

Abstract
Dipetalogaster maximais a blood-sucking Hemiptera that inhabits sylvatic areas in Mexico. It usually takes its blood meal from lizards, but following human population growth, it invaded suburban areas, feeding also on humans and domestic animals. Hematophagous insect salivary glands produce potent pharmacologic compounds that counteract host hemostasis, including anticlotting, antiplatelet, and vasodilatory molecules. To obtain further insight into the salivary biochemical and pharmacologic complexity of this insect, a cDNA library from its salivary glands was randomly sequenced. Salivary proteins were also submitted to one- and two-dimensional gel electrophoresis (1DE and 2DE) followed by mass spectrometry analysis. We present the analysis of a set of 2728 cDNA sequences, 1375 of which coded for proteins of a putative secretory nature. The saliva 2DE proteome displayed approximately 150 spots. The mass spectrometry analysis revealed mainly lipocalins, pallidipins, antigen 5-like proteins, and apyrases. The redundancy of sequence identification of saliva-secreted proteins suggests that proteins are present in multiple isoforms or derive from gene duplications. Supplemental files can be downloaded from http://exon.niaid.nih.gov/transcriptome/D_maxima/Dm-S1-web.xls and http://exon.niaid.nih.gov/transcriptome/D_maxima/Dm-S2-web.xls.
Keywords: Saliva, hematophagy, transcriptome, proteome, triatomine, D. maxima
Introduction
Dipetalogaster maxima (Hemiptera: Reduviidae), the largest triatomine known, lives in the southern area of Baja California Sur, Mexico. Although more closely related to the genus Triatoma, the gross anatomy and physiology of D. maxima (Dm) is practically identical to the phylogenetically more distant, but well studied bug Rhodnius prolixus, except for its size, which can reach 5 cm long as adults; each instar can be about six times larger than the corresponding one in Rhodnius.1,2 It resides in dry rocky areas, and its large size allows it to store a greater amount of blood and to survive fasting in semi-desert weather.3 It is an aggressive biter, attacking avidly when hungry, day or night, feeding on any available vertebrate—including humans—within the environment.4,5 Although Dm is typically from sylvatic environments where it feeds on lizards, its adaptation to humans has been documented in La Paz, the capital city of Baja California Sur.6 Many populations of this blood-sucking bug are naturally infected with the protozoan parasite Trypanosoma cruzi (American trypanosomiasis), the etiologic agent of Chagas’ disease, but its role in transmission of T. cruzi to the human host is unknown.3,7
Blood-feeding arthropods show a variety of antihemostatic compounds in their salivary glands (SGs) that help them obtain the blood meal.8,9 Hematophagous insects are able to counteract host hemostatic responses triggered to prevent blood loss following tissue injury, such as vasoconstriction, blood coagulation, and platelet aggregation.10 The sialome (set of RNA message + set of proteins found in SGs) of blood-sucking arthropods previously described11–13 has revealed a more complex composition than expected and the presence of many proteins to which we cannot yet ascribe a function. There has been no detailed report of Dm saliva and SG content, even though it, like other triatomines, 14 is able to avoid host hemostasis.
In this work, we present the analysis of a set of 2,728 SG cDNA sequences, 1375 of which code for proteins of a putative secretory nature. Most salivary proteins were described as lipocalins, corresponding to 93% of the transcripts coding for putative secretory proteins. Lipocalins are a large and heterogenous group of proteins that play various roles, mainly as carriers of small ligands in vertebrates and invertebrates.15 A great array of SG proteins belonging to the lipocalin family has generated a large number of different molecules having antihemostatic functions while maintaining the fundamental structure of the protein fold.16 Lipocalins were found in the saliva of other blood-sucking triatomine bugs such as Rhodnius prolixus,17 Triatoma brasiliensis,18 Triatoma infestans (Ti),19,20 and also in tick saliva.21
We expect this work will contribute with new salivary transcripts that could help us understand the role of salivary molecules in host/vector interactions and the evolutionary mechanisms leading to insect adaptation to this feeding habit.
Materials and Methods
Dm and SG cDNA Library Construction
Dm were reared in an insectary room in the Tropical Medicine Department, University of Brasilia. They were kept at 27°C ± 1.0°C with a relative humidity of 70% to 75% and a 16 h:8 h light:dark photoperiod. SGs were dissected from Vth instar nymphs and adults 7 days after a blood meal and transferred to RNA-Later (Ambion, Austin, TX, USA) solution in 1.5-ml polypropylene vials. SGs were kept at −20°C for isolating polyA+ RNA.
Dm SG mRNA was isolated from 15 SG pairs from Vth instar nymphs and adults using the Micro-FastTrack mRNA isolation kit (Invitrogen, San Diego, CA, USA). The PCR-based cDNA library was made following the instructions for the SMART (switching mechanism at 5′ end of RNA transcript) cDNA library construction kit (Clontech, Palo Alto, CA, USA). This kit provides a method for producing high-quality, full-length cDNA libraries from nanogram quantities of polyA+ or total RNA. It utilizes a specially designed oligonucleotide named SMART IV™ in the first-strand synthesis to generate high yields of full-length, double-stranded cDNA. Dm SG polyA+ RNA was used for reverse transcription to cDNA using Moloney murine leukemia virus reverse transcriptase (Clontech), SMART IV oligonucleotide, and CDS III/3′ primer (Clontech). Trehalose was added to the reaction that was carried out at 60°C for 1 h, then at 42°C for 40 min. Second-strand synthesis was performed according to a long-distance PCR-based protocol using the 5′ PCR primer and CDS III/3′ primer as sense and antisense primers, respectively. These two primers also create Sfi1A and B restriction enzyme sites at the end of the cDNA. Advantage™ Taq polymerase mix (Clontech) was used to carry out long-distance PCR reaction on a GeneAmp® PCR system 9700 (Perkin Elmer Corp., Foster City, CA, USA). The PCR conditions were: 95°C for 1 min; 14 cycles of 95°C for 10 s, 68°C for 6 min. An aliquot of the cDNA was analyzed on a 1.1% agarose/EtBr (0.1 μg/ml) gel to check for the quality and range of the synthesized cDNA. Double-stranded cDNA was immediately treated with proteinase K (0.8 μg/ml) at 45°C for 20 min and washed three times with water using Amicon filters with a 100-kDa cutoff (Millipore, Bedford, MA, USA). The clean double-stranded cDNA was then digested with SfiI restriction enzyme at 50°C for 2 h followed by size fractionation on a ChromaSpin–400 drip column (Clontech). The profiles of the fractions were checked on a 1.1% agarose/EtBr (0.1 μg/ml), and fractions containing cDNA were pooled in three different groups according to size: large, medium, or small sequences. Each group was concentrated and washed three times with water using an Amicon filter with a 100-kDa cutoff. Concentrated cDNA was then ligated into a λ TriplEx2 vector (Clontech), and the resulting ligation mixture was packaged using GigaPack® Gold III Plus packaging extract (Stratagene, La Jolla, CA, USA) according to the manufacturer’s instructions. The packaged library was plated by infecting log-phase XL1-Blue Escherichia coli cells (Clontech). The percentage of recombinant clones was determined by blue-white selection screening on LB/MgSO4 plates containing X-gal/IPTG.
Sequencing of the Dm cDNA Library
The Dm SG cDNA library was plated on LB/MgSO4 plates containing X-gal/IPTG to an average of 250 plaques per 150-mm Petri plate. Recombinant (white) plaques were randomly picked up and transferred to 96-well MicroTest ™ U-bottom plates (BD BioScience, San José, CA, USA) containing 75 μl of H2O per well. The phage suspension was either immediately used for PCR or stored at 4°C until use.
To amplify the cDNA using a PCR reaction, 4 μl of the phage sample were used as a template. The primers were sequences from the λ TriplEx2 vector and named PT2F1 (5′-AAG TACTCTAGCAATTGTGAGC-3′) and PT2R1 (5′-CTCTTCGCTATTACGCCAGCTG-3′), positioned at the 5′ and 3′ end of the cDNA insert, respectively. The reaction was carried out in MicroAmp 96-well PCR plates (Applied Biosystems, Inc., Fullerton, CA, USA) using FastStart PCR Master (Roche Molecular Biochemicals, Indianapolis, IN, USA) on a GeneAmp® PCR system 9700 (Perkin Elmer Corp.). The PCR conditions were: 1 hold of 75°C for 3 min, 1 hold of 94°C for 4 min, 33 cycles of 94°C for 1 min, 49°C for 1 min, and 72°C for 1 min 20 s. The amplified products were analyzed on a 1.2% agarose/EtBr gel. cDNA library clones (2880 clones) were PCR amplified, and those showing a single band were selected for sequencing. The PCR products were used as a template for a cycle-sequencing reaction using a DTCS labeling kit (Beckman Coulter, Fullerton, CA, USA). The primer used for sequencing (PT2F3) is upstream of the inserted cDNA and downstream of the PT2F1 primer. The sequencing reaction was performed on a Perkin Elmer 9700 thermocycler. Conditions were 1 hold of 75°C for 2 min, 1 hold of 94°C for 4 min, and 30 cycles of 96°C for 20 s, 50°C for 20 s, and 60°C for 4 min. After cycle-sequencing the samples, a cleaning step was performed using the multiscreen 96-well plate cleaning system (Millipore). The 96-well multiscreening plate was prepared by adding a fixed amount (manufacturer’s specification) of Sephadex-50 (Amersham Pharmacia Biotech, Piscataway, NJ, USA) and 300 μl of deionized water. After partially drying the Sephadex in the multiscreen plate, the entire cycle-sequencing reaction was added to the center of each well, centrifuged at 2,500 rpm for 5 min, and the clean sample was collected on a sequencing microtiter plate (Beckman Coulter). The plate was then dried on a Speed-Vac SC110 model with a microtiter plate holder (Savant Instruments Inc., Holbrook, NY, USA). Dried samples were immediately resuspended with 25 μl of formamide, and one drop of mineral oil was added to the top of each sample. Samples were either sequenced immediately on a CEQ 2000 DNA sequencing instrument (Beckman Coulter) or stored at −30°C. A total of 2728 cDNA library clones was sequenced.
Proteomic Characterization Using One-Dimension Gel Electrophoresis (1DE) and tandem mass spectrometry (MS)
Both the soluble and insoluble (pellet following centrifugation at 16,000 ×g) protein fractions from SG homogenates from Dm corresponding to three pairs of SGs (approximately 500 μg of protein per gland) were brought up in reducing Laemmli gel-loading buffer. The samples were boiled for 10 min, and approximately 100 μg of protein were resolved on a NuPAGE 4–12% Bis-Tris precast gel. Separated proteins were visualized by staining with SimplyBlue (Invitrogen). Gels were sliced into 20 (soluble) or 24 (pellet) individual sections (Figure 1) that were destained and digested overnight with trypsin at 37°C. Peptides were extracted and desalted using ZipTips (Millipore) and resuspended in 0.1% TFA prior to MS analysis.
Figure 1.
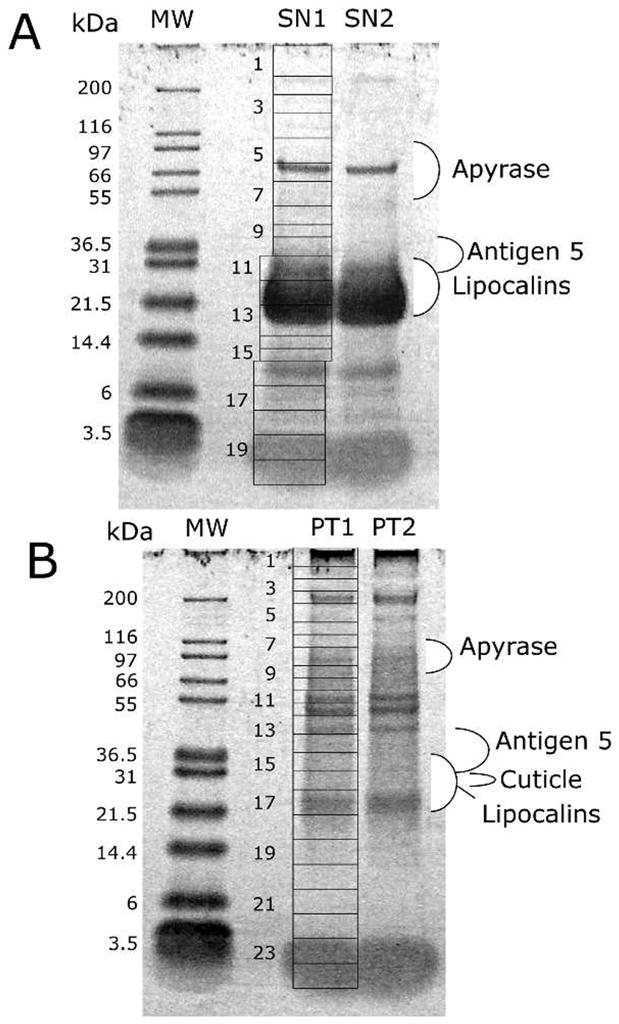
One-dimensional polyacrylamide gel electrophoresis separation of the soluble (A) and insoluble (B) fractions of Dipetalogaster maxima salivary proteins. The grids indicate how the duplicated gel bands containing the salivary proteins (SN1, SN2, PT1, and PT2) were cut for tryptic digestion and mass spectrometric detection of peptides following nano reversed-phase high-performance liquid chromatography. The left of the figures indicate the molecular weight of the markers (kDa) indicated in the MW lane. For more information, see text.
Nanoflow reverse-phase liquid chromatography tandem MS was performed using an Agilent 1100 nanoflow LC system (Agilent Technologies, Palo Alto, CA, USA) coupled online with a linear ion-trap (LIT) mass spectrometer (LTQ, ThermoElectron, San José, CA, USA). NanoRPLC columns were slurry packed in-house with 5 μm, 300-Å pore size C-18 phase (Jupiter, Phenomenex, CA) in a 75-μm i.d. × 10-cm fused silica capillary (Polymicro Technologies, Phoenix, AZ) with a flame-pulled tip. After sample injection, the column was washed for 30 min with 98% mobile phase A (0.1% formic acid in water) at 0.5 μl/min, and peptides were eluted using a linear gradient of 2% mobile phase B (0.1% formic acid in acetonitrile [ACN]) to 42% mobile phase B in 40 min at 0.25 μl/min, then to 98% B for an additional 10 min. The LIT-MS was operated in a data-dependent MS/MS mode in which each full MS scan was followed by seven MS/MS scans where the seven most abundant molecular ions were dynamically selected for collision-induced dissociation (CID) using a normalized collision energy of 35%. Dynamic exclusion was applied to minimize repeated selection of peptides previously selected for CID.
Tandem mass spectra were searched using SEQUEST on a 20-node Beowulf cluster against a Dm proteome database with methionine oxidation included as dynamic modification. Only tryptic peptides with up to two missed cleavage sites meeting a specific SEQUEST scoring criteria [delta correlation (ΔCn) ≥ 0.08 and charge state-dependent cross correlation (Xcorr) ≥ 1.9 for [M+H]1+, ≥ 2.2 for [M+2H]2+ and ≥ 3.5 for [M+3H]3+] were considered as legitimate identifications. The peptides identified by MS were converted to Prosite block format 22 by a program written in Visual Basic. This database was used to search matches in the Fasta-formatted database of salivary proteins, using the Seedtop program, which is part of the BLAST package. The result of the Seedtop search was piped into the hyperlinked spreadsheet to produce a hyperlinked text file with details of the match. Note that the ID lines indicate, for example, sDGM12_80, which means that one match was found for fragment number 80 from gel band 12. Because the same tryptic fragment can be found in many gel bands, another program was written to count the number of fragments for each gel band, displaying a summarized result in an Excel table, for example on cell AX4 of Supplemental Table S2. The summary in the form of sDGM12 → 5|sDGM13 → 4 indicates that five fragments were found in band 12, while four peptides were found in band 13. Furthermore, this summary included a protein identification only when two or more peptide matches to the protein were obtained from the same gel slice. The summary program also produces additional spreadsheet cells with the larger number of peptides found in a single gel band and the percent amino acid (aa) sequence coverage of the sum of the peptide matches, thus facilitating data analysis.
Proteomic Characterization Using Two-Dimensional Gel Electrophoresis (2DE) and peptide mass fingerprinting (PMF)
SGs obtained from several bug adults dissected at 7 to 9 days following the blood meal were punctured, and intraluminal fluids were harvested by centrifugation (2,000 × g, 5 min, 5°C). Salivary proteins were quantified using the Plus One 2D Quant Kit (GE Healthcare, Uppsala, Sweden) according to the manufacturer’s instructions. Saliva proteins (80 μg) resuspended in 350 μL of 2DE sample buffer (7 M urea, 2 M thiourea, 2.5% v/v Triton X-100, 85 mM DTT, 0.5% v/v Pharmalyte 3–10, 10% isopropanol) were applied to 18 cm pH 3–10 IPG gel strips, and submitted to isoelectrofocusing using an Ettan IPGphor3 Unit (GE-Healthcare, Piscataway, NJ, USA), no current for 6 h and 30 V for 6 h, followed by 500 V for 1 h, 1000 V for 1 h, and, 8000 V for 4 h, with a maximum current of 50 μA per strip, as optimized 19. Before SDS-PAGE, IPG strips were subjected to reduction in equilibrium buffer (50 mM Tris, pH 8.8, 6 M urea, glycerol 30%, 4% SDS), adding with 125 mM DTT for 40 min at room temperature and alkylation in equilibrium buffer, and with 300 mM acrylamide for an extended 20-min period. Subsequently, the proteins were separated by 12% SDS-PAGE on a Protean II system (BioRad, Richmond, CA, USA), and silver-stained as adapted from Blum et al.23 After washing with ultrapure H2O, the gels were scanned with the Image Scanner (PowerLook 1120; Amersham Biosciences) at 300-dpi resolution and stored in 1% acetic acid. Digitized images were analyzed with Image Master 2D Platinum 5.0 software (GE Healthcare) to count protein spots.
Protein identification was achieved by tryptic peptide fingerprinting using matrix-assisted laser desorption/ionization (MALDI)-time-of-flight (TOF)/TOF MS. Protein spots were cut from 2DE gels and processed as adapted from Garaguso and Borlak 24. Briefly, spots were destained for 10 min in a fresh solution of 15 mM potassium ferrocyanide and 50 mM sodium thiosulphate. After destaining, gel pieces were washed with three cycles of 200 μl ultrapure H2O followed by 50% ACN, then another two cycles in 50 mM NH4HCO3 for 5 min followed by washing in ACN for 5 min. During the final washing with ACN, gel fragments were pistil macerated and vacuum dried in a Speed Vacuum for 20 min. Dried gels were rehydrated with 5–10 μl of 25 mM NH4HCO3, 2.5 mM CaCl2, 12.5 ng/μl trypsin (Promega, Madison, WI, USA) and incubated in ice for 45 min. Immediately after incubation, the remaining solution was removed; to it was added, depending on the size of the gel piece, 5–10 μl of the same digestion buffer without enzyme, and it was incubated overnight at 37°C. The following day, 1 μl of 1% TFA was added to solution covering the gel pieces which contains the digestion product, and 1 μl of this acidified sample was loaded onto a 600-nm AnchorChip™ target plate (Bruker Daltonics, Karlsruhe, Germany) and allowed to dry completely. Afterward, 0.5 μl of 5-μg/μl DHB (2,5-dihydroxybenzoic acid) matrix in 30% ACN and 0.1% TFA was applied, allowed to dry completely, and subjected to PMF analysis. PMF analysis was performed using a MALDI-TOF/TOF mass spectrometer (Autoflex II; Bruker Daltonics). Mass spectra were processed using Flex Analysis 2.2 and Biotools 2.2 software (Bruker Daltronics).
Protein identification was performed using MASCOT software against the database of proteins predicted from the Dm cDNA library in our server. Spectra containing autodigested trypsin peptides were internally calibrated. The following parameters were used for database searches: monoisotopic mass accuracy up to 100 ppm for internally calibrated spectra and up to 200 ppm for uncalibrated spectra; up to one missed cleavage site; propionamide of cysteine as fixed modification; and oxidation of methionine and protein N-acetylation as variable modifications.
Bioinformatic Tools and Procedures
Expressed sequence tags (ESTs) were trimmed of primer and vector sequences, clusterized, and compared with other databases as previously described.25 The BLAST tool,26 CAP3 assembler,27 ClustalW,28 and TreeView software29 were used to compare, assemble, and align sequences and to visualize alignments. For functional annotation of the transcripts, we used blastx30 to compare the nucleotide sequences with the non-redundant (NR) protein database of the National Center of Biological Information (NCBI) and to the Gene Ontology (GO) database.31 Rpsblast 32 was used to search for conserved protein domains in the Pfam,33 Smart,34 Kog,35 and conserved domains (CDD) databases.36 We also compared the transcripts with other subsets of mitochondrial and rRNA nucleotide sequences.
Segments of the three-frame translations of the EST (because the libraries were unidirectional, we did not use six-frame translations) starting with a methionine in the first 100 predicted aa—or the predicted protein translation, in the case of complete coding sequences—were submitted to the SignalP server37 to help identify translation products that could be secreted. O-glycosylation sites on the proteins were predicted with the program NetOGlyc (http://www.cbs.dtu.dk/services/NetOGlyc/).38 Functional annotation of the transcripts was based on all the comparisons above. Following inspection of all results, transcripts were classified as either secretory (S), housekeeping (H), or of unknown (U) function, with further subdivisions based on function and/or protein families. Sequence alignments were done with the ClustalX software package.39 Phylogenetic analysis and statistical neighbor-joining bootstrap tests (10,000 iterations) of the phylogenies were performed with the Mega package.40 Hyperlinked Excel spreadsheets of the assembled ESTs and of the salivary protein database are supplied as Supplemental spreadsheets S1 and S2 at the journal site.
Results
Description of Clusterized Data Set/cDNA Library Characteristics
A total of 2728 sequences were used to assemble a clusterized database, yielding 1199 clusters of related sequences, 1014 of which contained only one EST. The consensus sequence of each cluster is named either a contig (deriving from two or more sequences) or a singleton (deriving from a single sequence). In this paper, we use the denomination contig to address sequences deriving from both consensus sequences and from singletons. The 1199 contigs were compared by the program blastx, blastn, or rpsblast 30 to the NR protein database of the NCBI, to the gene ontology database,31 to the conserved domains database of the NCBI,36 and to a custom-prepared subset of the NCBI nucleotide database containing either mitochondrial or rRNA sequences. Because the libraries are unidirectional, the three-frame translations of the dataset were also derived, and open reading frames starting with methionine and longer than 40-aa residues were submitted to SignalP server37 to help identify putative secreted proteins. The EST assembly, BLAST, and signal peptide results were transferred into an Excel spreadsheet for manual annotation.
Six categories of expressed genes derived from the manual annotation of the contigs (Table 1). The S category contained 13.4% of the clusters and 50.4% of the sequences, with an average number of 8.5 sequences per cluster. The H category had 8.9% and 7.6% of the clusters and sequences, respectively, and an average of 1.9 sequences per cluster. Forty-one percent of the sequences, corresponding to 75.8% of all contigs, were classified as U, because no assignment for their function could be made; most of those consisted of singletons. Possible transposable elements originated 20 clusters, mostly singletons, including both class I and class II elements, including a mariner transposase. We have also identified a microbial and a viral transcript in our dataset. These data can be downloaded as Supplemental Table S1 for the EST data.
Table 1.
Functional Classification of Transcripts Originating from the Salivary Gland Transcriptome of Dipetalogaster maxima
| Class | Number of Contigs | Number of ESTs | ESTs/Contig |
|---|---|---|---|
| Secreted | 161 | 1375 | 8.5 |
| Housekeeping | 107 | 207 | 1.9 |
| Viral | 1 | 1 | 1.0 |
| Microbial | 1 | 1 | 1.0 |
| Transposable element | 20 | 21 | 1.1 |
| Unknown | 909 | 1123 | 1.2 |
| Total | 1199 | 2728 |
Proteomic Assays
To attempt identification of the proteins expressed in the SGs of Dm, we separated the proteins by 1DE (Figure 1) and 2DE (Figure 2) followed by mass spectrometry identification, as indicated in Methods. The transcriptome and extracted coding sequences (CDS) provided for the database needed for assigning peptide hits. These hits are indicated in Figures 1 and 2 as well as in the Supplemental spreadsheets S1 and S2.
Figure 2.

Two-dimensional gel electrophoresis map of Dipetalogaster maxima secreted salivary proteins. IEF was performed from approximately 80 μg of saliva protein under denaturing conditions using a 3–10 linear IPG strip and following by 12% SDS-PAGE in a second dimension. The gel was silver stained. The dashed circles correspond to localizations of the different families of D. maxima identified proteins by MALDI-MS against the transcriptomic data. The spot numbers correspond to the identified proteins shown in supplemental table 1.
2DE analysis of Dm saliva was performed using isoelectrofocusing within a wide pH range (3–10) followed by 12% SDS-PAGE as optimized for the 2DE-based Ti proteomic analysis.19 The average number of silver-stained spots detected from the Dm 2DE profile was approximately 150 (Figure 2) versus about 200 from Ti saliva19 under the same experimental conditions. The majority of Dm saliva secreted proteins were visualized between 20 to 30 kDa on both 1DE and 2DE gels. Despite the absence of Dm-secreted saliva protein sequences available in the NCBI NR database, conventional database searching based on protein identification by peptide mass fingerprinting and peptide fragmentation fingerprinting was attempted without success. The identification was, however, successfully performed directly from the extensive Dm cDNA library reported here, although several predicted protein sequences are partial and consequently experimental and theoretical molecular masses and isoelectric points for several identified spots were very different (supplemental table 1)
The following sections will describe the protein families observed in the transcriptome analysis in context with the proteome assays reported in Figures 1 and 2. It was interesting to note that both 1DE- and 2DE-based-proteome analyses allowed assignment of regions of gels corresponding to the identified secreted protein families (Figures 1 and 2).
Transcripts Coding for Putative Secreted Proteins in Dm SGs
The S group of ESTs was further classified into six groups (Table 2) including lipocalins, antigen 5, apyrase, immunity-related products, hemolysins, and other putative secreted polypeptides, as described in more detail below.
Table 2.
Functional Classification of Transcripts Attributed to Secreted Products from the Sialotranscriptome of Dipetalogaster maxima
| Class | Number of ESTs |
|---|---|
| Lipocalins | 1285 |
| Antigen 5 family | 67 |
| Apyrase/5′ nucleotidase family | 9 |
| Immunity-related products | 2 |
| Hemolysin family | 1 |
| Other putative secreted polypeptides | 11 |
| Total | 1375 |
Lipocalins
The most abundant group of putative secreted proteins in Dm SGs is the lipocalins, corresponding to 93% of the transcripts in the S class. Similarly, the sialotranscriptome of T. brasiliensis revealed a high content of lipocalins in its SGs, with 93.8% of the transcripts coding for putative secreted proteins.18 Lipocalins are widely distributed and heterogeneous proteins occurring in animals, plants, and bacteria. An interesting feature of the lipocalins is their well conserved three-dimensional structure, contrasting with their primary sequence that shows a low percentage of similarity when comparing randomly selected members of this family.41 They are typically small extracellular proteins sharing several common molecular recognition properties: the binding of small, principally hydrophobic molecules, and binding to specific cell-surface receptors, forming covalent and non-covalent complexes with other soluble macromolecules. Although they have been classified mainly as transport proteins, it is now clear that members of the lipocalin family fulfill a wide variety of functions.15 Efforts to discover the role of salivary lipocalins in blood-sucking insects through functional genomics and proteomic studies have identified anticoagulants, antiplatelets, anti-complement, and vasodilatory molecules.42
We found lipocalins similar to salivary proteins of other species of Triatoma such as pallidipin, an inhibitor of collagen-induced platelet aggregation from Triatoma pallidipennis,43 and procalin, a salivary allergen from Triatoma protracta.44 The South American insect Ti was also shown to contain salivary cDNA sequences similar to these previously described lipocalins.20 Lipocalins have also been found in tick saliva and in Rhodnius performing similar functions, such as histamine and serotonin binding.17,21,45
Supplemental spreadsheet S2 presents the coding sequences for 68 lipocalins. Several of these are possible alleles of the same gene, because they are >90 % identical at the aa level. If we exclude those within this group, 45 sequences are still obtained, indicating a vast expansion of this protein family.
Lipocalin sequences from Dm were aligned with sequences from other Triatominae, and a neighbor-joining phylogenetic tree was constructed (Figure 3). The diverse nature of lipocalins has been noted before, and the phylogenetic tree shows divergent groups of proteins, suggesting that these sequences have evolved beyond recognition of a common ancestor; however, three groups of Dipetalogaster proteins with more than 2 sequences each are clearly discerned in the phylogenetic tree. Group I (Figure 3) appears to be a large Dipetalogaster gene expansion that might have occurred by multiple gene duplication events. This group contains the Procalin antigen precursor of T. protracta. Group II also represents a gene expansion and is related to the Pallidipin family of platelet aggregation inhibitors, and, more distantly to the triplatins, also platelet inhibitors from T. infestans. Group III is more closely related to the Triafestin family which are inhibitors of the plasma kallikrein-kinin system, and also to Triatin, a thrombin inhibitor.
Figure 3.

Dendrogram of the Dipetalogaster maxima salivary lipocalins with other insect salivary lipocalins. The sequences derived from the nonredundant protein database of the National Center for Biotechnology Information (NCBI) are represented by six letters followed by the NCBI gi| accession number. The six letters derive from the first three letters of the genus and the first three letters from the species name. The protein sequences were aligned by the Clustal program,39 and the dendrogram was made with the Mega package40 after 10,000 bootstraps with the neighbor-joining algorithm. The bar at the top represents 20% amino acid substitution. The colorful squares indicate each insect species whose sequences were used.
Proteomic experiments revealed the expression of most lipocalins, as indicated in Supplemental spreadsheet S2. These lipocalins were identified in gel fractions 11, 12, and 13 (Figure 1A), and fractions 15, 16, and 17 (Figure 1B) at regions corresponding to MW markers ranging from 21 to 30 kDa and coincident with the strongest gel band co-migrating with the 21.5 kDa marker, in agreement with the estimated mature molecular mass of these proteins. The 2DE gels presented the same lipocalins spread over different pIs, mostly to the basic side of the gel, providing good resolution of the various polypeptides that appear as a single blob in Figure 1. Of note is the set of spots that migrated in the bottom of the gel (around 20 kDa) that revealed many lipocalins. The lipocalins annotated as Dm-96 and Dm-58 are the most abundantly transcribed and have been assembled from 138 and 122 ESTs, respectively. Dm-96 is identified with neighboring spots 36 and 37 in the 2D gel, whereas six fragment ions were obtained in each of the spots, while Dm-58 was identified in several spots, including the strong spots 32 (10 fragment ions), 43 (12 ions) and 11 (10 ions), this last spot near the 40-kDa marker, and same pI as spot 32, indicating a possible dimer or posttranslational modifications. These abundant lipocalins are candidates for antagonizing histamine or serotonin, where micromolar amounts of these 20-kDa proteins are needed to bind and neutralize the near micromolar amounts of the biogenic amines that accumulate in inflamed tissues. Less abundant lipocalins may act by binding to agonists of inflammation that occur at lower concentrations, such as thromboxane A2 and leukotrienes, which are the targets of several tick lipocalins.46,47 The intense spots numbered 23, 20, 24, and 25 or gel regions consistent with a molecular mass of 25 kDa produce ion matches to members of the Pallidipin clade. Spots 22 and 21, on the extreme basic side of the gel, provide matches to Dm-1189 and Dm-99, respectively.
Apyrase/5′ nucleotidase family
Apyrases are ATP-diphosphohydrolases that catalyze hydrolysis of both ATP and ADP to AMP, facilitating blood feeding by degradation of ADP, a mediator of platelet aggregation and inflammation,8 and ATP, a mediator of neutrophil activation.48,49 Apyrases are very commonly found in the SGs of blood-feeding insects9 and ticks,50 the activity being higher in mammal feeders and lower in bird or lizard feeders.51–53 The salivary apyrase of Ti was cloned and shown to be a member of the 5′ nucleotidase family.54,55 Partial coding sequences (truncated in the 5′ region) for members of the 5′ nucleotidase family were found in the sialotranscriptome of Dm (Supplemental spreadsheets S1 and S2).
5′-nucleotidases are typically membrane bound and extracellular enzymes. A glycophosphatidyl-inositol (GPI) anchor in the carboxyterminal domain is associated with binding to the membrane and is indicated by the box in the alignment in Figure 4; indeed, 5′ nucleotidases from bovine liver,56 electric ray,57 human,58 and rat liver59 have GPI anchor motifs, according to the PredGPI prediction server.60 Apyrases of the 5′-nucleotidase family lacking the aa sequence required for a GPI anchor are secreted,61 which suggests that apyrases from Dm are probably secreted, although the amino-terminally truncated sequences does not permit verification of a signal peptide for secretion. The contig Dm-68 has an extension characteristic of mucins, rich in serine and threonine, when compared with other apyrases/5′-nucleotidases (Figure 4), suggesting that this protein can be highly glycosylated. In both cases of Dm, no GPI anchor region was found, indicating that the proteins are secreted.
Figure 4.

Clustal alignment of members of the apyrase/5′ nucleotidase family deriving from salivary glands of D. maxima and from other organisms. The D. maxima proteins are Dm-60, Dm-68, and Dm-1003. The remaining sequences are named with the first three letters from the genus name followed by two letters from the species name and by their NCBI protein accession number. The box shows the membrane anchor region present in the salivary 5′ nucleotidases. The symbols above the alignment indicate: (*) identical sites; (:) conserved sites; (.) less conserved sites. For more details, see text.
The 1DE proteomic experiment (Figure 1) identifies apyrase fragments on fractions 6 (where a strong gel band exists, just above the 66-kDa marker) and fraction 7 of the soluble protein gel (Figure 1A), and in fractions 8 and 9 of the insoluble protein gel (Figure 1B). The 2DE experiment (Figure 2) identifies apyrase isoforms from the unique contig Dm-68 in the vicinal spots 1, 2, 3, and 4, each having between three and five ion matches. These bands are located under the 97-kDa marker, in a slightly acidic pI, in a region of the gel consistent with the 79-kDa molecular mass of Triatoma apyrases.54. Furthermore, Ti saliva would have a multigene apyrase family with at least three to five forms and N-glycosylation.62 In 2DE-based-proteomic analysis, five Ti apyrase forms displayed a typical pattern of isoforms for each form.19 The same 2DE pattern was observed in the case of the Dm identified apyrases, suggesting that posttranslational modifications might also be a source of apyrase heterogeneity in this saliva.
Antigen 5 family
This is a family of secreted proteins that belong to the CAP family (cysteine-rich secretory proteins; antigen 5 proteins of insects; pathogenesis-related protein 1 of plants).63 The CAP family is related to venom allergens in social wasps and ants.64,65 Antigen 5 is the most studied member of a conserved family of proteins, and together with hyaluronidase and phospholipase A1 is one of the three major venom proteins acting as allergens in vespids.66,67 Members of this protein family are found in the SGs of most blood-sucking insects. 11, 13, 20, 68–70 However, the function of most antigen 5 proteins in the saliva of blood-sucking arthropod is still unknown. Exceptionally, a tabanid protein of this family incorporated a disintegrin domain and acquired a platelet aggregation inhibitory function 71, and another family member expressed in the SGs of the stable fly binds immunoglobulins and could serve as inhibitor of the classical pathway of complement activation 72.
We here report five salivary members of the antigen 5 family found in Dm SGs (Supplemental spreadsheet S2), two of which are probable alleles (Dm-124 and Dm-127). Alignment and phylogenetic analysis of insect members of this family indicate that Dm salivary antigen 5 protein clusters with other reported antigen 5 proteins from R. prolixus, Ti, and vespids (Figure 5). The hymenoptera protein, from vespids, has an additional domain after the signal peptide, indicating that its evolution probably diverged at some point. Like Rhodnius and Triatoma proteins, antigen 5 contigs from Dm have a poly-Lys-rich basic tail, which may direct these proteins to negatively charged membranes such as activated platelet surfaces. This binding/interaction on the platelet surface could interfere or block platelets’ physiologic function, disrupting, then, the host hemostasis process.
Figure 5.
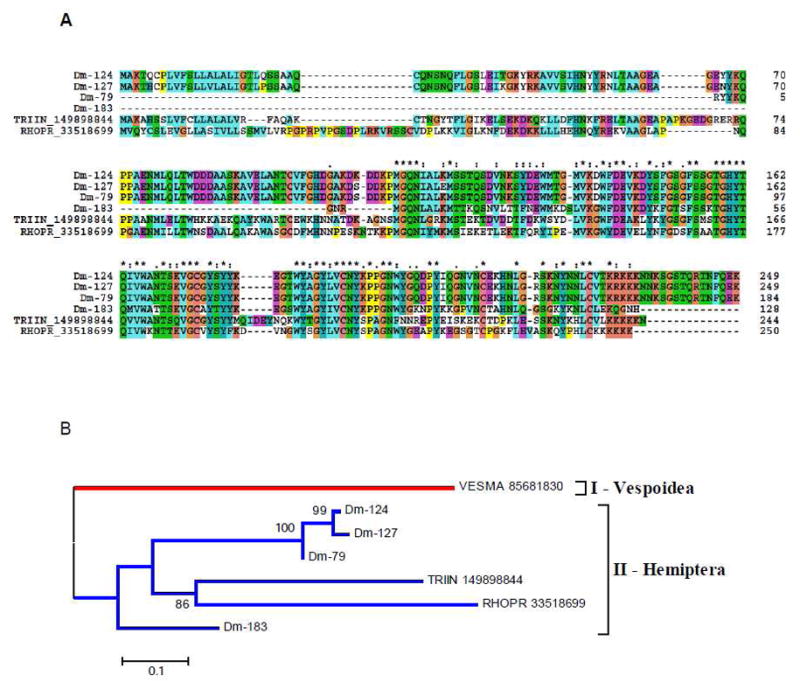
(A) Clustal alignment of D. maxima salivary Antigen-5 proteins with members of the same family from other Triatominae. (B) Phylogenetic tree of selected members of the antigen-5 family of proteins from Hemiptera and Vespoidea, obtained by the NJ method after 10,000 bootstraps. The numbers in the phylogram nodes indicates percent bootstrap support for the phylogeny. The bar at the bottom indicates 10% amino acid divergence in sequences.
The 1DE experiment (Figure 1) shows most of the ions hitting antigen 5 proteins located in band 11 of the soluble protein gel and an equivalent location in the insoluble protein gel (Figure 1A). The 2DE experiment indicates that the strongly stained spot 14—as well as the vicinal spots 13, 15, 16, and 17—produce hits to the antigen 5 family. In all cases, the gel location is consistent with the MW of this protein family, between 25 and 30 kDa.
Hemolysin family
Hemolysins are secreted, water-soluble proteins that form transmembrane polymeric channels. They are capable of permeating target cells, forming pores in cytoplasmic membranes of erythrocytes, leukocytes, and other cells, causing modification of cellular functions and/or lysis of host cells. This family of proteins is more often described in microorganisms such as Escherichia coli, Staphylococcus aureus, and Bacillus cereus,73–75 being considered a virulence factor. Collectively, they are members of the RTX (repeats in toxin) protein family.76 The presence of amphipathic and hydrophilic domains confers to the protein an overall amphiphilic character, which would explain its ability to be secreted and to interact with membranes.77–79
We found one contig with similarity to salivary-secreted hemolysin-like proteins (Supplemental figure 1), previously described in the sialotranscriptome of Ti. These proteins could act as cytolytic proteins, causing erythrocyte lysis in their saliva and helping the early steps of the digestion process, or having antimicrobial activity. A single ion with sequence A-T-S-Y-[IL]-G-N-[KQ]-V-[IL]-D-[KQ] was found matching Dm-594 in gel band 17 of the insoluble protein experiment (Figure 1B). This is not a promiscuous match, having been found solely to this protein.
Immunity-related products
One contig from the Dm cDNA library showed similarity to a portion of the carboxyterminal region of hemolectin (Supplemental figure 2), a multidomain protein described in Drosophila. Hemolectin is the most abundant insect clot protein, and it shares conserved domains with von Willebrand factor and coagulation and complement factors.80 Clotting of hemolymph in arthropods is important to promote wound healing and limit loss of hemolymph. It also plays a role in immune defense, forming a secondary barrier to infection.81 Also, loss of hemolectin in Drosophila was associated with more susceptibility to infection by E. coli, suggesting it could be part of an integrated defense mechanism to prevent bacterial contamination82 or, alternatively, being exapted for an antihemostatic function. A single ion corresponding to the sequence [KQ]-E-S-V-[KQ]-T-C-[KQ]-[KQ]-T-C-[KQ] and found in band 9 of the insoluble protein gel matches Dipetalogaster hemolectin, in a region of the gel consistent with proteins in the range of 70–80 kDa. Hemolectins are large proteins with over 3000 aa, suggesting the match may be spurious or related to a protein fragment.
Proteins containing Gly-Tyr repeats have been commonly found in sialotranscriptomes of blood-sucking arthropods. These proteins resemble nematode peptides identified as having an immune function83 but are also similar to cuticular proteins. No matches to this predicted protein were found in the proteomic experiments.
Other putative secreted polypeptides
Histidine-rich peptide precursor
This EST sequence found in Dm shows similarity to secreted histidine-rich peptide precursors (Supplemental figure 3). Polypeptides rich in histidine are described as both small and large molecules. Histatins, a family of small histidine-rich basic salivary proteins, exhibit antimicrobial activity against bacteria and fungi (perhaps by their Cu or Ni or Zn ion chelating properties), similar to many other small basic polypeptides, being considered a component in the host defense system.84 Histidine-rich glycoprotein is a plasma glycoprotein that has a multidomain structure and interacts with many ligands including Zn+2, tropomyosin, heparin, plasminogen, plasmin, fibrinogen, thrombospondin, IgG and complement. This protein could act as an extracellular adaptor, bringing together different ligands on cell surfaces.85 The histidine-rich region in these proteins is highly responsible to Zn+2 and low pH, conditions associated with sites of tissue injury.86 Metal binding by histidine-rich proteins could have important physiologic functions related to microbial growth.84 For the triatomine Dm, the ability to interact with molecules related to coagulation and immune response through a histidine-rich protein could be an advantage, helping the acquisition of the blood meal and avoiding the host response. Also, they could play a role in the innate defense system. No matches to this protein were found in the proteomic experiments.
Larval cuticle protein
This cDNA library also revealed a contig with similarity to larval cuticle protein, and the alignment shows the presence of a chitin-binding domain (Supplemental figure 4). Arthropod cuticles are composed of fiber of chitin, a biopolymer that contributes strength and rigidity when in fibrillar form. One of the common functions for chitin is its structural role, serving as mechanical support and protective barriers in the exoskeleton of arthropods.87,88 This protein may be related to the structure of the salivary duct or tracheoli. The 1D proteomic experiment with the insoluble protein fraction revealed six ions matching Dm-850 in band 16, located between the 21.5- and 31-kDa markers (Figure 1B).
Housekeeping (H) Genes
The 107 gene contigs (comprising 207 ESTs) attributed to H genes expressed in the SGs of Dm were further characterized into 16 groups consistent to their possible function (Table 3). According to an organ specialized in secreting polypeptides and as observed in previous sialotranscriptomes,11,17,70,89 the two larger sets were associated with protein synthesis machinery (102 ESTs in 33 contigs) and with energy metabolism (20 contigs containing 43 ESTs). We have also included in this category a group of 10 ESTs that grouped into 10 contigs and represent conserved proteins of unknown function presumably associated with cellular metabolism. Other sequences with homology to housekeeping proteins include those coding for ribosomal proteins, cytochrome, and NADH-dehydrogenase, among other molecules (Table 3 and Supplemental Spreadsheet S1). Several of these housekeeping gene products were identified by the proteomic experiments. They can be verified in Supplemental spreadsheets S1 and S2, and are not marked in Figures 1 and 2.
Table 3.
Functional Classification of Transcripts Attributed to the Housekeeping Class Originating from the Sialotranscriptome of Dipetalogaster maxima
| Class | Number of Contigs | Number of ESTs | ESTs/Contig |
|---|---|---|---|
| Protein synthesis machinery | 33 | 102 | 3.1 |
| Metabolism, energy | 20 | 43 | 2.2 |
| Unknown, conserved | 10 | 10 | 1.0 |
| Transcription machinery | 7 | 7 | 1.0 |
| Signal transduction | 7 | 10 | 1.4 |
| Protein export machinery | 6 | 6 | 1.0 |
| Protein modification machinery | 5 | 9 | 1.8 |
| Transporters/storage | 4 | 4 | 1.0 |
| Cytoskeletal | 4 | 4 | 1.0 |
| Transcription factor | 2 | 2 | 1.0 |
| Proteasome machinery | 2 | 2 | 1.0 |
| Oxidant metabolism/detoxification | 2 | 2 | 1.0 |
| Metabolism, carbohydrate | 2 | 2 | 1.0 |
| Nuclear regulation | 1 | 2 | 2.0 |
| Metabolism, intermediate | 1 | 1 | 1.0 |
| Extracellular matrix/cell adhesion | 1 | 1 | 1.0 |
| Total | 107 | 207 |
Conclusions
In an attempt to improve our understanding of the variety of proteins and transcripts expressed in Dm SGs, we have made a cDNA library and both 1D and 2D-gel electrophoreses using mRNA and proteins from this same tissue. We described the set of cDNA present in the SGs of Dm. Comparison of this set with other triatomine sialomes indicates that Dipetalogaster has a less complex sialome when compared to Triatoma. In addition, the 2DE map of the Dm saliva presented a protein number considerably inferior to that of Ti saliva, 150 versus 200, respectively, under the same experimental conditions. The lesser salivary proteome complexity of the Dm in comparison with Ti could be correlated with the quantity of SG pairs, one versus three, respectively. For example, the following protein families are missing when compared with the Ti sialome:19,20 trialysin, triatox, kazal, pheromone-binding, inositol phosphatase, and serine protease. Perhaps these extra protein families in Triatoma reflect its adaptation to feed on mammals, while Dipetalogaster mainly feeds on lizards; however, we cannot exclude the possible that more intense sequencing of the Dipetalogaster sialotranscriptome may reveal transcription of these families, although these Triatoma-secreted proteins were not identified by proteomic studies. On the other hand, Dipetalogaster homologs were found for previously orphan protein families from Ti, such as the hemolysins family, indicating the ancient status of this family within Triatomines before the Dipetalogaster/Triatoma branch. Most of these proteins have unknown function. Expression and bioassay of the novel proteins will ultimately characterize the salivary pharmacologic complexity from Dm evolution to blood feeding.
This cDNA library will facilitate our continuing efforts to understanding the evolution of blood sucking in vector arthropods and the discovery of novel pharmacologically active compounds.
Supplementary Material
Acknowledgments
This work was supported by the Intramural Research Program of the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organization imply endorsement by the government of the United States of America. This work was supported by Brazilian Grants from CNPq, FINEP, FAP-DF, CAPES, and PRONEX.
Because T.C.F.A., S.C., P.B.M.S., I.M.B.F., C.N.A., V.M.P., R.M.L.Q., C.A.R., J.M.S., and J.M.C.R. are government employees and this is a government work, the work is in the public domain in the United States. Notwithstanding any other agreements, the NIH reserves the right to provide the work to PubMedCentral for display and use by the public, and PubMedCentral may tag or modify the work consistent with its customary practices. You can establish rights outside of the U.S. subject to a government use license.
Abbreviations
- 2D
two-dimensional
- aa
amino acid
- ACN
acetonitrile
- Dm
Dipetalogaster maxima
- EST
expressed sequence tags
- H
putative housekeeping transcripts
- MALDI
matrix-assisted laser desorption/ionization
- MS
mass spectrometry
- NCBI
National Center of Biological Information
- NR
non-redundant
- PMF
peptide mass fingerprinting
- S
putative secreted transcripts
- SG
salivary glands
- Ti
Triatoma infestans
- TOF
time-of-flight
- U
unknown function transcripts
Footnotes
• Supplemental Spreadsheet S1: Excel spreadsheet of assembled ESTs
• Supplemental Spreadsheet S2: Excel spreadsheet of the salivary protein database
Supplemental materials are available free of charge via the Internet at http://pubs.acs.org.
Contributor Information
Teresa C. F. Assumpção, Email: assumpcaot@niaid.nih.gov.
Sébastien Charneau, Email: charneau@unb.br.
Paula B. M. Santiago, Email: paulabatriz@hotmail.com.
Ivo M. B. Francischetti, Email: ifrancischetti@niaid.nih.gov.
Zhaojing Meng, Email: mengz@ncifcrf.gov.
Carla N. Araújo, Email: cnunes@unb.br.
Van M. Pham, Email: vpham@niaid.nih.gov.
Rayner M. L. Queiroz, Email: raynermyr@gmail.com.
Cleudson Nery de Castro, Email: cleudson@unb.br.
Carlos André Ricart, Email: ricart@unb.br.
Jaime M. Santana, Email: jsantana@unb.br.
José M. C. Ribeiro, Email: jribeiro@niaid.nih.gov.
References
- 1.Salazar Schettino PM, de Haro Arteaga I, Uribarren Berrueta T. Chagas disease in Mexico. Parasitol Today. 1988;4(12):348–352. doi: 10.1016/0169-4758(88)90004-x. [DOI] [PubMed] [Google Scholar]
- 2.Nijhout HF. Abdominal stretch reception in Dipetalogaster maximus (Hemiptera: Reduviidae) J Insect Physiol. 1984;30(8):629–633. [Google Scholar]
- 3.Guzmán-Bracho C. Epidemiology of Chagas disease in Mexico: an update. Trends Parasitol. 2001;17(8):372–376. doi: 10.1016/s1471-4922(01)01952-3. [DOI] [PubMed] [Google Scholar]
- 4.Zeledón R, Rabinovich JE. Chagas’ disease: an ecological appraisal with special emphasis on its insect vectors. Annu Rev Entomol. 1981;26:101–133. doi: 10.1146/annurev.en.26.010181.000533. [DOI] [PubMed] [Google Scholar]
- 5.Zárate LG. Comportamiento de los triatomineos en relacion a su potencial transmisor de la enfermedad de Chagas (Hemiptera: Reduviiidae) Fol Entomol Mex. 1984;61:257–271. [Google Scholar]
- 6.Jiménez ML, Palacios C. Incidencia de la chinche piedrera (Dipetalogaster maximus) (Hemiptera:Heteroptera:Reduviidae) vector de Trypanosoma cruzi en zonas urbanas de La Paz, Baja California Sur, Mexico. Ann del Inst de Biol UNAM, Ser Zoologia. 1999;70(2):215–221. [Google Scholar]
- 7.Jiménez ML, Llinas J, Palacios C. Infection rates in Dipetalogaster maximus (Reduviidae: Triatominae) by Trypanosoma cruzi in the Cape Region, Baja California Sur, Mexico. J Med Entomol. 2003;40(1):18–21. doi: 10.1603/0022-2585-40.1.18. [DOI] [PubMed] [Google Scholar]
- 8.Ribeiro JM, Francischetti IM. Role of arthropod saliva in blood feeding: sialome and post-sialome perspectives. Annu Rev Entomol. 2003;48:73–88. doi: 10.1146/annurev.ento.48.060402.102812. [DOI] [PubMed] [Google Scholar]
- 9.Ribeiro JMC, Arca B. From sialomes to the sialoverse: an insight into the salivary potion of blood feeding insects. Adv Insect Physiol. 2009;37:59–118. [Google Scholar]
- 10.Ribeiro JM. Blood-feeding arthropods: live syringes or invertebrate pharmacologists? Infect Agents Dis. 1995;4(3):143–152. [PubMed] [Google Scholar]
- 11.Francischetti IM, Valenzuela JG, Pham VM, Garfield MK, Ribeiro JM. Toward a catalog for the transcripts and proteins (sialome) from the salivary gland of the malaria vector Anopheles gambiae. J Exp Biol. 2002;205(Pt 16):2429–2451. doi: 10.1242/jeb.205.16.2429. [DOI] [PubMed] [Google Scholar]
- 12.Valenzuela JG, Francischetti IM, Pham VM, Garfield MK, Mather TN, Ribeiro JM. Exploring the sialome of the tick Ixodes scapularis. J Exp Biol. 2002;205(Pt 18):2843–2864. doi: 10.1242/jeb.205.18.2843. [DOI] [PubMed] [Google Scholar]
- 13.Valenzuela JG, Pham VM, Garfield MK, Francischetti IM, Ribeiro JM. Toward a description of the sialome of the adult female mosquito Aedes aegypti. Insect Biochem Mol Biol. 2002;32(9):1101–1122. doi: 10.1016/s0965-1748(02)00047-4. [DOI] [PubMed] [Google Scholar]
- 14.Ribeiro JM, Schneider M, Isaias T, Jurberg J, Galvao C, Guimaraes JA. Role of salivary antihemostatic components in blood feeding by triatomine bugs (Heteroptera) J Med Entomol. 1998;35(4):599–610. doi: 10.1093/jmedent/35.4.599. [DOI] [PubMed] [Google Scholar]
- 15.Flower DR, North AC, Sansom CE. The lipocalin protein family: structural and sequence overview. Biochim Biophys Acta. 2000;1482(1–2):9–24. doi: 10.1016/s0167-4838(00)00148-5. [DOI] [PubMed] [Google Scholar]
- 16.Montfort WR, Weichsel A, Andersen JF. Nitrophorins and related antihemostatic lipocalins from Rhodnius prolixus and other blood-sucking arthropods. Biochim Biophys Acta. 2000;1482(1–2):110–118. doi: 10.1016/s0167-4838(00)00165-5. [DOI] [PubMed] [Google Scholar]
- 17.Ribeiro JM, Andersen J, Silva-Neto MA, Pham VM, Garfield MK, Valenzuela JG. Exploring the sialome of the blood-sucking bug Rhodnius prolixus. Insect Biochem Mol Biol. 2004;34(1):61–79. doi: 10.1016/j.ibmb.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 18.Santos A, Ribeiro JM, Lehane MJ, Gontijo NF, Veloso AB, Sant’ Anna MR, Nascimento Araujo R, Grisard EC, Pereira MH. The sialotranscriptome of the blood-sucking bug Triatoma brasiliensis (Hemiptera, Triatominae) Insect Biochem Mol Biol. 2007;37(7):702–712. doi: 10.1016/j.ibmb.2007.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Charneau S, Junqueira M, Costa CM, Pires DL, Fernandes ES, Bussacos AC, Sousa MV, Ricart CAO, Shevchenko A, Teixeira ARL. The saliva proteome of the blood-feeding insect Triatoma infestans is rich in platelet-aggregation inhibitors. Int J Mass Spectrom. 2007;268(2–3):265–276. [Google Scholar]
- 20.Assumpção TC, Francischetti IM, Andersen JF, Schwarz A, Santana JM, Ribeiro JM. An insight into the sialome of the blood-sucking bug Triatoma infestans, a vector of Chagas’ disease. Insect Biochem Mol Biol. 2008;38(2):213–232. doi: 10.1016/j.ibmb.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paesen GC, Adams PL, Nuttall PA, Stuart DL. Tick histamine-binding proteins: lipocalins with a second binding cavity. Biochim Biophys Acta. 2000;1482(1–2):92–101. doi: 10.1016/s0167-4838(00)00168-0. [DOI] [PubMed] [Google Scholar]
- 22.Hulo N, Bairoch A, Bulliard V, Cerutti L, De Castro E, Langendijk-Genevaux PS, Pagni M, Sigrist CJ. The PROSITE database. Nucleic Acids Res. 2006;34(Database issue):D227–230. doi: 10.1093/nar/gkj063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blum H, Beier H, Gross HJ. Improved silver staining of plant-proteins, RNA and DNA in polyacrylamide gels. Electrophoresis. 1987;8(2):93–99. [Google Scholar]
- 24.Garaguso I, Borlak J. Matrix layer sample preparation: an improved MALDI-MS peptide analysis method for proteomic studies. Proteomics. 2008;8(13):2583–2595. doi: 10.1002/pmic.200701147. [DOI] [PubMed] [Google Scholar]
- 25.Valenzuela JG, Francischetti IM, Pham VM, Garfield MK, Ribeiro JM. Exploring the salivary gland transcriptome and proteome of the Anopheles stephensi mosquito. Insect Biochem Mol Biol. 2003;33(7):717–732. doi: 10.1016/s0965-1748(03)00067-5. [DOI] [PubMed] [Google Scholar]
- 26.Altschul SF, Gish W. Local alignment statistics. Methods Enzymol. 1996;266:460–480. doi: 10.1016/s0076-6879(96)66029-7. [DOI] [PubMed] [Google Scholar]
- 27.Huang X, Madan A. CAP3: A DNA sequence assembly program. Genome Res. 1999;9(9):868–877. doi: 10.1101/gr.9.9.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thompson JD, Higgins DG, Gibson TJ. ClustalW: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Page RD. TreeView: an application to display phylogenetic trees on personal computers. Comput Appl Biosci. 1996;12(4):357–358. doi: 10.1093/bioinformatics/12.4.357. [DOI] [PubMed] [Google Scholar]
- 30.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schaffer AA, Aravind L, Madden TL, Shavirin S, Spouge JL, Wolf YI, Koonin EV, Altschul SF. Improving the accuracy of PSI-BLAST protein database searches with composition-based statistics and other refinements. Nucleic Acids Res. 2001;29(14):2994–3005. doi: 10.1093/nar/29.14.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bateman A, Birney E, Durbin R, Eddy SR, Howe KL, Sonnhammer EL. The Pfam protein families database. Nucleic Acids Res. 2000;28(1):263–266. doi: 10.1093/nar/28.1.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Letunic I, Goodstadt L, Dickens NJ, Doerks T, Schultz J, Mott R, Ciccarelli F, Copley RR, Ponting CP, Bork P. Recent improvements to the SMART domain-based sequence annotation resource. Nucleic Acids Res. 2002;30(1):242–244. doi: 10.1093/nar/30.1.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov DM, Mazumder R, Mekhedov SL, Nikolskaya AN, Rao BS, Smirnov S, Sverdlov AV, Vasudevan S, Wolf YI, Yin JJ, Natale DA. The COG database: an updated version includes eukaryotes. BMC Bioinformatics. 2003;4:41. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marchler-Bauer A, Panchenko AR, Shoemaker BA, Thiessen PA, Geer LY, Bryant SH. CDD: a database of conserved domain alignments with links to domain three-dimensional structure. Nucleic Acids Res. 2002;30(1):281–283. doi: 10.1093/nar/30.1.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nielsen H, Engelbrecht J, Brunak S, von Heijne G. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 1997;10(1):1–6. doi: 10.1093/protein/10.1.1. [DOI] [PubMed] [Google Scholar]
- 38.Hansen JE, Lund O, Tolstrup N, Gooley AA, Williams KL, Brunak S. NetOglyc: prediction of mucin type O-glycosylation sites based on sequence context and surface accessibility. Glycoconj J. 1998;15(2):115–130. doi: 10.1023/a:1006960004440. [DOI] [PubMed] [Google Scholar]
- 39.Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The Clustal_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25(24):4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kumar S, Tamura K, Nei M. MEGA3: Integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform. 2004;5(2):150–163. doi: 10.1093/bib/5.2.150. [DOI] [PubMed] [Google Scholar]
- 41.Flower DR. Multiple molecular recognition properties of the lipocalin protein family. J Mol Recognit. 1995;8(3):185–195. doi: 10.1002/jmr.300080304. [DOI] [PubMed] [Google Scholar]
- 42.Andersen JF, Gudderra NP, Francischetti IM, Ribeiro JM. The role of salivary lipocalins in blood feeding by Rhodnius prolixus. Arch Insect Biochem Physiol. 2005;58(2):97–105. doi: 10.1002/arch.20032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Noeske-Jungblut C, Kratzschmar J, Haendler B, Alagon A, Possani L, Verhallen P, Donner P, Schleuning WD. An inhibitor of collagen-induced platelet aggregation from the saliva of Triatoma pallidipennis. J Biol Chem. 1994;269(7):5050–5053. [PubMed] [Google Scholar]
- 44.Paddock CD, McKerrow JH, Hansell E, Foreman KW, Hsieh I, Marshall N. Identification, cloning, and recombinant expression of procalin, a major triatomine allergen. J Immunol. 2001;167(5):2694–2699. doi: 10.4049/jimmunol.167.5.2694. [DOI] [PubMed] [Google Scholar]
- 45.Sangamnatdej S, Paesen GC, Slovak M, Nuttall PA. A high affinity serotonin- and histamine-binding lipocalin from tick saliva. Insect Mol Biol. 2002;11(1):79–86. doi: 10.1046/j.0962-1075.2001.00311.x. [DOI] [PubMed] [Google Scholar]
- 46.Mans BJ, Ribeiro JM. Function, mechanism and evolution of the moubatin-clade of soft tick lipocalins. Insect Biochem Mol Biol. 2008;38(9):841–852. doi: 10.1016/j.ibmb.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mans BJ, Ribeiro JM. A novel clade of cysteinyl leukotriene scavengers in soft ticks. Insect Biochem Mol Biol. 2008;38(9):862–870. doi: 10.1016/j.ibmb.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kuroki M, Minakami S. Extracellular ATP triggers superoxide production in human neutrophils. Biochem Biophys Res Comm. 1989;162:377–380. doi: 10.1016/0006-291x(89)92007-x. [DOI] [PubMed] [Google Scholar]
- 49.O’Flaherty J, Cordes JF. Human neutrophil degranulation responses to nucleotides. Lab Invest. 1994;70:816–821. [PubMed] [Google Scholar]
- 50.Francischetti IMB, Sá-Nunes A, Mans BJ, Santos IM, Ribeiro JMC. The role of saliva in tick feeding. Frontiers Biosci. 2009;14:2051–2088. doi: 10.2741/3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ribeiro JMC, Modi GB, Resh RB. Salivary apyrase activity of some Old World phlebotomine sand flies. Insect Biochem. 1989;19:409–412. [Google Scholar]
- 52.Ribeiro JM. Blood-feeding in mosquitoes: probing time and salivary gland anti- haemostatic activities in representatives of three genera (Aedes, Anopheles, Culex). Med Vet Entomol. 2000;14(2):142–148. doi: 10.1046/j.1365-2915.2000.00227.x. [DOI] [PubMed] [Google Scholar]
- 53.Calvo E, Sanchez-Vargas I, Favreau AJ, Barbian KD, Pham VM, Olson KE, Ribeiro JM. An insight into the sialotranscriptome of the West Nile mosquito vector, Culex tarsalis. BMC Genomics. 2010;11(1):51. doi: 10.1186/1471-2164-11-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Faudry E, Lozzi SP, Santana JM, D’Souza-Ault M, Kieffer S, Felix CR, Ricart CA, Sousa MV, Vernet T, Teixeira AR. Triatoma infestans apyrases belong to the 5′-nucleotidase family. J Biol Chem. 2004;279(19):19607–19613. doi: 10.1074/jbc.M401681200. [DOI] [PubMed] [Google Scholar]
- 55.Faudry E, Santana JM, Ebel C, Vernet T, Teixeira AR. Salivary apyrases of Triatoma infestans are assembled into homo-oligomers. Biochem J. 2006;396(3):509–515. doi: 10.1042/BJ20052019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Suzuki K, Furukawa Y, Tamura H, Ejiri N, Suematsu H, Taguchi R, Nakamura S, Suzuki Y, Ikezawa H. Purification and cDNA cloning of bovine liver 5′-nucleotidase, a GPI-anchored protein, and its expression in COS cells. J Biochem. 1993;113(5):607–613. doi: 10.1093/oxfordjournals.jbchem.a124090. [DOI] [PubMed] [Google Scholar]
- 57.Volknandt W, Vogel M, Pevsner J, Misumi Y, Ikehara Y, Zimmermann H. 5′-nucleotidase from the electric ray electric lobe. Primary structure and relation to mammalian and procaryotic enzymes. Eur J Biochem. 1991;202(3):855–861. doi: 10.1111/j.1432-1033.1991.tb16443.x. [DOI] [PubMed] [Google Scholar]
- 58.Misumi Y, Ogata S, Ohkubo K, Hirose S, Ikehara Y. Primary structure of human placental 5′-nucleotidase and identification of the glycolipid anchor in the mature form. Eur J Biochem. 1990;191(3):563–569. doi: 10.1111/j.1432-1033.1990.tb19158.x. [DOI] [PubMed] [Google Scholar]
- 59.Misumi Y, Ogata S, Hirose S, Ikehara Y. Primary structure of rat liver 5′-nucleotidase deduced from the cDNA. Presence of the COOH-terminal hydrophobic domain for possible post-translational modification by glycophospholipid. J Biol Chem. 1990;265(4):2178–2183. [PubMed] [Google Scholar]
- 60.Pierleoni A, Martelli PL, Casadio R. PredGPI: a GPI-anchor predictor. BMC Bioinformatics. 2008;9:392. doi: 10.1186/1471-2105-9-392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Champagne DE, Smartt CT, Ribeiro JM, James AA. The salivary gland-specific apyrase of the mosquito Aedes aegypti is a member of the 5′-nucleotidase family. Proc Natl Acad Sci U S A. 1995;92(3):694–698. doi: 10.1073/pnas.92.3.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Faudry E, Rocha PS, Vernet T, Lozzi SP, Teixeira AR. Kinetics of expression of the salivary apyrases in Triatoma infestans. Insect Biochem Mol Biol. 2004;34(10):1051–1058. doi: 10.1016/j.ibmb.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 63.Megraw T, Kaufman TC, Kovalick GE. Sequence and expression of Drosophila Antigen 5-related 2, a new member of the CAP gene family. Gene. 1998;222(2):297–304. doi: 10.1016/s0378-1119(98)00489-2. [DOI] [PubMed] [Google Scholar]
- 64.Hoffman DR. Allergens in Hymenoptera venom. XXV: The amino acid sequences of antigen 5 molecules and the structural basis of antigenic cross-reactivity. J Allergy Clin Immunol. 1993;92(5):707–716. doi: 10.1016/0091-6749(93)90014-7. [DOI] [PubMed] [Google Scholar]
- 65.King TP, Spangfort MD. Structure and biology of stinging insect venom allergens. Int Arch Allergy Immunol. 2000;123(2):99–106. doi: 10.1159/000024440. [DOI] [PubMed] [Google Scholar]
- 66.Hoffman DR. Hymenoptera venom allergens. Clin Rev Allergy Immunol. 2006;30(2):109–128. doi: 10.1385/criai:30:2:109. [DOI] [PubMed] [Google Scholar]
- 67.Fang KS, Vitale M, Fehlner P, King TP. cDNA cloning and primary structure of a white-face hornet venom allergen, antigen 5. Proc Natl Acad Sci U S A. 1988;85(3):895–899. doi: 10.1073/pnas.85.3.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li S, Kwon J, Aksoy S. Characterization of genes expressed in the salivary glands of the tsetse fly, Glossina morsitans morsitans. Insect Mol Biol. 2001;10(1):69–76. doi: 10.1046/j.1365-2583.2001.00240.x. [DOI] [PubMed] [Google Scholar]
- 69.Arcà B, Lombardo F, Valenzuela JG, Francischetti IM, Marinotti O, Coluzzi M, Ribeiro JM. An updated catalogue of salivary gland transcripts in the adult female mosquito, Anopheles gambiae. J Exp Biol. 2005;208(Pt 20):3971–3986. doi: 10.1242/jeb.01849. [DOI] [PubMed] [Google Scholar]
- 70.Calvo E, Dao A, Pham VM, Ribeiro JM. An insight into the sialome of Anopheles funestus reveals an emerging pattern in anopheline salivary protein families. Insect Biochem Mol Biol. 2007;37(2):164–175. doi: 10.1016/j.ibmb.2006.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xu X, Yang H, Ma D, Wu J, Wang Y, Song Y, Wang X, Lu Y, Yang J, Lai R. Toward an understanding of the molecular mechanism for successful blood feeding by coupling proteomics analysis with pharmacological testing of horsefly salivary glands. Mol Cell Proteomics. 2008;7(3):582–590. doi: 10.1074/mcp.M700497-MCP200. [DOI] [PubMed] [Google Scholar]
- 72.Wang X, Ribeiro JM, Broce AB, Wilkerson MJ, Kanost MR. An insight into the transcriptome and proteome of the salivary gland of the stable fly, Stomoxys calcitrans. Insect Biochem Mol Biol. 2009;39(9):607–614. doi: 10.1016/j.ibmb.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bhakdi S, Mackman N, Nicaud JM, Holland IB. Escherichia coli hemolysin may damage target cell membranes by generating transmembrane pores. Infect Immun. 1986;52(1):63–69. doi: 10.1128/iai.52.1.63-69.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gouaux E. alpha-Hemolysin from Staphylococcus aureus: an archetype of beta-barrel, channel-forming toxins. J Struct Biol. 1998;121(2):110–122. doi: 10.1006/jsbi.1998.3959. [DOI] [PubMed] [Google Scholar]
- 75.Andreeva ZI, Nesterenko VF, Yurkov IS, Budarina ZI, Sineva EV, Solonin AS. Purification and cytotoxic properties of Bacillus cereus hemolysin II. Protein Expr Purif. 2006;47(1):186–193. doi: 10.1016/j.pep.2005.10.030. [DOI] [PubMed] [Google Scholar]
- 76.Welch RA. RTX toxin structure and function: a story of numerous anomalies and few analogies in toxin biology. Curr Top Microbiol Immunol. 2001;257:85–111. doi: 10.1007/978-3-642-56508-3_5. [DOI] [PubMed] [Google Scholar]
- 77.Soloaga A, Veiga MP, Garcia-Segura LM, Ostolaza H, Brasseur R, Goni FM. Insertion of Escherichia coli alpha-haemolysin in lipid bilayers as a non-transmembrane integral protein: prediction and experiment. Mol Microbiol. 1999;31(4):1013–1024. doi: 10.1046/j.1365-2958.1999.01225.x. [DOI] [PubMed] [Google Scholar]
- 78.Schindel C, Zitzer A, Schulte B, Gerhards A, Stanley P, Hughes C, Koronakis V, Bhakdi S, Palmer M. Interaction of Escherichia coli hemolysin with biological membranes. A study using cysteine scanning mutagenesis. Eur J Biochem. 2001;268(3):800–808. doi: 10.1046/j.1432-1327.2001.01937.x. [DOI] [PubMed] [Google Scholar]
- 79.Hyland C, Vuillard L, Hughes C, Koronakis V. Membrane interaction of Escherichia coli hemolysin: flotation and insertion-dependent labeling by phospholipid vesicles. J Bacteriol. 2001;183(18):5364–5370. doi: 10.1128/JB.183.18.5364-5370.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Goto A, Kumagai T, Kumagai C, Hirose J, Narita H, Mori H, Kadowaki T, Beck K, Kitagawa Y. A Drosophila haemocyte-specific protein, hemolectin, similar to human von Willebrand factor. Biochem J. 2001;359(Pt 1):99–108. doi: 10.1042/0264-6021:3590099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Scherfer C, Karlsson C, Loseva O, Bidla G, Goto A, Havemann J, Dushay MS, Theopold U. Isolation and characterization of hemolymph clotting factors in Drosophila melanogaster by a pullout method. Curr Biol. 2004;14(7):625–629. doi: 10.1016/j.cub.2004.03.030. [DOI] [PubMed] [Google Scholar]
- 82.Lesch C, Goto A, Lindgren M, Bidla G, Dushay MS, Theopold U. A role for hemolectin in coagulation and immunity in Drosophila melanogaster. Dev Comp Immunol. 2007;31(12):1255–1263. doi: 10.1016/j.dci.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 83.Couillault C, Pujol N, Reboul J, Sabatier L, Guichou JF, Kohara Y, Ewbank JJ. TLR-independent control of innate immunity in Caenorhabditis elegans by the TIR domain adaptor protein TIR-1, an ortholog of human SARM. Nat Immunol. 2004;5(5):488–494. doi: 10.1038/ni1060. [DOI] [PubMed] [Google Scholar]
- 84.Gusman H, Lendenmann U, Grogan J, Troxler RF, Oppenheim FG. Is salivary histatin 5 a metallopeptide? Biochim Biophys Acta. 2001;1545(1–2):86–95. doi: 10.1016/s0167-4838(00)00265-x. [DOI] [PubMed] [Google Scholar]
- 85.Blank M, Shoenfeld Y. Histidine-rich glycoprotein modulation of immune/autoimmune, vascular, and coagulation systems. Clin Rev Allergy Immunol. 2008;34(3):307–312. doi: 10.1007/s12016-007-8058-6. [DOI] [PubMed] [Google Scholar]
- 86.Jones AL, Hulett MD, Parish CR. Histidine-rich glycoprotein: a novel adaptor protein in plasma that modulates the immune, vascular and coagulation systems. Immunol Cell Biol. 2005;83(2):106–118. doi: 10.1111/j.1440-1711.2005.01320.x. [DOI] [PubMed] [Google Scholar]
- 87.Cohen E. Chitin synthesis and inhibition: a revisit. Pest Manag Sci. 2001;57(10):946–950. doi: 10.1002/ps.363. [DOI] [PubMed] [Google Scholar]
- 88.Rebers JE, Willis JH. A conserved domain in arthropod cuticular proteins binds chitin. Insect Biochem Mol Biol. 2001;31(11):1083–1093. doi: 10.1016/s0965-1748(01)00056-x. [DOI] [PubMed] [Google Scholar]
- 89.Ribeiro JM, Charlab R, Pham VM, Garfield M, Valenzuela JG. An insight into the salivary transcriptome and proteome of the adult female mosquito Culex pipiens quinquefasciatus. Insect Biochem Mol Biol. 2004;34(6):543–563. doi: 10.1016/j.ibmb.2004.02.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.