

Abstract
Skin cancer is the most common cancer in the United States. Ultraviolet B (UVB) radiation in sunlight is the major environmental factor causing skin cancer. p21, a p53-inducible protein, plays an important role in cell cycle, DNA repair, and apoptosis. Here we have investigated the effect of UVB radiation on p21 and its molecular mechanisms and function in human HaCaT keratinocytes, which we used as a premalignant cellular model because normal skin harbors numerous clones of p53-mutated keratinocytes. We found that in human HaCaT keratinocytes UVB induces rapid p21 down-regulation via a proteasomal degradation mechanism. In p53-defective HaCaT cells, the p21 protein levels remain decreased at a later time post-UVB, but in normal human and mouse epidermal keratinocytes with wild-type p53 the p21 levels are initially reduced but later increase post-UVB. These findings indicate that loss of p53 function leads to sustained p21 down-regulation in response to UVB damage. Degradation of p21 following UVB radiation does not require ATR, ATM, or both, because either the ATR/ATM inhibitor caffeine or siRNA knockdown of ATR, ATM, or both failed to reverse p21 degradation. However, inhibiting MDM2 or GSK3β partially reduced UVB-induced p21 degradation, while inhibiting both enzymes completely prevented it. Restoring the p21 protein levels in UVB-irradiated keratinocytes reduced apoptosis. Although at the molecular level increasing p21 expression has no effect on the protein levels of the Bcl-2 family members, it enhances the activation of AKT, a critical survival pathway to protect cells from apoptosis. Our results suggest a distinct mechanism of p21 degradation in keratinocytes by UVB, and this p21 degradation may significantly enhance UVB-induced apoptosis of premalignant keratinocytes with a p53 defect to eliminate damaged cells and therefore prevent skin cancer development.
Introduction
Skin cancer is the most common cancer in the United States with a lifetime risk nearly equal to that of all other cancers combined. More than one million new cases will be diagnosed each year. Ultraviolet (UV) radiation in sunlight is the major environmental factor causing skin cancer. UV radiation in sunlight reaching the Earth’s surface is composed of UVB (280–315 nm) and UVA (315–400 nm), whereas UVC (100–280 nm) is blocked completely by the ozone layer. UVB causes DNA damage; surviving cells with incomplete repair of DNA damage pose an increased risk for gene mutations and ultimately for cancer development in the skin. Therefore apoptosis of keratinocytes is critical for eliminating damaged cells to prevent skin carcinogenesis.
The cyclin-dependent kinase inhibitor p21 belongs to the CIP/KIP family of cyclin-dependent kinase inhibitors and is best known as an inhibitor of cell proliferation. Recently it has been shown that p21 plays a complex role in apoptosis, differentiation, and in cancer etiology and treatment.1,2 This protein is short lived, with a half-life of ~30 min, and its level is tightly regulated by proteasome-mediated protein degradation. Both SKP2-dependent and -independent pathways have been indicated in the turnover of p21 protein.3,4
Proteasomal degradation of p21 has been demonstrated upon radiation with UVB as well as UVC, a shorter wavelength range of UV radiation, and this degradation plays a critical role in the cellular response to DNA damage.3,5–8 In addition, UVB activates p53, which is well-known to up-regulate p21 depending on UVB doses.9,10 Recent studies have shown that ablation of p21 expression enhances the capacity of p53-deficient human colon cancer cells to repair UVB-induced DNA damage.11 However, the mechanism that leads to modification of p21 in epidermal keratinocytes in responses to UVB radiation and the function of such p21 regulation in p53-defective cells are still poorly understood. In the present study, we have examined the effect of UVB radiation on the p21 protein levels in HaCaT cells, non-tumorigenic human keratinocytes with UV-type p53 mutations resulting in a defect in its DNA-binding domain. In normal humans, p53 mutations are common in sun-exposed noncancerous epidermis.12–16 Thus HaCaT cells are relevant to UV-induced initiated keratinocytes, and this study yields new insights into the molecular and cellular responses of premalignant epidermal cells to additional UVB damage. Our findings indicate that, in contrast to UVC, UVB induces p21 degradation independent of the ATR/ATM pathway. It appears that the MDM2 and GSK3β pathways are involved in UVB-induced p21 degradation, and restoring p21 reduces UVB-induced apoptosis of keratinocytes.
Materials and methods
Cell culture
Human HaCaT keratinocytes (obtained from Professor N. Fusenig) were maintained in a monolayer culture in 95% air/5% CO2 at 37°C in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 units per mL penicillin, and 100 mg per mL streptomycin (Invitrogen, Carlsbad, California, USA). Normal human epidermal keratinocytes (NHEK) were purchased from Invitrogen and cultured in Epi-Life medium. Primary mouse keratinocytes were isolated and cultured as described previously.17
UVB irradiation
Cells were grown overnight to approximately 70–80% confluence in 60 mm dishes (for Western blot, cell cycle, or apoptosis analysis). Cells were cultured in medium containing 0.1% fetal bovine serum (FBS) for 24–36 h to eliminate serum effect or in 10% FBS as a comparison. Cells were washed twice with PBS, and then exposed to UVB radiation (UV stratalinker 2400 with UVB bulbs, Strategene) at different doses (0, 5, 10, 20, 30, or 40 mJ cm−2) with PBS added. Control samples were sham-irradiated under the same conditions. Cells were provided with medium and collected at predetermined time points. The spectral outputs of the lamps (Fig. 1) were determined using a spectroradiometer (Luzchem Research, Inc, Ontario, Canada), and the UVB doses were measured using a Goldilux UV meter equipped with a UVB detector (Oriel Instruments, Stratford, CT, USA). This UV system comprises 0% UVC, 51% UVB, and 49% UVA. The UVB flux was 1.0 mW cm−2 at a distance of 13 cm. For most of the studies, we used 20 mJ cm−2, because (1) our central goal is to determine the mechanisms of UVB-induced p21 degradation and its function in apoptosis, and (2) this UVB dose but not lower ones causes apoptosis in HaCaT keratinocytes.18 This UVB dose took approximately 21 s.
Fig. 1.
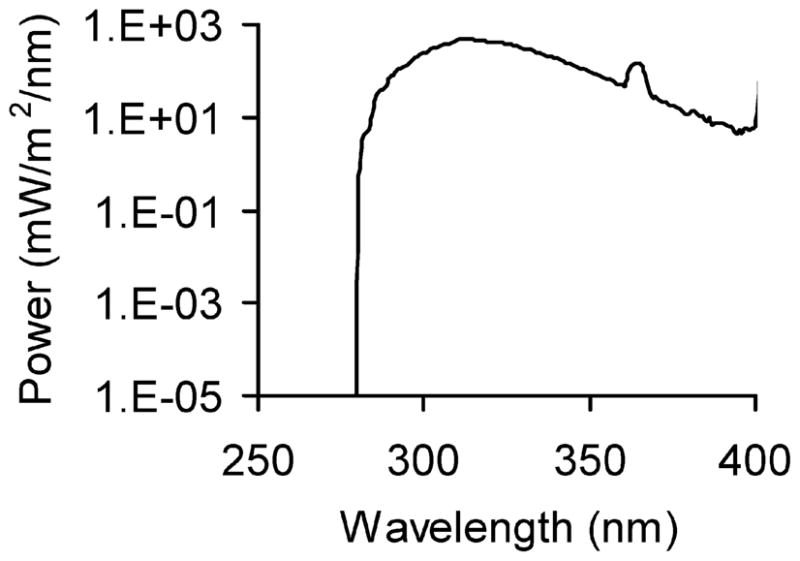
The spectral output of the UVB system.
Cytosol and nuclear protein extraction
Cytosol and nuclear protein were isolated using Chemicon’s Nuclear Extraction Kit according to the manufacturer’s instructions (Chemicon).
Determination of apoptosis by flow cytometry
Apoptosis was determined by staining cells with Annexin-V/propidium iodide (PI) using TACS™ annexin V kits according to the manufacturer’s instructions (Trevigen, Gaithersburg, MD, USA) as described in our recent studies.18,19 Cells positive for annexin V-fluorescein isothiocyanate (apoptotic cells) were quantified by flow cytometry using a BD Calibur (BD Biosciences).
Cell cycle analysis
Cell cycle analysis were performed as described previously.20,21 Briefly, at predetermined time points after treatment, the cells were harvested and fixed with 5 mL of ice cold 70% ethanol. The fixed cells were incubated with propidium iodide (20 μg mL−1) containing RNase A (1 mg mL−1; Sigma Chemical). The DNA content was determined by flow cytometry (Beckman Coulter) and ModFitLT software (Verity Software House, Inc.).
Western blotting
Protein concentrations were determined using the BCA assay (Pierce, Rockford, IL, USA). Equal amounts of protein were subjected to electrophoresis. Western blotting was performed as described previously.18,19 Antibodies used included p21, p53, PARP, caspase-3, p-GSK3β, GSK3β, cyclin D1, ATM, ATR, p-AKT (serine 473), AKT, p-ERK, ERK, p-EGFR (Tyrosine 1173), β-actin (Santa Cruz), EGFR (NeoMarker), p-p38, p38, anti-apoptotic Bcl-2 family, and pro-apoptosis Bcl-2 family (Cell Signaling Technology).
SiRNA transfection
Cells were transfected with negative control (NC) or siRNA (ON-TARGETplus SMARTpool, Dharmacon) targeting ATR, ATM, or the combination of ATR and ATM, using Amaxa Nucleofector according to the manufacturer’s instructions as described previously.20 Briefly, cells at ~90% confluency were trypsinized and electroporated with siRNA. Five million HaCaT cells were electroporated in 100 μL Nucleofector Solution V containing 1.5 μg siRNA using program U-20. After transfection, cells were seeded in 2 mL prewarmed medium in 60 mm dishes and then treated as indicated. Knockdown efficiency was confirmed by Western blotting analysis.
Adenovirus infection
The day after the cells were seeded, adenoviral vectors expressing red fluorescent protein (RFP) (Ad-RFP, Ad-Vector), or RFP and p21 (Ad-p21) (kind gifts from Tong-Chuan He, University of Chicago) were added into the cells in the presence of Polybrene (Sigma, 8 μg ml−1) in a multiplicity of infection (MOI) of 2.5, 5, 10, or 20. Fluorescence intensity and immunoblot analysis of p21 were used to confirm the overexpression of p21. After 14 h incubation with the virus, cells were washed and given fresh medium. At 36 h post infection, cells were irradiated with UVB.
Statistical analysis
Data were expressed as the mean of three independent experiments and were analyzed by Student’s t-test and two-way ANOVA. A two-sided value of P < 0.05 was considered significant in all cases.
Results
UVB induces p21 degradation in human HaCaT keratinocytes
In attempt to determine the effect of UVB radiation on p21 levels in human HaCaT keratinocytes, we starved the cells for 24 h to 36 h with low serum (0.1%) to achieve a quiescent state, as in actively dividing cells p21 is an unstable protein.1 These cells were then exposed to UVB at different doses (0, 5, 10, 20, 30, or 40 mJ cm−2) and collected at 1.5 h. The p21 protein levels were significantly down-regulated by UVB (Fig. 2A and 2B; p < 0.05, Student’s t-test for each UVB dose versus sham irradiation). To determine whether in actively cycling cells p21 is down-regulated by UVB, we irradiated HaCaT cells cultured in 10% FBS with UVB (20 mJ cm−2) and analyzed the p21 levels at 0.5 h and 1.5 h post-UVB. The p21 protein levels were reduced in UVB-irradiated cells at both time points as compared with that in sham-irradiated cells (Fig. 2C), indicating that p21 is down-regulated independent of serum levels. It is noteworthy that p21 levels are lower in cells cultured in normal serum than in cells cultured in low serum (data not shown), likely due to the cell-cycle-dependent p21 degradation and regulation.1 This may be due to the presence of about 54% of cells at the G1 phase, although it is lower than in serum-starved cells with 86% of cells at the G1 phase (Fig. 2D).
Fig. 2.

UVB irradiation reduces the p21 protein levels as determined by Western analysis. (A) HaCaT cells were irradiated with 5, 10, 20, 30, or 40 mJ cm−2 and collected at 1.5 h post-UVB for analysis of p21 protein levels using a specific anti-p21 antibody. β-Actin was used as an equal loading control. (B) Quantification of the p21 protein levels in A. (C) Same as in A, except that HaCaT cells were cultured in 10% FBS. (D) Cell cycle analysis of HaCaT cells cultured in 0.1% or 10% FBS. Numbers indicate the percent (%) of cells at the S, G1, and G2 phases. (E) Cells were exposed to UVB (20 mJ cm−2) and then incubated for different times. The p21 and p53 protein levels were determined as in A. (F) Quantification of the p21 protein levels in C. (G) Cells were exposed to UVB (20 mJ cm−2) and collected at 1.5 h post-UVB. Cytosolic and nuclear protein was isolated and p21 protein levels were determined as in A. Caspase-3 (cytosolic marker) and PARP (nuclear marker) were used as isolation controls for successful cytosolic and nuclear fractionation and equal loading controls. (H) Quantification of the p21 protein levels in E. (I) Same as in C except that normal human epidermal keratinocytes were used and collected at 1.5, 6 and 9 h post-UVB (20 mJ cm−2). (J) Same as in I, except that primary mouse keratinocytes were used. * in B and D, p < 0.05, significant differences from sham-irradiated cells.
As the inhibitory role of p21 in cell cycle arrest in response to UVB radiation has been well studied,1 we focused in this study on the new role of p21 regulation in UVB-induced apoptosis. We selected this UVB dose in this and several other figures, because (1) this UVB dose caused both significant p21 degradation (Fig. 2A) and apoptosis,18 allowing us to determine the mechanisms of p21 degradation and its function in UVB responses. To determine whether p21 decrease is sustained or transient, cells were exposed to UVB (20 mJ cm−2) and collected at different time points. The p21 protein levels were significantly down-regulated as early as 1.5 h up to 9 h following UVB radiation (Fig. 2E and 2F; p < 0.05, Student’s t-test for each time point, and two-way ANOVA for all time points). In comparison, p53 levels remained unaltered (Fig. 2E), consistent with previous studies.22
The function of p21 is associated with its subcellular localization. The growth-inhibitory functions of p21 are linked with its nuclear localization; however, the anti-apoptotic or oncogenic activities of p21 are frequently associated with its accumulation in the cytoplasm.1 To determine whether a UVB-induced decrease in p21 is specific to p21 in the cytosol or the nucleus, we isolated cytosolic and nuclear protein following UVB radiation (20 mJ cm−2). In sham-irradiated cells, p21 was mainly localized in the cytosol rather than in the nucleus. Upon UVB radiation (20 mJ cm−2), p21 levels were significantly reduced in both cytosolic and nuclear fractions (Fig. 2G and 2H; p < 0.05, Student’s t-test). The absence of caspase-3 in the nuclear fraction and the absence of PARP in the cytosolic fraction confirmed the successful isolation of cytosolic and nuclear proteins. Thus these findings indicate that UVB induces rapid p21 down-regulation in both the cytosol and the nucleus.
UVB induces p21 degradation via a proteasomal pathway
To investigate whether the decreased p21 protein levels were caused by reduced protein stability, we measured the half-life of p21 in UVB-irradiated HaCaT cells. The cells were treated with the protein synthesis inhibitor cycloheximide (CHX, 50 μg ml−1) post-UVB (20 mJ cm−2) or post-sham irradiation. While CHX significantly reduced the p21 protein levels as early as 0.5 h in sham-irradiated cells, UVB (20 mJ cm−2) further reduced p21 levels substantially in CHX-treated cells (Fig. 3A and 3B; p < 0.05, two-way ANOVA over the time course). Thus UVB significantly accelerates p21 degradation (p < 0.01, two-way ANOVA). To determine the half-life of the p21 protein in cells with or without UVB exposure, the immunoblot data were semi-quantified by ImageJ and expressed in a logarithmic scale (Fig. 3B). The half-life of p21 was approximately 24 min in HaCaT cells kept in the dark. However, the half-life of p21 in cells exposed to UVB was reduced to 4 min.
Fig. 3.

UVB irradiation reduces p21 protein stability via proteasome-mediated degradation. The p21 protein levels were determined as in Fig. 2A. (A) HaCaT cells were exposed to UVB (20 mJ cm−2) and then treated with CHX (50 μg ml−1). Cells were collected at different time points as indicated. (B) Quantification of the p21 protein levels in A. (C) HaCaT cells were treated with MG132 (MG, 10 μM) after UVB irradiation (5 or 20 mJ cm−2) and collected at 1.5 h post-UVB for immunoblot analysis of p21 protein levels. (D) HaCaT cells were treated with MG132 (MG, 10 μM) after UVB irradiation (20 mJ cm−2) and collected at 1.5 h post radiation. Cytosolic and nuclear protein was fractionated and the p21 protein levels were determined as in Fig. 2A. Caspase-3 (cytosolic marker) and PARP (nuclear marker) were used as isolation controls for successful cytosolic and nuclear fractionation and equal loading controls for these two fractions. *, significant differences from cells kept in the dark (p < 0.05).
To further examine whether UVB causes p21 degradation through a proteasomal pathway, we exposed the cells to UVB (20 mJ cm−2) and then treated the cells with the proteasome inhibitor MG 132 (MG, 10 μM). MG not only increased the p21 protein levels slightly in sham-irradiated cells, but also prevented UVB-induced p21 reduction (Fig. 3C; p < 0.05, Student’s t-test). In addition, both cytosolic and nuclear p21 degradation were inhibited by MG treatment (Fig. 3D; p < 0.05, Student’s t-test). These data indicated that UVB-induced p21 reduction in HaCaT cells is caused by increased proteasomal degradation.
UVB induced p21 degradation in human HaCaT keratinocytes is independent of ATR/ATM
Upon DNA damage, ataxia-telangiectasia mutated (ATM), and ATM and RAD3-related (ATR) are activated to coordinate cell-cycle transitions, DNA replication, DNA repair, and apoptosis.23,24 Activation of the ATR pathway has been shown to mediate p21 degradation caused by UVC as demonstrated by the complete inhibition of p21 degradation by caffeine and ATR deletion.3 To determine whether ATR is required for UVB-induced p21 degradation in HaCaT keratinocytes, we pretreated the cells without or with 1 or 5 mM caffeine, the ATR/ATM inhibitor that inhibits UVC-induced p21 degradation.3 As shown in Fig. 4A, caffeine at these concentrations failed to prevent p21 degradation following UVB radiation (20 mJ cm−2). To further examine the role of ATM, ATR, or the combination of both, we use siRNA to knock down ATR, ATM, or both. The siRNA targeting ATM or ATR knocked down these targets effectively (Fig. 4B). However, knockdown of ATR, ATM, or the combination of both had no effect on p21 degradation by 5 or 20 mJ cm−2 UVB (Fig. 4C). These data demonstrate that UVB-induced p21 degradation is ATR/ATM independent.
Fig. 4.
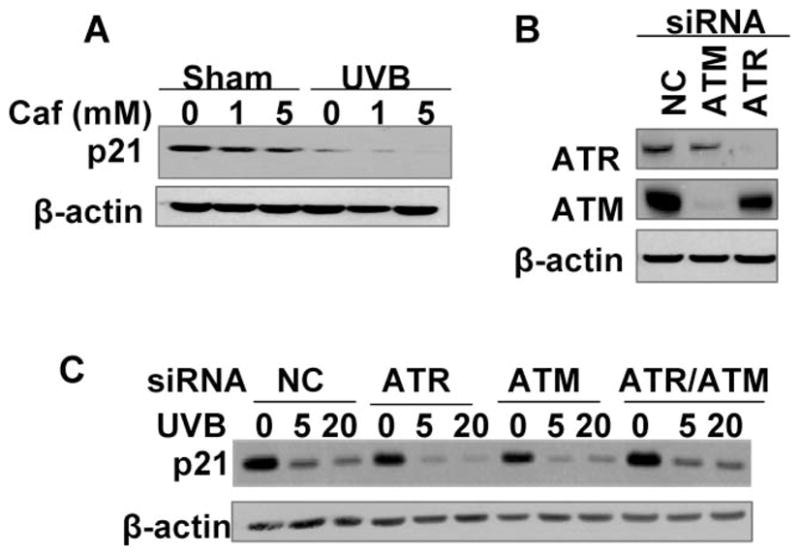
UVB-induced p21 degradation is independent of ATR/ATM pathways. p21 protein levels were determined as in Fig. 2A. (A) Cells were pretreated with caffeine (1 or 5 mM) for 30 min and then exposed to UVB radiation (20 mJ cm−2). Cells were then collected at 1.5 h post-UVB. (B) Cells were transfected with siRNA targeting ATR or ATM. Non-targeting siRNA was used as a negative control (NC) for siRNA transfection. The protein levels of ATR or ATM were determined by Western blotting using specific anti-ATR and anti-ATM antibodies. (C) Cells were transfected with siRNA targeting ATR, ATM, the combination of both (ATR/ATM), or NC, and then exposed to UVB (5 and 20 mJ cm−2). Cells were collected at 1.5 h post-UVB.
MDM2 and GSK3β are involved in UVB-induced p21 degradation
Recent studies have shown that both glycogen synthase kinase 3beta (GSK3β) and murine double minute (MDM2) play important roles in p21 degradation.5,25,26 To investigate whether MDM2, GSK3β, or both mediate p21 degradation upon UVB radiation, we first ascertained whether UVB affects GSK3β activity. We measured two endpoints: (1) phosphorylation of GSK3β at serine 9 (p-GSK3β), an inhibitory modification;27 and (2) cyclin D1 degradation, which is known to be GSK3β-mediated via phosphorylation during the cell cycle.28
First, UVB radiation (20 mJ cm−2) reduced GSK3β phosphorylation at serine 9, suggesting that UVB increases GSK3β activity (Fig. 5A). Furthermore, UVB (20 mJ cm−2) attenuated this GSK3β phosphorylation in both cytosolic and nuclear fractions (Fig. 5B). Second, UVB radiation (20 mJ cm−2) significantly down-regulated cyclin D1 at 1.5 and 3 h post-UVB (Fig. 5C; p < 0.05, Student’s t-test for each time point). Proteasome inhibitor MG132 (MG, 10 μM) blocked cyclin D1 down-regulation post-UVB (20 mJ cm−2), indicating that UVB-induced cyclin D1 down-regulation is mediated by the proteasome degradation pathway (Fig. 5D). Cyclin D1 degradation was inhibited by GSK3β inhibitor BIO (1 μM) (Fig. 5D; p < 0.05, Student’s t-test), indicating that UVB-induced cyclin D1 degradation requires GSK3β activation. These findings confirm that UVB activates GSK3β.
Fig. 5.

Inhibiting MDM2 and GSK3β inhibits UVB-induced p21 degradation. p21 protein levels were determined as in Fig. 2A. (A) Cells were exposed to UVB (20 mJ cm−2) and then collected at 1.5 h post-UVB. Phosphorylation of GSK3β at serine 9 (p-GSK3β) was determined by Western analysis using a specific anti-p-GSK3β antibody. (B) Same as in A except that cytosol and nuclear protein was isolated and p-GSK3β was determined in these fractions. Caspase-3 (cytosolic marker) and PARP (nuclear marker) were used as isolation controls. (C) Cells were exposed to UVB as in A and then collected at indicated times. Cyclin D1 levels were determined by Western analysis using a specific anti-cyclin D1 antibody. β-actin was used as an equal loading control. (D) Cells were pretreated with MG 132 (MG, 10 μM) for 1 h or BIO (1 μM) for 24 h. Cells were exposed to UVB (20 mJ cm−2) and then collected at 1.5 h post-UVB. Cyclin D1 levels were determined as in C. (E) Cells were pretreated with the GSK3β inhibitor BIO at different concentrations as indicated for 24 h and then exposed to UVB radiation (20 mJ cm−2). (F) Cells were incubated with the MDM2 inhibitor Nutlin at different concentrations as indicated for 24 h and then exposed to UVB radiation (20 mJ cm−2). p21 and p53 levels were analyzed by immunoblotting. (G) Cells were treated with the combination of BIO (1 μM) and Nutlin (10 μM) for 24 h and then exposed to UVB radiation. (H) Quantification of the p21 protein levels in E, F, and G. *, p < 0.05, and **, p < 0.01; significant differences from UVB-irradiated cells without inhibitor treatment. #, p < 0.05; significant differences from UVB-irradiated cells treated with either BIO or Nutlin alone. NS, not statistically significant.
To determine whether MDM2, GSK3β, or both, mediates p21 degradation upon UVB radiation, we pretreated cells with the MDM2 inhibitor Nutlin, the GSK3β inhibitor BIO, or the combination of both, and then exposed the cells to UVB radiation (20 mJ cm−2). Either BIO alone or Nutlin alone partially reduced p21 degradation following UVB radiation (Fig. 5E, 5F, and 5H; p < 0.05, Student’s t-test). In comparison, the combination of BIO and Nutlin more effectively prevented UVB-induced p21 degradation than either BIO or Nutlin alone (Fig. 5G and 5H; p < 0.05, Student’s t-test between indicated inhibitor treatment groups in UVB-irradiated cells); in addition, it completely blocked UVB-induced p21 degradation (Fig. 5G and 5H; p > 0.8, student t-test; not significant between sham- and UVB irradiated in the presence of BIO and Nutlin). These data indicate that both MDM2 and GSK3β play important roles in UVB-induced p21 degradation.
p21 degradation enhances keratinocyte apoptosis induced by UVB radiation
Recent evidence suggests a critical and complex role of p21 in apoptosis.1 Whether UVB-induced p21 degradation plays an active role in keratinocyte apoptosis has been unclear. To determine whether restoring p21 protein levels in UVB-exposed cells affects UVB-induced apoptosis, we infected HaCaT cells with an adenoviral vector expressing wild-type p21 (Ad-p21) or vector alone (Ad-RFP). A unique feature of the Ad-p21 adenoviral vector is that it contains a built-in red fluorescent protein (RFP) expression cassette that allows tracking the adenoviral infection and gene expression in the cells (Fig. 6A). A recombinant adenoviral vector that only expressed RFP was used as a vector control.
Fig. 6.

Reconstituting p21 protein levels inhibits UVB-induced apoptosis. (A) HaCaT cells were infected with adenoviral vectors expressing red fluorescent protein (RFP) (Ad-RFP) only, p21 and RFP (Ad-p21), or mock infected without viruses. Fluorescence showed efficient adenoviral infection and expression of RFP. (B) Cells were infected as in A and then exposed to UVB radiation (20 mJ cm−2). Cells were treated with or without MG following UVB radiation. The p21 protein levels were determined at 1.5 h upon UVB exposure. (C) Cells were infected as in A and then exposed to apoptosis-inducing UVB radiation (20 or 30 mJ cm−2). Cleaved PARP (PARP), an apoptosis marker, was determined at 6 h post-UVB. (D) Cells were infected and exposed to UVB as in B. Apoptosis was determined and quantified using Annexin V-FITC/propidium iodide staining followed by flow cytometric analysis. (E) Percentage (%) of apoptotic cells (Annevin V positive) from histograms in D. *, p < 0.05; significant differences from UVB-irradiated vector-infected cells. (F) Immunoblot analysis of p21, Bax, Bad, Bik, Bim, Bid, MCL1, Bcl-XL, Bcl-2, and β-actin in HaCaT cells infected with an adenoviral vector expressing RFP alone or p21-RFP. (G) Immunoblot analysis of p21, p-AKT (serine 473), AKT, p-ERK, ERK, p-EGFR (Tyrosine 1197), EGFR, p-p38, p38, and β-actin in cells as in F.
To determine the infection efficiency of the adenoviral vectors, both Ad-p21 and Ad-RFP were used to infect HaCaT cells. Then the cells were examined under a fluorescence microscope to assess the RFP levels, which should be co-expressed in Ad-p21 infected cells. As demonstrated in Fig. 6A, a comparable infection efficiency and RFP expression at a MOI of 5 between the Ad-RFP and Ad-p21 vectors were observed, followed by confirmation by fluorescence intensity determined by microplate reader (data not shown). Overexpression of p21 was detected in the cells infected with Ad-p21 compared to those with Ad-RFP control (Fig. 6B). Adenoviral infection with Ad-RFP had no effect on UVB-induced p21 degradation (Fig. 6B). In Ad-p21 infected cells, p21 levels were increased as compared to Ad-RFP infected cells with or without UVB. The proteasome inhibitor MG prevented UVB-induced p21 degradation in cells without adenoviral infection, or infected with Ad-RFP or Ad-p21, suggesting that reduction of p21 in these cells is mediated by proteasomal degradation (Fig. 6B). Through testing different infection MOIs, we determined that infection with adenoviral vector at a MOI of 5 restores p21 protein levels in UVB-irradiated (Fig. 6B; p = 0.85, Student’s t-test between Ad-p21-infected UVB-irradiated cells versus Ad-RFP-infected sham-irradiated cells). The same infection conditions were used to determine the role of p21 in UVB-induced apoptosis.
Apoptosis eliminates damaged cells to prevent tumorigenic transformation. To determine whether UVB-induced p21 degradation plays an active role in apoptosis of keratinocytes, cells were infected with Ad-RFP or Ad-p21 and then exposed to UVB at 20 or 30 mJ cm−2. Six hours after UVB radiation, cleaved PARP, an apoptosis marker, was detected in UVB exposed cells (Fig. 6C). The PARP cleavage increased with increasing UVB dose. However, increasing the p21 levels by infecting HaCaT cells with Ad-p21 inhibited PARP cleavage (Fig. 6C), indicating that reconstituting p21 in UVB-exposed cells reduces UVB-induced apoptosis. To further confirm and quantify the effect of p21 increase in UVB-induced apoptosis in HaCaT keratinocytes, we infected and then irradiated HaCaT cells with UVB as in Fig. 6C and cells were collected for flow cytometric analysis following staining with Annexin-V/propidium iodide. UVB at 20 or 30 mJ cm−2 induced apoptosis in HaCaT cells (Fig. 6D). Increasing p21 protein levels significantly inhibited apoptosis caused by either dose of UVB used (Fig. 6D and 6E; p < 0.05, Student’s t-test for each UVB dose). These data indicate that p21 degradation increases UVB-induced apoptosis of keratinocytes.
To determine the mechanisms by which p21 inhibits UVB-induced apoptosis, we determined the effect of reconstituting the p21 protein levels on several survival pathways in keratinocytes including the expression of the Bcl-2 family members 29–31 and the activation of the AKT, ERK, or EGFR pathway.32–34 Over-expressing p21 had no effect on the protein levels of the pro-apoptotic members including Bax, Bad, Bik, Bim, Bid and the anti-apoptotic members including MCL1, Bcl-XL and Bcl-2 (Fig. 6F). However, increasing the p21 protein levels substantially increased the activation of AKT and EGFR while it only slightly increased the activation of ERK (Fig. 6G). However, no similar difference was detected in p38 activation (Fig. 5G). These data suggest that compensating the p21 protein levels in HaCaT keratinocytes selectively increases AKT activation.
Discussion
In this study, we have explored the mechanisms of p21 regulation and its function in keratinocytes in response to UVB radiation. Recent studies have shown that UVC-induced direct or indirect p21 down-regulation is mediated by and critical for the DNA repair process in non-keratinocyte cell types.3,6,8 Whether ubiquitylation is involved in UVC-induced nuclear p21 degradation is still under debate.3,5,6 Here we have found that UVB induces proteasomal p21 degradation in both cytosol and nucleus in human HaCaT keratinocytes. The degradation of p21 requires the activation of MDM2 and GSK3β but is independent of the ATR/ATM pathways. Reconstituting the p21 protein levels in HaCaT cells prevents UVB-induced apoptosis. Based on our findings and previous studies,8,35 we propose that UVB activates MDM2 and GSK3β, which induces p21 degradation, which sensitizes keratinocytes to apoptosis to eliminate damaged cells.
UVB radiation induces rapid and dose-dependent p21 proteasomal degradation in human HaCaT keratinocytes. We found that in keratinocytes, most of the p21 protein is localized in the cytosol while a small fraction of this protein is in the nucleus. Upon UVB radiation, p21 in the cytosol and the nucleus decreases rapidly in a time-dependent manner. This trend of p21 down-regulation was not seen in cells exposed to UVB radiation (30 and 70 mJ cm−2) at later time points,10 suggesting that p21 down-regulation in human keratinocytes is specific for the early time points and the defective p53 status in HaCaT cells used in our studies. Indeed, in normal human and mouse keratinocytes with functional p53, p21 down-regulation was only found at 1.5 h post-UVB, while p21 levels were increased at much later time points (6 and 9 h post-UVB, Fig. 2I and 2J).
p53 plays a crucial role in the orchestration of a cell’s response to UV-induced damage and skin cancer.13,14,36,37 In response to UV-induced damage, it is known that induction of p21 via transcriptional activation 38 is critical for G1 checkpoint control.9,39,40 In HaCaT cells with UV-type p53 mutations in both alleles,41 the mutated p53 is stabilized 22 and has been suggested to play a role in apoptosis and differentiation.42,43 In these cells, however, UV radiation fails to induce p21 expression,22 which may be responsible for sustained p21 down-regulation.
In addition, this p53 defect may play a role in the dispensable roles of the ATR and ATM pathways in UVB-induced p21 degradation, distinct from UVC-induced p21 degradation, which is mediated via the ATR pathway and prevented by caffeine.3,5 This discrepancy could be due to the different physical and biological properties of UVB and UVC, cell type-specific mechanisms of p21 degradation, or both. The ATR and ATM pathways activate p53 to coordinate DNA repair, cell cycle arrest, and apoptosis.22,44–46 More investigation is needed to determine whether p53 pathway plays a role in the ATR-mediated p21 degradation found in previous studies.3,5
The MDM2 and GSK3β pathways play important roles in UVB-induced p21 degradation. It appears that MDM2 and GSK3β additively mediate p21 down-regulation upon UVB treatment. Although ubiquitylation has been shown to play an important role in PCNA-dependent p21 degradation,47 MDM2 and GSK3β promote p21 proteasomal degradation without ubiquitylating it.5,25,26 In addition, MDM2- or GSK3β-mediated p21 degradation is independent of p53.5,26 These findings points to a complex regulatory machinery among p21, MDM2, and GSK3β. It is possible that UVB-induced p21 degradation does not require p21 ubiquitylation.
It is likely that, post-UVB-induced DNA damage, the early steps of DNA repair through recruitment and activation of UV-damaged DNA-binding protein (UV-DDB), which is composed of two subunits DDB1 and DDB2 (XPE, xeroderma pigmentosum E),48 lead to the activation of MDM2 and GSK3β. Indeed, recent studies have shown that DDB1 plays a critical role in proteolytic degradation of MDM2, whereas DDB2 targets p21 for degradation,35,49 suggesting that p21 degradation is a consequence of DNA repair processes, but not of ATR/ATM, the DNA damage response pathways that require recognition and processing of the DNA lesions by DNA repair for activation.50 Recent studies have demonstrated that, along with Cul4A and DDB1, proliferating cell nuclear antigen (PCNA) is required for p21 degradation during the S phase and after UV irradiation.6,47 More investigation is needed to further determine the molecular mechanisms of the complex interactions among DNA repair, MDM2, and GSK3β in UVB-induced p21 degradation in keratinocytes.
Recent studies have indicated that genetic p21 depletion enhances repair of UVB-induced DNA damage in p53-null colon carcinoma cells,11 and that p21 inhibits repair of UV-type DNA damage while deletion of p21 restores the repair capacity in Ddb2−/− mouse embryonic fibroblast cells.8 Indeed p21 is a known inhibitor of the short-gap filling activity of proliferating cell nuclear antigen (PCNA) during DNA repair, and p21 down-regulation has been shown to play a role in efficient PCNA ubiquitylation after UV irradiation and thus promotes PCNA-dependent repair.51
UVB-induced p21 degradation facilitates apoptosis of HaCaT keratinocytes, in line with previous findings.11 This anti-apoptotic function of p21 has been supported in several other systems.2 In addition to inhibiting cyclin-dependent kinase (CDK) at the G1/S and G2/M interfaces to regulate cell cycle checkpoints, recent studies have demonstrated that p21 inhibits apoptosis, which may counteract its tumor-suppressive function as a growth inhibitor in tumor cells.1,2 We found that increasing p21 levels enhances AKT activation, a pathway shown to protect keratinocytes from apoptosis.52,53 Further studies are needed to elucidate the mechanisms of enhanced AKT activation mediated by p21 and to gain a complete understanding of the anti-apoptotic effect of p21 in response to UVB radiation. It is possible that tumor cells with up-regulated p21 levels use the same mechanism to promote cell survival and escape apoptosis induced by chemotherapy. This anti-apoptotic function of p21 may be affected upon p53 inactivation. Nevertheless, as in many human cancer cells, UV-exposed premalignant epidermal keratinocytes harbor inactivating p53 mutations;12–16 thus our findings support the conclusion that UVB-induced p21 degradation plays an active role in eliminating damaged premalignant cells.
In summary, in human HaCaT keratinocytes, proteasome-mediated p21 degradation caused by UVB radiation involves the MDM2 and GSK3β pathways but is independent of ATR/ATM pathways. UVB-induced p21 down-regulation increases apoptosis, critical for eliminating damaged cells. As p21 has complex roles in cancer and can be an oncogene in skin cancer,1 our findings suggest that in the non-tumorigenic HaCaT keratinocytes with the UV type of defective p53 mutations,41 p21 degradation following UVB insults may provide a self-defense mechanism to prevent skin cancer development in premalignant cells after loss of p53 function.
Acknowledgments
This work was supported by the American Skin Association, Wendy Will Case Cancer Fund, the University of Chicago Comprehensive Cancer Center (P30 CA014599), the CTSA (NCRR, UL1 RR024999), and the University of Chicago “Friends of Dermatology” Endowment Fund. We are grateful to Dr Tong-Chuan He for the adenoviral vectors expressing RFP and p21-RFP. We thank Dr Ann Motten for her critical reading of this manuscript.
Abbreviations
- AKT
A serine-threonine kinase, downstream of PI3K, also called protein kinase B
- ATM
Ataxia-telangiectasia mutated
- ATR
Ataxia-telangiectasia and Rad3-related
- EGFR
Epidermal growth factor receptor
- ERK
Extracellular signal-regulated kinase
- GSK3β
Glycogen Synthase Kinase 3beta
- MAPK
Mitogen activated protein kinase
- MDM2
Murine double minute
- NMSC
Non-melanoma skin cancer
- PI3K
Phosphatidylinosital-3 kinase
- siRNA
Small interfering RNA
- UVA
Ultraviolet A (315–400 nm)
- UVB
Ultraviolet B (280–315 nm)
References
- 1.Abbas T, Dutta A. Nat Rev Cancer. 2009;9:400–414. doi: 10.1038/nrc2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gartel AL, Tyner AL. Mol Cancer Therapeut. 2002;1:639–649. [PubMed] [Google Scholar]
- 3.Bendjennat M, Boulaire J, Jascur T, Brickner H, Barbier V, Sarasin A, Fotedar A, Fotedar R. Cell. 2003;114:599–610. doi: 10.1016/j.cell.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 4.Lee H, Zeng SX, Lu H. J Biol Chem. 2006;281:26876–26883. doi: 10.1074/jbc.M605366200. [DOI] [PubMed] [Google Scholar]
- 5.Lee JY, Yu SJ, Park YG, Kim J, Sohn J. Mol Cell Biol. 2007;27:3187–3198. doi: 10.1128/MCB.01461-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nishitani H, Shiomi Y, Iida H, Michishita M, Takami T, Tsurimoto T. J Biol Chem. 2008;283:29045–29052. doi: 10.1074/jbc.M806045200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Soria G, Speroni J, Podhajcer OL, Prives C, Gottifredi V. J Cell Sci. 2008;121:3271–3282. doi: 10.1242/jcs.027730. [DOI] [PubMed] [Google Scholar]
- 8.Stoyanova T, Yoon T, Kopanja D, Mokyr MB, Raychaudhuri P. Mol Cell Biol. 2008;28:177–187. doi: 10.1128/MCB.00880-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ponten F, Berne B, Ren ZP, Nister M, Ponten J. J Invest Dermatol. 1995;105:402–406. doi: 10.1111/1523-1747.ep12321071. [DOI] [PubMed] [Google Scholar]
- 10.Abd Elmageed ZY, Gaur RL, Williams M, Abdraboh ME, Rao PN, Raj MH, Ismail FM, Ouhtit A. J Invest Dermatol. 2009;129:175–183. doi: 10.1038/jid.2008.208. [DOI] [PubMed] [Google Scholar]
- 11.Therrien JP, Loignon M, Drouin R, Drobetsky EA. Cancer Res. 2001;61:3781–3786. [PubMed] [Google Scholar]
- 12.Jonason AS, Kunala S, Price GJ, Restifo RJ, Spinelli HM, Persing JA, Leffell DJ, Tarone RE, Brash DE. Proc Natl Acad Sci U S A. 1996;93:14025–14029. doi: 10.1073/pnas.93.24.14025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Gruijl FR. Exp Dermatol. 2002;11:37–39. doi: 10.1034/j.1600-0625.11.s.1.9.x. [DOI] [PubMed] [Google Scholar]
- 14.de Gruijl FR, Rebel H. Photochem Photobiol. 2008;84:382–387. doi: 10.1111/j.1751-1097.2007.00275.x. [DOI] [PubMed] [Google Scholar]
- 15.Ling G, Persson A, Berne B, Uhlen M, Lundeberg J, Ponten F. Am J Pathol. 2001;159:1247–1253. doi: 10.1016/S0002-9440(10)62511-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakazawa H, English D, Randell PL, Nakazawa K, Martel N, Armstrong BK, Yamasaki H. Proc Natl Acad Sci U S A. 1994;91:360–364. doi: 10.1073/pnas.91.1.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.He YY, Huang JL, Block ML, Hong JS, Chignell CF. J Invest Dermatol. 2005;125:560–566. doi: 10.1111/j.0022-202X.2005.23851.x. [DOI] [PubMed] [Google Scholar]
- 18.Ming M, Han W, Maddox J, Soltani K, Shea CR, Freeman DM, He YY. Oncogene. 2010;29:492–502. doi: 10.1038/onc.2009.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He YY, Pi J, Huang JL, Diwan BA, Waalkes MP, Chignell CF. Oncogene. 2006;25:3680–3688. doi: 10.1038/sj.onc.1209384. [DOI] [PubMed] [Google Scholar]
- 20.He YY, Council SE, Feng L, Chignell CF. Cancer Res. 2008;68:3752–3758. doi: 10.1158/0008-5472.CAN-07-6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Han W, He YY. Photochem Photobiol. 2009 doi: 10.1111/j.1751-1097.2008.00531.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoon K, Smart RC. Mol Cell Biol. 2004;24:10650–10660. doi: 10.1128/MCB.24.24.10650-10660.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cimprich KA, Cortez D. Nat Rev Mol Cell Biol. 2008;9:616–627. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou BB, Elledge SJ. Nature. 2000;408:433–439. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 25.Jin Y, Lee H, Zeng SX, Dai MS, Lu H. EMBO J. 2003;22:6365–6377. doi: 10.1093/emboj/cdg600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Z, Wang H, Li M, Agrawal S, Chen X, Zhang R. J Biol Chem. 2004;279:16000–16006. doi: 10.1074/jbc.M312264200. [DOI] [PubMed] [Google Scholar]
- 27.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 28.Diehl JA, Cheng M, Roussel MF, Sherr CJ. Genes Dev. 1998;12:3499–3511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chao DT, Korsmeyer SJ. Annu Rev Immunol. 1998;16:395–419. doi: 10.1146/annurev.immunol.16.1.395. [DOI] [PubMed] [Google Scholar]
- 30.Youle RJ, Strasser A. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 31.Cory S, Adams JM. Nat Rev Cancer. 2002;2:647–656. doi: 10.1038/nrc883. [DOI] [PubMed] [Google Scholar]
- 32.Bode AM, Dong Z. Science’s STKE. 2003;2003:2re. doi: 10.1126/stke.2003.167.re2. [DOI] [PubMed] [Google Scholar]
- 33.Bowden GT. Nat Rev Cancer. 2004;4:23–35. doi: 10.1038/nrc1253. [DOI] [PubMed] [Google Scholar]
- 34.He YY, Huang JL, Chignell CF. J Biol Chem. 2004;279:53867–53874. doi: 10.1074/jbc.M405781200. [DOI] [PubMed] [Google Scholar]
- 35.Stoyanova T, Roy N, Kopanja D, Bagchi S, Raychaudhuri P. Proc Natl Acad Sci U S A. 2009;106:10690–10695. doi: 10.1073/pnas.0812254106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu X, Lane DP. Cell. 1993;75:765–778. doi: 10.1016/0092-8674(93)90496-d. [DOI] [PubMed] [Google Scholar]
- 37.Brash DE, Rudolph JA, Simon JA, Lin A, McKenna GJ, Baden HP, Halperin AJ, Ponten J. Proc Natl Acad Sci U S A. 1991;88:10124–10128. doi: 10.1073/pnas.88.22.10124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 39.Deng C, Zhang P, Harper JW, Elledge SJ, Leder P. Cell. 1995;82:675–684. doi: 10.1016/0092-8674(95)90039-x. [DOI] [PubMed] [Google Scholar]
- 40.Brugarolas J, Chandrasekaran C, Gordon JI, Beach D, Jacks T, Hannon GJ. Nature. 1995;377:552–557. doi: 10.1038/377552a0. [DOI] [PubMed] [Google Scholar]
- 41.Lehman TA, Modali R, Boukamp P, Stanek J, Bennett WP, Welsh JA, Metcalf RA, Stampfer MR, Fusenig N, Rogan EM, et al. Carcinogenesis. 1993;14:833–839. doi: 10.1093/carcin/14.5.833. [DOI] [PubMed] [Google Scholar]
- 42.Henseleit U, Zhang J, Wanner R, Haase I, Kolde G, Rosenbach T. J Invest Dermatol. 1997;109:722–727. doi: 10.1111/1523-1747.ep12340708. [DOI] [PubMed] [Google Scholar]
- 43.Paramio JM, Segrelles C, Lain S, Gomez-Casero E, Lane DP, Lane EB, Jorcano JL. Mol Carcinog. 2000;29:251–262. doi: 10.1002/1098-2744(200012)29:4<251::aid-mc1007>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 44.Chehab NH, Malikzay A, Appel M, Halazonetis TD. Genes Dev. 2000;14:278–288. [PMC free article] [PubMed] [Google Scholar]
- 45.Shieh SY, Ahn J, Tamai K, Taya Y, Prives C. Genes Dev. 2000;14:289–300. [PMC free article] [PubMed] [Google Scholar]
- 46.Ou YH, Chung PH, Sun TP, Shieh SY. Mol Biol Cell. 2005;16:1684–1695. doi: 10.1091/mbc.E04-08-0689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Abbas T, Sivaprasad U, Terai K, Amador V, Pagano M, Dutta A. Genes Dev. 2008;22:2496–2506. doi: 10.1101/gad.1676108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sugasawa K. DNA Repair. 2009;8:969–972. doi: 10.1016/j.dnarep.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 49.Banks D, Wu M, Higa LA, Gavrilova N, Quan J, Ye T, Kobayashi R, Sun H, Zhang H. Cell Cycle. 2006;5:1719–1729. doi: 10.4161/cc.5.15.3150. [DOI] [PubMed] [Google Scholar]
- 50.Marini F, Nardo T, Giannattasio M, Minuzzo M, Stefanini M, Plevani P, Muzi Falconi M. Proc Natl Acad Sci U S A. 2006;103:17325–17330. doi: 10.1073/pnas.0605446103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Soria G, Podhajcer O, Prives C, Gottifredi V. Oncogene. 2006;25:2829–2838. doi: 10.1038/sj.onc.1209315. [DOI] [PubMed] [Google Scholar]
- 52.Saegusa J, Hsu DK, Liu W, Kuwabara I, Kuwabara Y, Yu L, Liu FT. J Invest Dermatol. 2008;128:2403–2411. doi: 10.1038/jid.2008.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Decraene D, Van Laethem A, Agostinis P, De Peuter L, Degreef H, Bouillon R, Garmyn M. J Invest Dermatol. 2004;123:207–212. doi: 10.1111/j.0022-202X.2004.22702.x. [DOI] [PubMed] [Google Scholar]