaB 0.725 μM (hexamers) and 2.175 μM native DnaG (monomers) was resolved by analytical gel filtration through a Superdex S200 10/30 column as described in Experimental procedures (A). DnaB elutes as a hexamer (303 kDa) while DnaG elutes as a monomer (67 kDa). Samples were taken from the positions of the elution profiles indicated by numbers 1–5 and analysed by SDS-PAGE. Control peaks 1 and 2 show DnaB and DnaG respectively. Peak 3 shows the DnaB-DnaG complex, whereas peak 5 indicates the excess DnaG that did not bind to DnaB. In all the experiments, the numbers on the peaks correspond to the numbers of the lanes in the SDS-PAGE gel. The same experiment was repeated with the Y548A (B), R555A (C) and R495A (D) mutant DnaG proteins. R495A and R555A formed a stable complex with DnaB comparable to that formed by the wild-type DnaG. The amount of complex formed between Y548A and DnaB was much reduced compared with that formed by the wild-type DnaG, indicating a defect in the stability of the complex. For the defective Y548A mutant protein, more extensive SDS-PAGE analysis of all the fractions is shown. Molecular weight markers (lanes marked M) are shown in all gels.
Control elution profiles with just DnaB or DnaG were obtained first, followed by the elution profile of the DnaB plus DnaG mixture (Fig. 5A). Samples from all the peaks (1, 2, 3, 5) and the region between the peaks (labelled 4) were analysed by SDS-PAGE. As expected, DnaG was found with DnaB in peak 3, indicating that it was complexed with DnaB and separated from the excess DnaG in peak 5. The presence of DnaB or DnaG in the control peaks 1 and 2 respectively, was also verified by SDS-PAGE (Fig. 5A).
The same experiments were repeated with the Y548A protein (Fig. 5B). SDS-PAGE analysis of samples from all the fractions of the Y548A + DnaB elution profile revealed no binding to DnaB. There was no Y548A detected in peak 3; it was only present in peak 5 well separated from the DnaB in peak 3. In similar experiments, R555A was found to interact normally with DnaB (stable complex shown by SDS-PAGE in peak 3) in a manner comparable to the native DnaG (compare A and C in Fig. 5). For comparison, the R495A protein, which retained its ability to stimulate the ATPase activity but was compromised in its ability to stimulate the helicase activity of DnaB, also interacted with DnaB in a manner comparable to the native DnaG (Fig. 5D). All the other DnaG mutant proteins formed a stable complex with DnaB (data not shown).
The failure to detect the Y548A–DnaB complex by gel filtration could be a consequence of no interaction or a weaker transient interaction that cannot be detected by gel filtration. Therefore, the Y548A–DnaB interaction was examined quantitatively using isothermal titration calorimetry (ITC). Under our experimental conditions at 25°C, native DnaG was found to bind to DnaB in an endothermic reaction (ΔH = 1.782 × 104 cal mol−1 and ΔS = 97.1 cal mol−1deg−1) with an association constant Ka = 1.42 × 108 M−1, a dissociation constant Kd = 7.04 nM and a molar ratio N = 0.287, close, but not identical, to the expected molar ratio of 0.5 corresponding to three DnaG molecules per DnaB hexamer (Fig. 6A). By comparison, Y548A interacted with DnaB with an association constant Ka = 1.2 × 107 M−1, a dissociation constant Kd = 83.3 nM and with a stoichiometry of 0.509 (Fig. 6B).
Fig. 6.
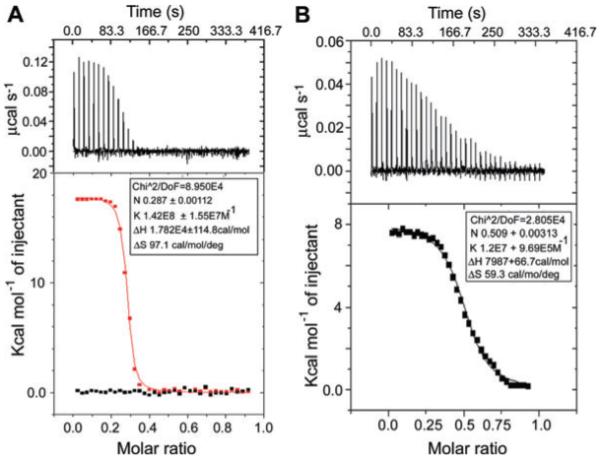
Examination of the DnaG–DnaB and Y548A–DnaB interactions in vitro by ITC.
A. The top panel shows the raw data for the DnaG–DnaB interaction in terms of μcal s−1 plotted against time in minutes, after the integration baseline has been subtracted. The bottom graph shows normalized integration data in terms of kcal mol−1 of injectant (black is buffer, red is DnaG) plotted against molar ratio. The two x axes are linked so that the integrated area for each peak appears directly below the corresponding peak in the raw data. The data fitted well into a ‘one set of sites’ built-in fitting model (red curve). The interaction is endothermic and the fitting parameters are displayed in the inset.
B. The top panel shows the raw data for the Y548A–DnaB interaction in terms of μcal s−1 plotted against time in minutes, after the integration baseline has been subtracted. The bottom graph shows normalized integration data in terms of kcal mol−1 of injectant (Y548A) plotted against molar ratio. The two x axes are linked so that the integrated area for each peak appears directly below the corresponding peak in the raw data. The data fitted well into a ‘one set of sites’ built-in fitting model (black curve). The interaction is endothermic and the fitting parameters are displayed in the inset.
Together, the gel filtration and ITC data suggest that binding of Y548A to DnaB is defective. Y548A has an order of magnitude weaker affinity than native DnaG for DnaB. The functional defects observed with Y548A can be simply explained by its defective binding to DnaB. Therefore, Y548 participates directly in the interaction with DnaB, whereas R555 is likely to be part of an allosteric network of residues that mediate the stimulatory effects of DnaG on the activity of DnaB.
Primase mutations that affect its ability to stimulate the ATPase but not the helicase activity of DnaB
The E464G and H494A mutations produced proteins, with somewhat defective ability to stimulate the DNA-independent ATPase activity (Fig. 7A), but differed in their effect on the DNA-dependent ATPase activity of DnaB. H494A was unable to stimulate, while E464G stimulated the DNA-dependent ATPase activity of DnaB (Fig. S1B). As expected, E464G fully stimulated the DnaB helicase activity but, unexpectedly, H494A also retained its ability to stimulate helicase activity despite its failure to stimulate both the DNA-dependent and -independent ATPase activities (Fig. 7B and Fig. S1B). One possibility may be that these mutations have weakened the affinity of DnaG–DnaB interactions but under our helicase assay conditions, with large excess of DnaG, this effect may be masked. In order to address this point, we carried out ITC experiments with both H494A and E464G to determine their affinities for DnaB. The ITC data revealed that both mutations did not affect the affinity of the primase for DnaB (Fig. 7C and D). Under our experimental conditions at 25°C, E464G and H494A were found to bind to DnaB in an endothermic reaction both with an association constant Ka = 1.18 × 108 M−1, and dissociation constant Kd = 8.5 nM, comparable to the parameters obtained for native DnaG (compare Figs 6A and 7C, D). We conclude that the functional effects observed with these mutants are not because of reduced binding affinity for DnaB but instead because of allosteric effects.
Fig. 7.
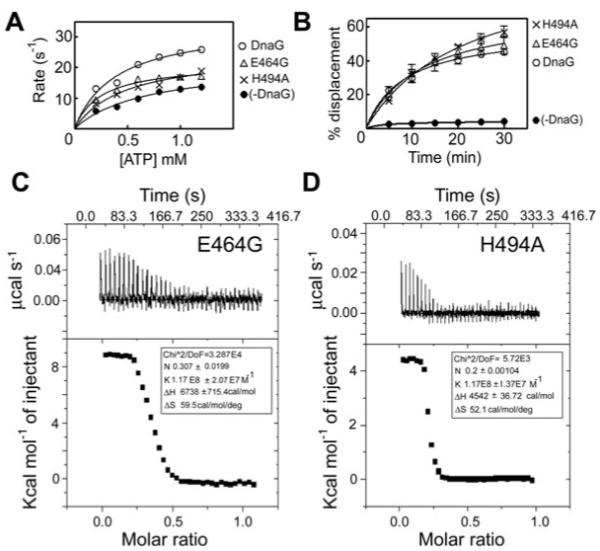
The E464G and H494A primase mutations affect the stimulation of the DnaB ATPase activity but do not affect the stimulation of the helicase activity and the affinity of DnaG for DnaB. The E464G and H494A mutant proteins exhibited defective ability to stimulate the ATPase (A) but not the helicase activity of DnaB (B), compared with native DnaG. ATPase reactions were carried out with 30 nM hexamers of DnaB in the presence or absence of 90 nM R495A, while helicase reactions were carried out with 37.5 nM hexamers of DnaB in the presence or absence of 675 nM E464G or H494A, as described in Experimental procedures. Both mutant proteins exhibited binding affinities to DnaB comparable to that of native DnaG, as shown by ITC experiments. C and D show the ITC data for E464G and H494A respectively. In both cases, the top panels show the raw data for the primase–helicase interaction in terms of μcal s−1 plotted against time in minutes, after the integration baseline has been subtracted. The bottom graphs show normalized integration data in terms of kcal mol−1 of injectant plotted against molar ratio. The two x axes are linked so that the integrated area for each peak appears directly below the corresponding peak in the raw data. The data fitted well into a ‘one set of sites’ built-in fitting model. The interaction is endothermic and the fitting parameters are displayed in the inset.
Discussion
The activity of the primosome (DnaB–DnaG) at the replication fork is modulated by dynamic interactions of the helicase and primase with other components of the replisome (Johnson and O’Donnell, 2005). For example, when the helicase is coupled to DNA synthesis through an interaction with τ, its unwinding increases to a rate that is comparable to the speed of the holoenzyme (Kim et al., 1996). The primer handoff from the primase to the DNA polymerase is accelerated by the χ subunit of the clamp loader, which competes with the primase for binding to SSB (Yuzhakov et al., 1999). In addition to interactions with other replisomal proteins, the function of the primosome is primarily regulated by the interaction of its two components, the helicase and the primase. Both proteins are engaged in mutual modulation of their activities when in complex. This interaction is mediated via the C-terminal p16 domain of the primase (Tougu and Marians, 1996b; Bird et al., 2000). A conserved putative network of amino acid residues may be important for allosteric modulation of the helicase activity (Soultanas, 2005; Syson et al., 2005).
Our data provide evidence that four residues (R555, E464, H494 and R495) from this network are involved in allosteric modulation of the helicase activity, one of these residues (E464) is also important in modulation of the primase activity and a fifth residue (Y548) participates directly in the interaction with DnaB. R555, E464, H494 and R495 form a patch on the same face of the C1 subdomain of p16 (Fig. 8A). This subdomain has been assigned as a functional modulator of helicase activity in the primosome (Syson et al., 2005). A peptide fragment (3HF) comprising the hairpin of C2 and the last helix of C1 exhibited strong binding to DnaB (Syson et al., 2005). Y548 is situated in the middle of the last helix of C1 and given that our data reveal that it participates directly in the interaction with the helicase, this is good evidence that C1 of DnaG is not only a functional modulator of helicase activity but that its last helix in the vicinity of C2 also participates structurally in the interaction with DnaB. Indeed, the crystal structure of the DnaB–p16 complex (Bailey et al., 2007) published during the preparation of this manuscript confirmed this and revealed that Y548 is at the interaction interface between the NTDs of an interacting DnaB molecule and p16 (Fig. 8A and B). The side-chain of Y548 appears to be in a hydrophobic pocket comprising residues L545, I549, L466, L467 from p16, all within a distance of less than 4 Å, and the phenolic hydroxyl group is oriented towards (and is within 3.53 Å) the amide oxygen of N101 in the NTD of an interacting DnaB molecule (Fig. 8B). Our data suggest that this is an important stabilizing factor in the DnaB–p16 interaction. A comparison of the binding constants of DnaG and Y548A to DnaB revealed that the latter has an order of magnitude less affinity, highlighting the important contribution of Y548 to the stability of the complex.
Fig. 8.
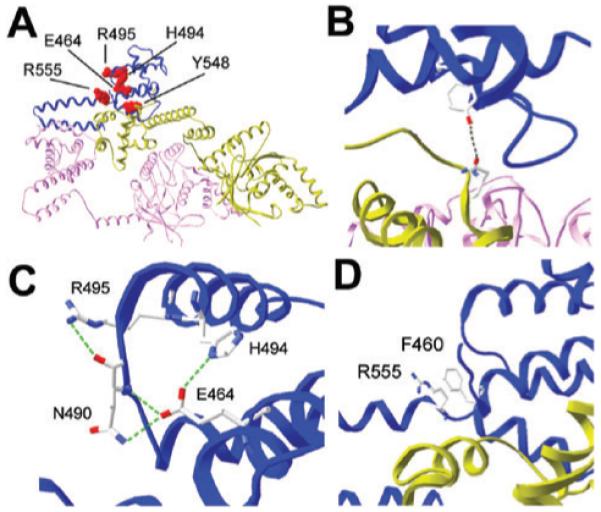
A. The structure of the B. stearothermophilus DnaB–p16 complex (pdb 2R6A). Two DnaB monomers, coloured pink and green, interact with one p16 molecule, coloured blue. The same colour profile is maintained in all panels. Three units of two DnaB molecules plus one p16 molecule are arranged in a ring structure. The other two units are not shown for clarity. The axis through the central hole of the ring is approximately perpendicular to the image shown. The five mutated p16 residues that exhibited functional defect are indicated in red and labelled.
B. The Y548A mutant exhibited defects in binding to DnaB and stimulating its ATPase and helicase activities. The Y548 residue of p16 is within 3.53 Å from the N101 residue in the interacting NTD of a DnaB molecule, as indicated.
C. E464, H494 and R495 together with N490 form a network of strong H-bonds. One strong H-bond (2.71 Å) forms between the imidazole ring of H494 and the carboxylic acid of E464. The carboxylic acid of E464 also forms strong H-bonds with the oxygen of the backbone amide of N490 (2.71 Å) and with the side-chain amide of N490 (2.76 Å). Another strong H-bond (3.12 Å) forms between the guanidinium of R495 and the oxygen of the backbone amide of N490. This H-bond network is characteristic of the DnaB–p16 complex and does not exist in the NMR structure of p16 (pdb 1Z8S), see Fig. S2.
D. R555 is close (~3.45 Å) to F460 from helix H1. The guanidinium and aromatic groups stack perfectly against each other and may participate in planar cation-π interactions that position helix H1 in an optimal spatial position relative to the C2 subdomain.
The E464G mutation has increased the primase activity both in the presence and absence of DnaB. E464 is situated near the N-terminus of the p16 domain which is distinct from the central p37 polymerization domain (Bird et al., 2000; Chintakayala et al., 2007). In the E. coli DnaG, a core fragment comprising residues 111–433 (DnaG-RNAP) retains the ability to transcribe RNA in vitro, although with reduced activity (Keck et al., 2000). This observation suggests that DnaG-RNAP retains some DNA-binding activity despite the fact that it lacks the Zinc-binding domain (ZBD) (Keck et al., 2000; Pan and Wigley, 2000) which is believed to interact in trans with the DNA during primer synthesis in vivo (Corn et al., 2005). The reduced polymerase activity of the DnaG-RNAP can be attributed to reduced affinity for DNA and it will be interesting to compare the primase activities of the ZBD-RNAP truncated protein and the native DnaG. Our data with the E464G mutant suggest that p16 is not only a structural element that mediates solely the interaction and the effects of the helicase on the activity of the primase, but may also act to auto-repress the polymerization activity of the DnaG-RNAP in the absence of the helicase. The E464G mutation has partly abolished this auto-regulatory effect. Such an effect is likely to be mediated allosterically, via an interaction between p16 and DnaG-RNAP. The fact that the E464A mutation results in an insoluble DnaG protein (data not shown) also suggests that the nature of the amino acid at this position affects the conformation of the protein.
The E464G and H494A mutations have also affected the ability of DnaG to stimulate the DNA-independent ATPase activity of DnaB. However, E464G was able to stimulate the DNA-dependent ATPase activity, in contrast to H494A that failed to do so. The presence of DNA alleviates the effect of the E464G mutation but not that of the H494A mutation. The affinities of both mutant primases for DnaB were comparable to the affinity of the native primase, suggesting that these functional defects are not due to defective binding to DnaB. These residues are found in different helices, E464 in helix H1 and H494 in helix H3 of p16, but they are spatially very close to each other in the DnaB–p16 complex. A strong H-bond (2.71 Å) is formed between the carboxylic acid of E464 and the imidazole ring of H494 (Fig. 8C). E464 is also spatially very close to N490 which is located in a loop that connects helix H2 and helix H3. The carboxylic acid of E464 also participates in strong H-bonds with the oxygen of the backbone amide (2.71 Å) and the side-chain imidazole (2.76 Å) of N490 (Fig. 8C). Mutations at the E464 or H494 positions would disrupt these H-bonds and could result in subtle repositioning of helices H1, H2 and H3, as well as the loop that connects H2 and H3 that could in turn mediate allosteric functional effects in the primosome. Interestingly, R495 also participates in this H-bond network. The guanidinium group of R495 forms a strong H-bond (3.12 Å) with the oxygen of the backbone amide of N490 (Fig. 8C). R495A interacts with DnaB normally but fails to stimulate helicase activity, despite the fact that it stimulates ATPase activity. It appears that the modulation of the two activities has been uncoupled in the R495A–DnaB complex. This coupling/uncoupling feature of the ATPase and helicase activities appears to be evident in the effects of the H494A mutation. Unexpectedly, the H494A mutation did not affect the helicase activity of DnaB as one would have expected from the reduced stimulation of the ATPase activity. One speculative explanation might be that the two activities are coupled more efficiently by the interaction of DnaB with H494A. By contrast, E464G stimulated the helicase activity as expected from the stimulation of the DNA-dependent ATPase activity.
Although in the DnaB–p16 complex, E464, H494, N490 and R495 are spatially close and form a network of strong H-bonds, in the NMR structure of p16 they are more than 6 Å away and do not appear to interact (Fig. S2). Clearly, these interactions are a feature of the complex. Our data indicate that they form an important H-bond network involved in the allosteric functional modulation of the primosome by determining the relative subtle repositioning of the H1, H2 and H3 helices and the loop between H2 and H3.
The role of R555 is not immediately obvious but an interesting observation is that the side-chains of R555 and F460 from helix H1 are close (~3.5 Å) and stack parallel against each other in the DnaB–p16 complex (Fig. 8D). It appears that the guanidinium group of R555 participates in planar π-facial hydrogen bonding with the aromatic ring of F460, thus helping to maintain an optimal spatial positioning of helix H1 in the complex. Indeed, in the NMR structure of p16, these two residues are too far away from each other to participate in an interaction (Fig. S2). Therefore, their proximity is a feature of p16 when in complex with DnaB. Cation-π interactions participate in inter- and intra-molecular protein interactions. In a recent study, 53% of the cation-π interactions were found to involve planar stacking of the guanidinium group and an aromatic ring, whereas 26% and 21% were found to be of the oblique or the orthogonal type respectively (Crowley and Golovin, 2005).
Experimental procedures
Site-directed mutagenesis
Site-directed mutagenesis was carried out using the QuikChange II-E Site-Directed Mutagenesis Kit (Stratagene), with appropriate mutagenic oligonucleotides (Fig. S3). All the correct mutations and the absence of spurious mutations were verified by sequencing. The numbering of mutated residues refers to the native DnaG. The equivalent numbers relative to the p16 sequence are shown in Fig. S4.
Protein purifications
Native DnaB was expressed in E. coli BL21 (DE3) and purified using a combination of Blue Sepharose, MonoQ, Hi-Trap heparin and Superdex S-200 chromatography, following the same protocol as described previously (Bird et al., 2000). Native DnaG and its mutant proteins were also expressed in E. coli BL21 (DE3) and purified using a combination of Hi-Trap heparin, Source-Q and Superdex S-75 chromatography, as described previously (Pan et al., 1999; Bird et al., 2000). All proteins were > 98% pure as assessed by SDS-PAGE analysis (an example is shown in Fig. S5). Protein concentrations were determined spectrophotometrically using specific absorbance values at 280 nm of 0.4215 and 0.6685 for native DnaB and DnaG respectively. Specific absorbance values for all proteins were calculated using the formula A280 (1 mg ml−1) = (5690 W + 1280Y + 120C)/M where W, Y and C are the numbers of Trp, Tyr and Cys residues in a polypeptide of mass M, and 5690, 1280 and 120 are the respective extinction coefficients for these residues (Aitken and Learmonth, 2002). For the Y548A protein, the specific absorbance value was adjusted to 0.6494 to account for the loss of a Tyr residue.
Helicase assays
The DNA substrate for helicase reactions was prepared by radiolabelling the oligonucleotide 5′-GTTATTGCATGAAAGCCCGGCTGACTCTAGAGGATCCCCGGGTACGTTATTGCATGAAAGCCCGGCTG-3′ (underlined bases are complementary to a unique sequence on M13mp18) at the 5′ end using [γ-32P]-ATP and T4 polynucleotide kinase (New England Biolabs) and annealing to ssM13mp18 to produce a 3′-5′-tailed DNA substrate. One molecule of DNA substrate is defined as one molecule of ssM13mp18 with one molecule of oligonucleotide annealed to it. Helicase reactions were carried out at 37°C in 50 mM Tris pH 7.4, 50 mM NaCl, 12.5 mM MgCl2, 2.5 mM ATP, 1 mM DTT, 0.5 nM DNA substrate and the appropriate proteins (37.5 nM hexamers of DnaB and 675 nM monomers of DnaG), as indicated for each experiment.
In mixing experiments, the appropriate proteins were mixed separately in the absence of the other reaction components and incubated at room temperature for 15 min to enhance complex formation. Reactions were initiated by the addition of 2.5 mM ATP. Samples (20 μl) were removed at appropriate time intervals, the reaction was terminated by the addition of 5 ml of stop buffer (0.4% w/v SDS, 40 mM EDTA, 8% v/v glycerol, 0.1% w/v bromophenol blue) and the samples were stored briefly at 4°C, prior to electrophoresis through a non-denaturing 10% v/v polyacrylamide gel. Gels were dried under vacuum. Imaging and quantitative analysis were carried out by a Molecular Imager FX (Bio-Rad) and associated software. All experiments were carried out twice and data were plotted as the average percentage of radiolabelled oligonucleotide displaced versus time.
ATPase assays
The DNA-independent and DNA-dependent steady-state ATPase activity of DnaB in the presence or absence of native or mutant DnaG proteins were assayed by monitoring the ATP-dependent oxidation of NADH to NAD at 340 nm, as described previously (Bird et al., 2000). Unless otherwise stated, all experiments were carried out in the same buffer used for the helicase assays, using 30 nM hexamers DnaB, in the presence or absence of 90 nM native or mutant DnaG monomers and in the presence or absence of 450 nM of a 42mer ssDNA oligonucleotide (5′-ATGGTTATTGCATGAAAGCCCGGCTGACTCTAGAGGATCCCC-3′), with varying concentrations of ATP (0.2–1.2 mM). In mixing experiments, the appropriate proteins were mixed separately in the absence of other reaction components and incubated at room temperature for 15 min to enhance complex formation. All experiments were carried out twice. Average turnover numbers were plotted as a function of ATP concentration.
Primase assays
Primase activity was determined using a primer synthesis assay. The primer production reactions were incubated at 56°C in nuclease-free buffer (50 mM HEPES, pH 7.5, 100 mM potassium glutamate and 10 mM DTT) containing 10 mM magnesium acetate, 400 μM of each ribonucleotide, 2 μM of a 23mer ssDNA template that contained the CTA initiation sequence (5′-CAGACACACACACACTACACACAC3′) (Integrated DNA Technologies, Coralville, IA) and primase either without or with helicase, as indicated. Following a 1 h incubation, RNA primer synthesis was quenched by desalting the reaction in a Sephadex G-25 spin column. The primase reaction products were then subjected to thermally denaturing HPLC, as previously described (Koepsell et al., 2004). The nucleic acids were separated by their size and hydrophobicity and quantified spectrophotometrically at 260 nm. Control single-stranded oligoribonucleotides and deoxyribonucleotides were used to correlate retention time on the column with the sequence of the nucleic acid species. All experiments were performed a minimum of two times.
Analytical gel filtration
Analytical gel filtration was carried out using 0.725 μM DnaB (hexamers) mixed with 2.175 μM DnaG (monomers), or its mutants, in 50 mM Tris pH 7.4, 1 mM EDTA, 1 mM DTT, 100 mM NaCl (TED/100). The protein mixture was incubated at room temperature for 10 min and then resolved through a Superdex S200 10/30 gel filtration column (GE Healthcare), equilibrated in TED/100. The flow rate was constant at 0.5 ml min−1 and 0.5 ml of fractions were collected while 20 μl of samples from these fractions were analysed by SDS-PAGE.
Isothermal titration calorimetry
Isothermal titration calorimetry was carried out using a VP-ITC instrument (MicroCal, LLC) at 25°C. A solution of DnaB (1.424 ml, 6.14 μM) in 50 mM Tris pH 7.4, 1 mM EDTA, 100 NaCl was used in the sample cell, while a solution of native DnaG (0.307 ml, 27 μM) or Y548A (0.307 ml, 26.4 μM) in the same buffer was used in the syringe. Thirty-five injections of 8 μl each of DnaG or Y548A were carried out and heat changes were recorded, using the VPViewer user interface. Data were analysed with the Origin v.7.0 software, by fitting into a ‘one set of sites’ built-in fitting model. An identical control experiment with injections of buffer was also carried out to obtain the baseline that was subsequently subtracted from the actual experiments. Similar experiments were carried out for the Y464G and H494A mutant primases.
Supplementary Material
Acknowledgements
We thank Nick Hopcroft for help with Fig. S2 and Jed Long for help with the ITC. This work was supported by a BBSRC Grant (BB/E004717/1) to P.S. and in part by a Grant from the Department of Defence, Defence Advanced Research Program Agency (W911NF0510275) to S.H.H.
Footnotes
Supplementary material
This material is available as part of the online article from: http://www.blackwell-synergy.com/doi/abs/10.1111/j.1365-2958.2008.06155.x
(This link will take you to the article abstract).
Please note: Blackwell Publishing is not responsible for the content or functionality of any supplementary materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Aitken A, Learmonth MP. Protein determination by UV absorption. In: Walker JM, editor. The Protein Protocols Handbook. 2nd edn. Human Press; Totowa, NJ: 2002. pp. 3–6. [Google Scholar]
- Bailey S, Eliason WK, Steitz TA. Structure of hexameric DnaB helicase and its complex with a domain of DnaG primase. Science. 2007;318:459–463. doi: 10.1126/science.1147353. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya S, Griep MA. DnaB helicase affects the initiation specifity of Escherichia coli primase on single-stranded DNA templates. Biochemistry. 2000;39:745–752. doi: 10.1021/bi991555d. [DOI] [PubMed] [Google Scholar]
- Bird LE, Pan H, Soultanas P, Wigley DB. Mapping protein–protein interactions within a stable complex of DNA primase and DnaB helicase from Bacillus stearothermophilus. Biochemistry. 2000;39:171–182. doi: 10.1021/bi9918801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bujalowski W, Klonowska MM, Jezewska MJ. Oligomeric structure of Escherichia coli primary replicative helicase DnaB protein. J Biol Chem. 1994;269:31350–31358. [PubMed] [Google Scholar]
- Chintakayala K, Larson MA, Grainger WH, Scott DJ, Griep MA, Hinrichs SH, Soultanas P. Domain swapping reveals that the C- and N-terminal domains of DnaG and DnaB, respectively, are functional homologues. Mol Microbiol. 2007;63:1629–1639. doi: 10.1111/j.1365-2958.2007.05617.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corn JE, Pease PJ, Hura GL, Berger JM. Crosstalk between primase subunits can act to regulate primer synthesis in trans. Mol Cell. 2005;200:391–401. doi: 10.1016/j.molcel.2005.09.004. [DOI] [PubMed] [Google Scholar]
- Crowley PB, Golovin A. Cation-π interactions in protein–protein interfaces. Proteins Struct Funct Bioinform. 2005;59:231–239. doi: 10.1002/prot.20417. [DOI] [PubMed] [Google Scholar]
- Fass D, Bogden CE, Berger JM. Crystal structure of the N-terminal domain of the DnaB hexameric helicase. Structure. 1999;7:691–698. doi: 10.1016/s0969-2126(99)80090-2. [DOI] [PubMed] [Google Scholar]
- Johnson A, O’Donnell M. Cellular DNA replicases: components and dynamics at the replication fork. Ann Rev Biochem. 2005;74:283–315. doi: 10.1146/annurev.biochem.73.011303.073859. [DOI] [PubMed] [Google Scholar]
- Johnson SK, Bhattacharyya S, Griep MA. DnaB helicase stimulates primer synthesis activity on short oligonucleotide templates. Biochem. 2000;39:736–744. doi: 10.1021/bi991554l. [DOI] [PubMed] [Google Scholar]
- Keck JL, Roche DD, Lynch AS, Berger JM. Structure of the RNA polymerase domain of E. coli primase. Science. 2000;287:2482–2486. doi: 10.1126/science.287.5462.2482. [DOI] [PubMed] [Google Scholar]
- Kim S, Dallmann HG, McHenry CS, Marians KJ. Coupling of a replicative polymerase and helicase: a τ–DnaB interaction mediates rapid replication fork movement. Cell. 1996;84:643–650. doi: 10.1016/s0092-8674(00)81039-9. [DOI] [PubMed] [Google Scholar]
- Koepsell SA, Bastola D, Hinrichs SH, Griep MA. Thermally denaturing high-performance liquid chromatography analysis of primase activity. Anal Biochem. 2004;332:330–336. doi: 10.1016/j.ab.2004.06.019. [DOI] [PubMed] [Google Scholar]
- Koepsell SA, Larson MA, Griep MA, Hinrichs SH. Staphylococcus aureus helicase but not Escherichia coli helicase stimulates S. aureus primase activity and maintains initiation specificity. J Bacteriol. 2006;188:4673–4680. doi: 10.1128/JB.00316-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J-B, Hite RK, Hamdan SM, Xie XS, Richardson CC, van Oijen AM. DNA primase acts as a molecular brake in DNA replication. Nature. 2006;439:621–624. doi: 10.1038/nature04317. [DOI] [PubMed] [Google Scholar]
- Lu YB, Ratnakar PVAL, Mohanty BK, Bastia D. Direct physical interaction between DnaG primase and DnaB helicase of Escherichia coli is necessary for optimal synthesis of primer RNA. Proc Nat Acad Sci USA. 1996;93:12902–12907. doi: 10.1073/pnas.93.23.12902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitkova AV, Khopde SM, Biswas SB. Mechanism and stoichiometry of interaction of DnaG primase with DnaB helicase of Escherichia coli in RNA primer synthesis. J Biol Chem. 2003;278:52253–52261. doi: 10.1074/jbc.M308956200. [DOI] [PubMed] [Google Scholar]
- Mott ML, Berger JM. DNA replication initiation: mechanisms and regulation in bacteria. Nat Rev Microbiol. 2007;5:343–354. doi: 10.1038/nrmicro1640. [DOI] [PubMed] [Google Scholar]
- Oakley AJ, Loscha KV, Schaeffer PM, Liepinsh E, Pintacuda G, Wilce MC, et al. Crystal and solution structures of the helicase-binding domain of Escherichia coli primase. J Biol Chem. 2005;280:11495–11504. doi: 10.1074/jbc.M412645200. [DOI] [PubMed] [Google Scholar]
- Pan H, Wigley DB. Structure of the zinc-binding domain of Bacillus stearothermophilus DNA primase. Structure. 2000;8:231–239. doi: 10.1016/s0969-2126(00)00101-5. [DOI] [PubMed] [Google Scholar]
- Pan H, Bird LE, Wigley DB. Cloning, expression, and purification of Bacillus stearothermophilus DNA primase and crystallization of the zinc-binding domain. Bioch Biophys Acta. 1999;1444:429–433. doi: 10.1016/s0167-4781(99)00025-1. [DOI] [PubMed] [Google Scholar]
- Patel SS, Picha KM. Structure and function of hexameric helicases. Ann Rev Biochem. 2000;69:651–697. doi: 10.1146/annurev.biochem.69.1.651. [DOI] [PubMed] [Google Scholar]
- Soultanas P. The bacterial helicase–primase interaction: a common structural/functional module. Structure. 2005;13:839–844. doi: 10.1016/j.str.2005.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soultanas P, Wigley DB. Site-directed mutagenesis rveals roles for conserved amino acid residues in the hexameric DNA helicase DnaB from Bacillus stearothermophilus. Nucleic Acids Res. 2002;30:4051–4060. doi: 10.1093/nar/gkf527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su X-C, Schaeffer PM, Loscha KV, Gan PHP, Dixon NE, Otting G. Monomeric solution structure of the helicase-binding doman of Escherichia coli DnaG primase. FEBS J. 2006;273:4997–5009. doi: 10.1111/j.1742-4658.2006.05495.x. [DOI] [PubMed] [Google Scholar]
- Syson K, Thirlway J, Hounslow AM, Soultanas P, Waltho JP. Solution structure of the helicase interaction domain of the primase DnaG: a model for helicase activation. Structure. 2005;13:609–616. doi: 10.1016/j.str.2005.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thirlway J, Soultanas P. In the B. stearothermophilus DnaB-DnaG complex the activities of the two proteins are modulated by distinct but overlapping networks of residues. J Bacteriol. 2006;188:1534–1539. doi: 10.1128/JB.188.4.1534-1539.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thirlway J, Turner IJ, Gibson CT, Gardiner L, Brady K, Allen S, et al. DnaG interacts with a linker region that joins the N- and C-domains of DnaB and induces the formation of 3-fold symmetric rings. Nucleic Acids Res. 2004;32:2977–2986. doi: 10.1093/nar/gkh628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tougu K, Marians KJ. The interaction between helicase and primase sets the replication fork clock. J Biol Chem. 1996a;271:21398–21405. doi: 10.1074/jbc.271.35.21398. [DOI] [PubMed] [Google Scholar]
- Tougu K, Marians KJ. The extreme C terminus of primase is required for interaction with DnaB at the replication fork. J Biol Chem. 1996b;271:21391–21397. doi: 10.1074/jbc.271.35.21391. [DOI] [PubMed] [Google Scholar]
- Weigelt J, Brown SE, Miles CS, Dixon NE, Otting G. NMR structure of the N-terminal domain of E. coli DnaB helicase: implications for structure rearrangements in the helicase hexamer. Structure. 1999;7:681–690. doi: 10.1016/s0969-2126(99)80089-6. [DOI] [PubMed] [Google Scholar]
- Yang SX, Yu XO, VanLoock MS, Jezewska MJ, Bujalowski W, Egelman EH. Flexibility of the rings: Structural asymmetry in the DnaB hexameric helicase. J Mol Biol. 2002;321:839–849. doi: 10.1016/s0022-2836(02)00711-8. [DOI] [PubMed] [Google Scholar]
- Yu X, Jezewska MJ, Bujalowski W, Egelman EH. The hexameric E. coli DnaB helicase can exist in different quarternary states. J Mol Biol. 1996;259:7–14. doi: 10.1006/jmbi.1996.0297. [DOI] [PubMed] [Google Scholar]
- Yuzhakov A, Kelman Z, O’Donnell M. Trading places on DNA-a three-point switch underlies primer handoff from primase to the replicative DNA polymerase. Cell. 1999;96:153–163. doi: 10.1016/s0092-8674(00)80968-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.