

Summary
Neurodegenerative tauopathies characterized by hyperphosphorylated tau include frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17) and Alzheimer's disease (AD). Reducing tau levels improves cognitive function in mouse models of AD and FTDP-17, but the mechanisms regulating the turnover of pathogenic tau are unknown. We found that tau is acetylated and that tau acetylation prevents degradation of phosphorylated tau (p-tau). Using two antibodies specific for acetylated tau, we showed that tau acetylation is elevated in patients at early and moderate Braak stages of tauopathy. Histone acetyltransferase p300 was involved in tau acetylation and the class III protein deacetylase SIRT1 in deacetylation. Deleting SIRT1 enhanced levels of acetylated-tau and pathogenic forms of p-tau in vivo, likely by blocking proteasome-mediated degradation. Inhibiting p300 with a small molecule promoted tau deacetylation and eliminated p-tau associated with tauopathy. Modulating tau acetylation could be a new therapeutic strategy to reduce tau-mediated neurodegeneration.
Keywords: post-translational modification, sirtuin, deacetylase, acetyltransferase, protein degradation, proteasome-mediated degradation, ubiquitine, tau phosphorylation, tauopathy, Alzheimer's disease, FTDP-17
Introduction
The microtubule binding protein tau (MAPT) promotes the assembly and stabilization of microtubules (Kar et al., 2003). Identification of tau mutations in frontotemporal dementia patients with parkinsonism linked to chromosome-17 (FTDP-17) provides evidence that tau malfunction can trigger neurodegeneration (Hutton et al., 1998; Spillantini et al., 1998). In the brains of neurodegenerative tauopathies, including Alzheimer's disease (AD) and FTDP-17, hyperphosphorylated tau accumulates as neurofibrillary tangles (NFTs), a diagnostic hallmark of late-stage disease (Cairns et al., 2007; Ludolph et al., 2009). However, the accumulation of soluble forms of aberrantly phosphorylated tau (p-tau) may underlie tau-mediated neurodegeneration by disrupting the ability of tau to bind microtubules and promoting tau aggregation and fibrillization (Ballatore et al., 2007). Suppression of endogenous soluble tau or transgenic tau improves cognitive function in mouse models of AD and FTDP-17, respectively (Roberson et al., 2007; Santacruz et al., 2005). Thus, reducing tau accumulation, especially aberrant p-tau, is a therapeutic approach for neurodegenerative tauopathies. However, the molecular mechanisms of the accumulation of hyperphosphorylated tau are unclear.
Accumulation of hyperphosphorylated tau could reflect insufficient clearance. Degradation of tau, especially p-tau, may involve the ubiquitin-proteasome system (UPS). Soluble tau isolated from paired helical filaments (PHF) from AD brains is polyubiquitinated (Cripps et al., 2006). The E3 ubiquitin ligase CHIP (carboxyl terminus of the Hsc70-interacting protein) is involved in polyubiquitination and degradation of p-tau (Petrucelli et al., 2004; Shimura et al., 2004). Deletion of CHIP significantly increased p-tau levels in the brain (Dickey et al., 2006). Ubiquitination of lysines can be precluded by lysine acetylation, which inhibits proteasome-mediated degradation of numerous proteins, including p53 (Ito et al., 2002), Runx (Jin et al., 2004), and Smad7 (Gronroos et al., 2002). It is unknown whether tau is acetylated and whether tau acetylation contributes to tau accumulation.
Lysine acetylation rivals phosphorylation in regulating diverse cellular functions, including energy metabolism, signaling from the plasma membrane, and cytoskeleton dynamics (Choudhary et al., 2009; Kouzarides, 2000; Yang and Seto, 2008). Enzymes that add an acetyl group to the protein are called histone acetyltransferase (HAT) or lysine acetyltransferase. Of four major classes of HATs, p300/CBP (protein of 300 kDa and CREB-binding protein) and pCAF (p300-associated and CBP-associated factor) are exclusively present in metazoans (Goodman and Smolik, 2000). Enzymes that remove an acetyl group from the protein are called histone deacetylase (HDAC) or lysine deacetylase. There are three classes of HDACs. The activities of HDACs in classes I and II (HADC1–11) depend on zinc as a cofactor; the activities of class III HDACs (sirtuins) depend on the relative levels of NAD+ and NADH (Haigis and Guarente, 2006; Michan and Sinclair, 2007). Of the seven members of mammalian sirtuins (SIRT1–7), SIRT1 is the most studied and is strongly implicated in aging-related diseases, including AD (Gan and Mucke, 2008). SIRT1 levels are reduced in AD brains, and the reduction correlates with the accumulation of hyperphosphorylated tau aggregates (Julien et al., 2009). Overexpressing SIRT1 protects against neuronal loss in the inducible p25 transgenic mouse, a model of AD and tauopathy (Kim et al., 2007). It is unclear how SIRT1 protects against tau-mediated neurodegeneration.
In the current study, we investigated tau acetylation in primary neurons, tauopathy mouse models, and AD brains by generating two specific antibodies against acetylated-tau (ac-tau). Using gene-silencing and pharmacological approaches, we show that p300 acetylates and SIRT1 deacetylates tau. Our study also provided evidence that SIRT1 deficiency leads to hyperacetylation of tau and accumulation of p-tau. In contrast, promoting tau deacetylation eliminates p-tau.
Results
Tau Is Acetylated in Vitro and in Vivo
To demonstrate that tau is acetylated, recombinant tau was incubated with recombinant acetyltransferase p300 or pCAF (p300/CBP-associated factor) with 14C-acetyl-coenzyme A. Incubation with p300, not pCAF, led to tau acetylation, while both p300 and pCAF were active in transferring acetyl groups to histones as expected (Figure 1A). Matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) spectrometry identified multiple lysines that were acetylated by p300 in vitro. A total of 23 putatively acetylated lysines were detected out of 383 residues (∼86.8% coverage; Table-S1) throughout the tau sequence (2N4R, 441 amino acids). A few putative acetylated lysines were in the N- and C- terminal regions; 13 were in microtubule-binding domains (Figure 1B and Table-S1). Putative acetylated N-terminal lysines (e.g., lysines 163, 174, and 180) appeared to be acetylated in all MS analyses. Those in the microtubule-binding domains appeared to be acetylated in a subset of MS analyses, suggesting variable acetylation at these sites in vitro (Table-S1).
Figure 1. Tau is Acetylated in Vitro and in Vivo.
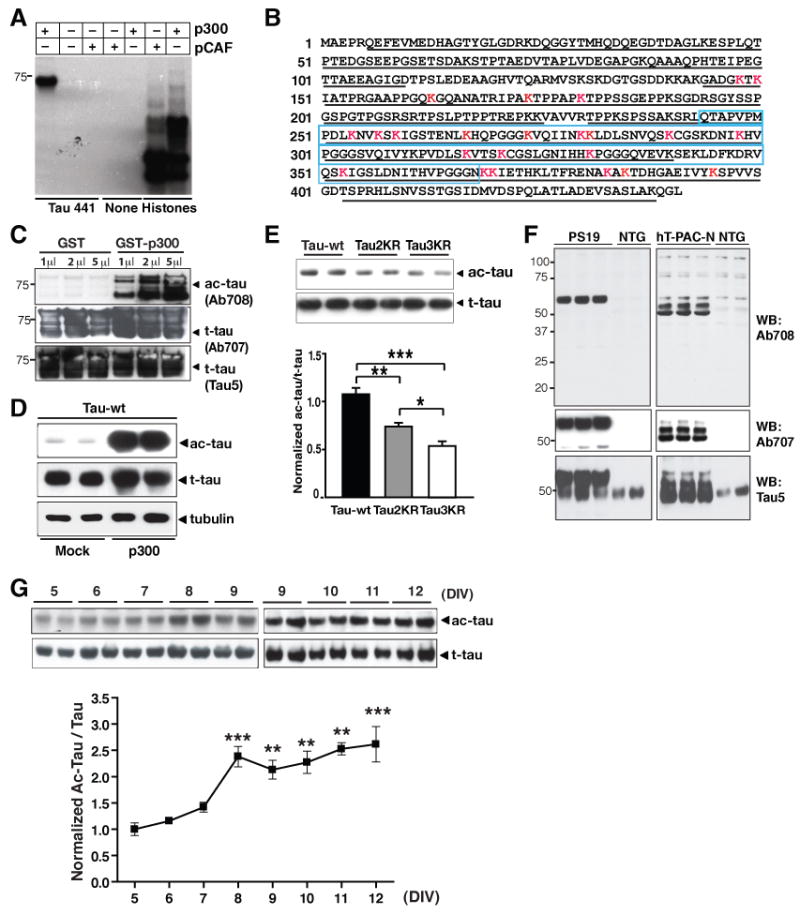
(A) Acetylation of h-tau (2N4R) by p300 but not pCAF under cell-free conditions, as shown by autoradiography.
(B) MALDI-TOF spectrometry identified ac-lysines on h-tau by p300 in vitro. Red: lysines (K) with acetyl group. Underlined: sequence covered by MS analysis. Blue box: microtubule-binding domains. See Table-S1 for full list and Figure S1 for MS-MS spectra.
(C–E) Ab708 specifically recognizes ac-tau. (C) Ab708 only recognized recombinant tau acetylated by GST-p300, not nonacetylated tau with GST alone. Similar levels of t-tau were detected with Ab707 and Tau 5 antibody. (D) Overexpressing p300 markedly enhanced ac-tau, detected with Ab708, in HEK293T cells. Levels of t-tau, detected with Tau 5, were similar with or without p300. Blots are representative of >5 experiments. (E) Putatively acetylated lysine sites recognized by Ab708. Ac-tau/t-tau levels in HEK293T cells expressing wildtype tau were set as 1. n = 4. *, P=0.012; ** P=0.003; ***, P=0.0003 (one-way ANOVA with Tukey-Kramer posthoc analysis).
(F) Ab708 recognizes human ac-tau in brains of PS19 or hT-PAC-N transgenic mice, not in NTG littermates. Human t-tau was detected with Ab707 antibody; human and mouse t-tau was detected with Tau 5 antibody. See Figure S2 for the sequence similarity among human, mouse and rat tau.
(G) Levels of Ab708-positive ac-tau were elevated in primary rat neurons as they matured in culture (DIV=5–12). n=2–7 from 2–3 independent experiments. ***, P<0.001 (DIV5 vs. DIV8 or DIV12); **, P<0.01 (DIV5 vs. DIV9–11). Values are means ± SEM (E, G).
To examine tau acetylation in vivo, we generated a polyclonal antibody (anti-ac-tau, Ab708) with a synthetic tau peptide (amino acids 160–182 for 2N4R tau isoform) containing acetylated lysines at positions 163 and 174 and 180 (Figure S1 for MS spectra). A control antibody was generated (anti-tau, Ab707) using the same peptide with nonacetylated lysines. To test the specificity of Ab708 against ac-tau, recombinant human tau (441; 2N4R isoform) was incubated with glutathione S-transferase (GST) alone or GST-p300. Immunoblotting with Ab708 detected strong tau signals after incubation with GST-p300, but not with GST alone (Figure 1C). In contrast, Ab707 or Tau 5 antibody detected similar levels of total tau (t-tau) with either GST or GST-p300 (Figure 1C). Thus, Ab708 specifically recognizes tau acetylated by p300 under cell-free conditions. In HEK293T cells transfected with tau, overexpression of p300 markedly elevated the levels of ac-tau detected with Ab708 while the increase in the levels of t-tau was modest, suggesting that Ab708 preferentially recognizes p300-induced ac-tau in cultured cells (Figure 1D). Mutation of lysines 163, 174 and 180 (Tau3KR) reduced ac-tau levels relative to t-tau levels in HEK293T cells (Figure 1E). A smaller yet still significant reduction was also observed when two lysines were mutated in Tau2KR(K174R/K180R) (Figure 1E). These findings suggest that Ab708 recognizes human tau acetylated at positions 163, 174 or 180 and possibly other acetylated lysines on tau, but not nonacetylated tau.
To detect ac-tau in vivo, we performed western blots with brain lysates from transgenic mice expressing human tau cDNA (1N4R) with P301S mutation (P19) (Yoshiyama et al., 2007) or from transgenic mice expressing the entire human wildtype MAPT (hT-PAC-N) with 0N3R and 0N4R as the two predominant tau isoforms (McMillan et al., 2008). Human and mouse tau differ at three positions in the region used to generate Ab708 and Ab707 (Figure S2). Ab708 detected specific signals in lysates from P19 and hT-PAC-N mice, but not those from nontransgenic (NTG) littermates (Figure 1F). These findings suggest that Ab708 recognizes various isoforms of human ac-tau, but not mouse ac-tau. The control antibody Ab707, which recognizes human t-tau, does not recognize mouse tau either. Endogenous tau in NTG mice was detected with Tau 5 antibody (Figure 1F).
Rat tau is more similar to human tau than mouse tau in the region used to generate Ab708 (Figure S2). Ab708 detected endogenous ac-tau in rat primary cortical neurons (Figure 1G). Levels of ac-tau/t-tau gradually increased as neurons matured from 5–12 days in vitro (DIV), suggesting that tau acetylation is regulated developmentally (Figure 1G). However, the isoforms of rat tau detected by Ab708 remain to be defined.
Acetylation of Tau by p300 Acetyltransferase
To determine the role of endogenous p300 or pCAF in tau acetylation, we transfected HEK293T cells expressing human tau cDNA (2N4R) with siRNAs targeting p300 or pCAF (Figure 2A) and assessed the effects on ac-tau or t-tau. Inhibiting p300 significantly reduced levels of ac-tau, but not t-tau (Figure 2B, 2C). In contrast, inhibiting pCAF had no effects (Figure 2B, 2C). These findings are consistent with the results of in vitro studies (Figure 1A). Next, we treated primary neurons with C646, a pyrazolone-containing small-molecule inhibitor of p300 with a Ki of 400 nM (Bowers et al., 2010). Under cell-free conditions, C646 at 10 μM inhibits p300 in a highly selective manner (86% inhibition vs. <10% for the six other acetyltransferases) (Bowers et al., 2010). Inhibition of p300 with C646 (20 μM) drastically reduced levels of ac-tau in primary neurons within 8 h. The levels of t-tau remained unchanged (Figure 2D). p300 is a transcriptional coactivator (Goodman and Smolik, 2000). However, C646 treatment for 8 h did not suppress tau transcripts as quantified with real-time RT-PCR (data not shown). Thus short-term (8 h) inhibition of p300 deacetylates tau without affecting t-tau levels. Extended treatment with C646 for 20 h lowered the levels of ac-tau relative to the t-tau (ac-tau/t-tau), but also those of t-tau (Figure 2E).
Figure 2. Tau Is Acetylated by p300 Acetyltransferase.
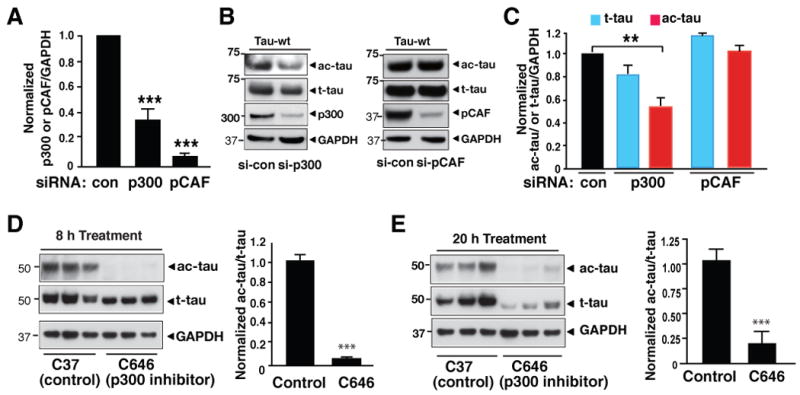
(A–C) Inhibiting p300, not pCAF, reduced ac-tau in HEK293T cells. (A) Inhibition of p300 or pCAF expression by siRNA transfections. Levels of p300/GAPDH or pCAF/GAPDH in control siRNA-transfected cells were set as 1. ***, P=0.0006 (p300 vs. control) or P<0.0001 (pCAF vs. control). (B) Representative western blots (from three experiments) showing levels of p300 or pCAF, ac-tau, t-tau, and GAPDH in cells transfected with control siRNA (CTRL) or siRNA targeting p300 or pCAF. (C) Inhibition of p300, not pCAF, reduced ac-tau levels. Levels of ac-tau/GAPDH or t-tau/GAPDH in control siRNA-transfected cells were set as 1. n=5–6. **, P=0.008 (paired t test).
(D) Inhibiting p300 acutely with C646 (20 μM for 8 h) eliminated ac-tau without affecting t-tau levels in primary rat cortical neurons. Left: Representative western blot from three experiments. Right: Ac-tau/t-tau levels in vehicle-treated cells were set as 1. n=3. ***, P=0.0001 (unpaired t test).
(E) Extended treatment with C646 (20 μM for 20 h) lowered t-tau in primary cortical neuron. Blots are representative of two experiments. Values are means ± SEM (A, C–E).
Deacetylation of Tau by SIRT1 in Cultures
To investigate the enzymes that deacetylate tau, we transfected an expression vector encoding FLAG-tagged SIRT1, SIRT2, HDAC5, or HDAC6 into HEK293T cells expressing human tau. All HDACs were expressed at high levels (Figure 3A). Although expressed at lower levels than SIRT1 and SIRT2, HDAC6 eliminated tubulin acetylation (Hubbert et al., 2002), suggesting sufficient expression (Figure 3B). Overexpression of SIRT1 reduced levels of Ab708-positive ac-tau. SIRT2 and HDAC6 overexpression also lowered ac-tau, although to lesser extents (Figure 3B, 3C). Levels of t-tau were also reduced in cells overexpressing SIRT1 and HDAC6. Nevertheless, the ac-tau/t-tau ratio was significantly reduced by SIRT1 overexpression (Figure 3D). The modest reduction in ac-tau/t-tau induced by HDAC6 or SIRT2 overexpression was not statistically significant (Figure 3D).
Figure 3. SIRT1 Deacetylates Tau in Culture.
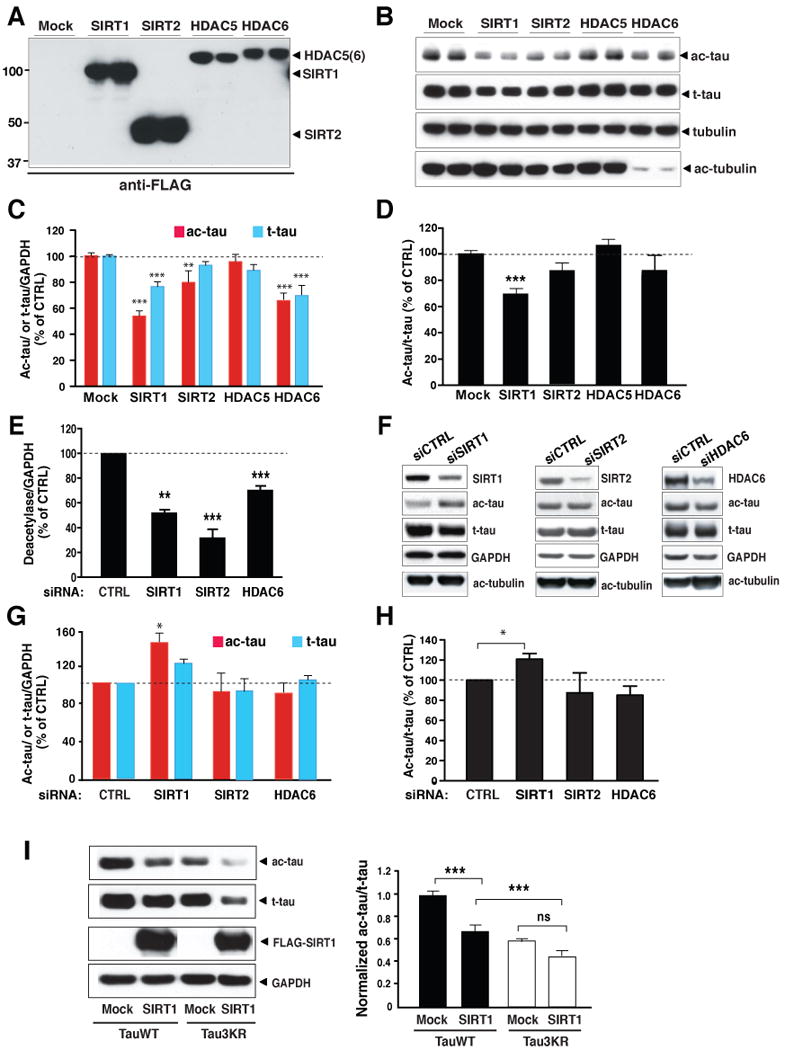
(A–D) SIRT1 overexpression lowered ac-tau levels in HEK293T cells. (A) Western blot showing expression of FLAG-tagged SIRT1, SIRT2, HDAC5, or HDAC6 with an anti-FLAG antibody. Blots are representative of 2–3 experiments. (B) Western blot showing levels of ac-tau, t-tau, tubulin, and ac-tubulin in cells overexpressing SIRT1, SIRT2, HDAC5, or HDAC6. Blots are epresentative of 2–3 experiments. (C) Overexpression of SIRT1, SIRT2 or HDAC6 significantly reduced levels of ac-tau/GAPDH. Levels of t-tau were also reduced by SIRT1 or HDAC6 overexpression. n=9–18 from 6–10 independent experiments. ***, P < 0.001 (Mock vs. SIRT1 or Mock vs. HDAC6); **, P < 0.01 (Mock vs. SIRT2) (two-way ANOVA and Bonferroni posthoc test). (D) Overexpression of SIRT1 significantly reduced ac-tau/t-tau. n=9–18 from 6–10 independent experiments, ***, P<0.001 (Mock vs. SIRT1) (one-way ANOVA and Tukey-Kramer posthoc test).
(E–H) Inhibition of SIRT1 elevated ac-tau in HEK293T cells. (E) Inhibition of SIRT1, SIRT2, or HDAC6 expression mediated by siRNA transfections. n=4–6 from 2–3 experiments. **, P =0.0015; ***, P=0.0001 (SIRT2 vs. control) or P=0.001 (HDAC6 vs. control) (paired t test). (F) Western blot showing levels of ac-tau, t-tau, tubulin, and ac-tubulin in cells transfected with control siRNA or siRNA targeting SIRT1, SIRT2, or HDAC6. Blots are representative of 2–3 experiments. (G–H) Inhibition of SIRT1, significantly elevated levels of ac-tau/GAPDH (G) or ac-tau/t-tau (H). n=4–6 from 2–3 experiments. *, P<0.05 (paired t test). Levels of deacetylase/GAPDH (E), ac-tau or t-tau/GAPDH (G), and ac-tau/t-tau (H) in control siRNA-transfected cells were set as 1. Also see Figure S3 for comparison of levels of ac-tau in MEFs with (SIRT1+/+) or without SIRT1 (SIRT1−/−).
(I) Deacetylation of Tau3KR by SIRT1. Left: Representative western blot showing levels of ac-tau, t-tau, FLAG-tagged SIRT1, and GAPDH. Right: Ac-tau/t-tau levels in mock-transfected cells expressing wildtype tau were set as 1. n=10–20 from 4–10 independent experiments. ***, P<0.001; ns, not significant (one-way ANOVA and Tukey-Kramer posthoc analysis). Values are means ± SEM (C–E, G–I)
To examine the effects of endogenous HDACs on ac-tau, we inhibited expression of SIRT1, SIRT2, or HDAC6 with siRNAs (Figure 3E). Relative to control siRNA, target siRNAs significantly reduced levels of SIRT1, SIRT2, and HDAC6. Despite modest inhibition, HDAC6 increased ac-tubulin levels (Figure 3F). However, only inhibition of SIRT1 increased ac-tau levels, suggesting the involvement of endogenous SIRT1 in deacetylating tau (Figure 3G). Consistent with the observation that SIRT1 overexpression reduced t-tau, SIRT1 inhibition led to a trend of increase in t-tau (Figure 3G). Nevertheless, inhibition of SIRT1, not SIRT2 or HDAC6, significantly elevated levels of ac-tau relative to t-tau (ac-tau/t-tau) (Figure 3H). These results provided direct support that SIRT1 is involved in tau deacetylation. Our findings so far do not support a prominent role of SIRT2 or HDAC6, which both deacetylate tubulin (Hubbert et al., 2002; North et al., 2003), in tau deacetylation. However, their involvement cannot be ruled out since only partial silencing of SIRT2 or HDAC6 was achieved with siRNA transfections.
To further investigate the role of SIRT1 in tau deacetylation, low-passage mouse embryonic fibroblasts (MEF) with (SIRT1+/+) or without SIRT1 (SIRT1−/−) were transfected with human tau cDNA (Figure S3). Deleting SIRT1 significantly raised ac-tau levels. The increase in t-tau did not reach statistical significance, suggesting SIRT1 deacetylates tau in MEFs. In HEK293T cells, when lysines 163, 174, and 180 were mutated to arginines (Tau3KR), levels of ac-tau were significantly reduced (Figure 3I). SIRT1 overexpression reduced ac-tau in TauWT cells, but the reduction was much attenuated in Tau3KR cells (Figure 3I). These results implicate SIRT1 in deacetylating lysines 163, 174, and 180. However, SIRT1 reduced ac-tau to lower levels in Tau3KR cells than in TauWT cells, indicating that SIRT1 could deacetylate additional lysine residues besides those at positions 163, 174, and 180 (Figure 3I).
SIRT1 Reduces Tau Acetylation in Primary Neurons and in Vivo
In primary neurons, ac-tau/t-tau increased as the neurons matured (Figure 1G) but levels of full-length SIRT1 decreased (Figure 4A). Consistent with the notion that SIRT1 deactylates tau, levels of SIRT1 negatively correlated with levels of ac-tau/t-tau in primary neurons during development (Figure 4B). To investigate if SIRT1 negatively regulates tau acetylation in neurons, we deleted SIRT1 in neurons by infecting neurons from SIRT1 conditional knockout mice (SIRT1F/F) (Chua et al., 2005) with a lentiviral vector expressing cre recombinase (Lenti-cre) (Figure 4C). Controls were infection with an empty vector (Lenti-con). SIRT1F/F neurons were also infected with a lentiviral vector expressing human tau. Deleting SIRT1 significantly elevated levels of acetylated human tau relative to t-tau (Figure 4C), indicating that SIRT1 deacetylates tau in neurons.
Figure 4. SIRT1 Reduces Tau Acetylation in Neurons and in Vivo.
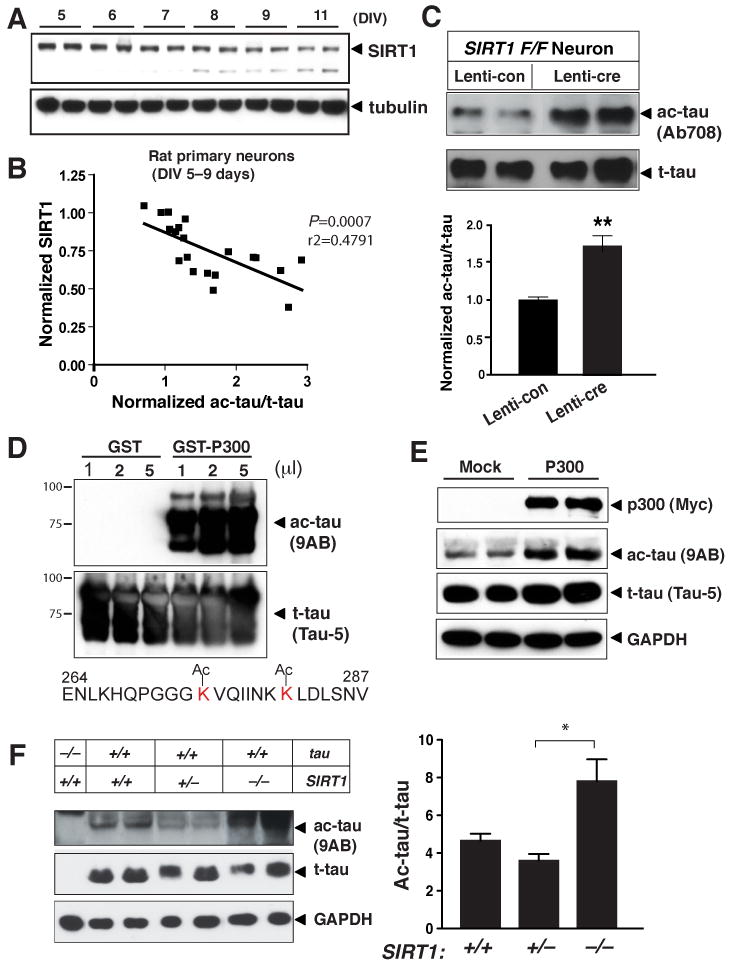
(A) Western blot showing expression of SIRT1 in primary cortical neurons during maturation in culture (DIV5–11). Blots are representative of 2–3 independent cultures.
(B) Levels of endogenous ac-tau relative to t-tau correlated negatively with levels of SIRT1 in primary rat neuronal cultures (DIV5–9). Levels of SIRT1 or ac-tau/t-tau at DIV=5 were set as 1. n=20 independent measurements. P=0.0007, Pearson correlation coefficient r2=0.4791.
(C) Deleting SIRT1 in neurons elevated levels of ac-tau relative to t-tau. Neurons cultured from SIRT1F/F mice were infected with control virus or virus expressing cre recombinase (Lenti-cre). Both cultures were also infected with a lentiviral vector expressing h-tau. n=8. P=0.001, (unpaired t test).
(D) Acetyl-specific antibody (9AB) recognized tau acetylated by GST-p300, but not non-ac-tau. Also shown is the sequence of the antigen used to generate 9AB.
(E) Overexpressing p300 enhanced 9AB-positive ac-tau in HEK293T cells. Levels of t-tau, detected with Tau 5, were similar with or without p300 overexpression. Blots are representative of three independent experiments.
(F) Deletion of SIRT1 elevated ac-tau relative to t-tau in the brain. Left: Representative western blots showing levels of ac-tau, t-tau, and GAPDH. Right: Levels of ac-tau/t-tau in SIRT1+/+, SIRT1+/−, and SIRT1−/− brains. n=3–6 mice/genotype. *, P=0.02 (SIRT1+/− vs. SIRT1−/−) (one-way ANOVA and Tukey-Kramer posthoc test). Values are means ± SEM (C, F)
To examine the effects of SIRT1 deletion on the acetylation of mouse tau in vivo, we developed another ac-tau-specific antibody targeting the microtubule-binding region (264–287), which is 100% conserved between mouse and human (Figure 4D). Recombinant tau was incubated with p300 to induce acetylation. Like Ab708, antibody 9AB recognized recombinant tau acetylated by GST-p300, but not tau incubated with GST alone, suggesting that 9AB does not cross-react with non-ac-tau. In HEK293T cells, overexpression of p300 markedly elevated levels of ac-tau detected with 9AB, but only modestly those of t-tau. Thus, 9AB also preferentially recognizes p300-induced ac-tau in cultured cells (Figure 4E). In mouse brains, 9AB detected low levels of ac-tau, which was absent in tau−/− mice. To delete SIRT1, we crossed SIRT1+/− mice on an outbred background, which partially rescued the embryonic lethality of SIRT1-null mice on the inbred background (Cheng et al., 2003). Deleting SIRT1 significantly enhanced levels of ac-tau in the brain, providing direct evidence that SIRT1 deacetylates tau in vivo (Figure 4F).
SIRT1 Interacts with Tau Directly
Although mainly localized in the nucleus, SIRT1 can be shuttled to the cytoplasm (Hisahara et al., 2008; Tanno et al., 2007). To determine if SIRT1 directly deacetylates tau, we performed in vitro deacetylation assays. Recombinant tau acetylated by p300 was incubated with SIRT1 immunoprecipitated from SIRT1-overexpressing HEK293T cells. Ac-tau/t-tau levels were significantly lower in the presence of immunoprecipitated SIRT1 (Figure 5A). To confirm that SIRT1 interacts with tau directly in vivo, we performed GST pull-down assays. Bead-bound GST-tau, not GST alone, interacted with FLAG-SIRT1 expressed in HEK293T cells or endogenous SIRT1 in nontransfected cells (Figure 5B). Moreover, in coimmunoprecipitation assays, after immunoprecipitation with an anti-FLAG antibody, endogenous SIRT1 was detected with an anti-SIRT1 antibody, and tau was detected with a pan-tau antibody (Tau 5) in HEK293T cells expressing FLAG-tagged tau (Figure 5C).
Figure 5. SIRT1 Interacts with Tau.
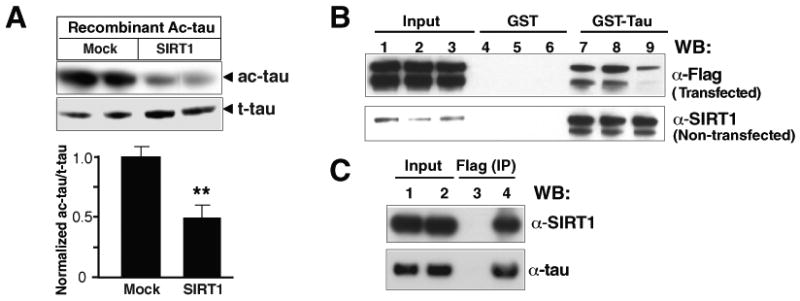
(A) SIRT1 directly deacetylated ac-tau in vitro. Ac-tau/t-tau levels in the absence of immunoprecipitated SIRT1 were set as 1. Ac-tau was detected with Ab708 antibody. n=5 from two experiments. **, P=0.0063 (unpaired t test).
(B) GST pull-down assays. GST-tau protein (lanes 7–9) or GST alone (lanes 4–6) was incubated with lysates of cells transfected with FLAG-tagged SIRT1 or of nontransfected cells. Lanes 1, 4 and 7: 0.1% Triton X-100; lanes 2, 5, and 8: 0.5% Triton X-100; lanes 3, 6, and 9: 0.5% NP-40. Data shown are representative of two experiments. The lower band is likely to represent the cleavage product of SIRT1 observed previously (Ohsawa and Miura, 2006).
(C) Coimmunoprecipitation assays. HEK293T cells were transfected with a plasmid encoding FLAG-tagged human tau. Cell lysates were collected 24 h later, immunoprecipitated with an anti-FLAG antibody, and immunoblotted with Tau 5 or an anti-SIRT1 antibody. Lanes 1–2: input; lane 3: no primary antibody; lane 4: anti-FLAG antibody. Values are means ± SEM (A).
SIRT1 Deficiency Increases Tau Acetylation and Suppresses Degradation of p-Tau
As a class III lysine deacetylase, SIRT1 supports and promotes longevity in diverse organisms. Besides regulating endocrine and behavioral responses to caloric restriction (Cohen et al., 2009), SIRT1 has been strongly implicated in neurodegenerative diseases (reviewed in Gan and Mucke, 2008). In AD brains, SIRT1 levels are significantly reduced, and the reduction appears to correlate with tau accumulation and aggregation (Julien et al., 2009). Thus, deficient SIRT1 activity may contribute to tauopathy. In primary neurons, inhibiting SIRT1 with a specific inhibitor EX527 (Napper et al., 2005) markedly increased ac-tau, AT8-positive p-tau, as well as t-tau (Figure 6A). Levels of ac-tau or p-tau relative to t-tau were significantly increased with EX527 treatments (Figure 6A). Thus, the increase in ac-tau induced by SIRT1 deficiency is accompanied by accumulation of pathogenic p-tau in primary neurons. In mouse brains, deleting SIRT1, which elevated ac-tau, also increased AT8-positive p-tau (Figure 6B).
Figure 6. Acetylation Slows Tau Turnover by Inhibiting Its Ubiquitination.
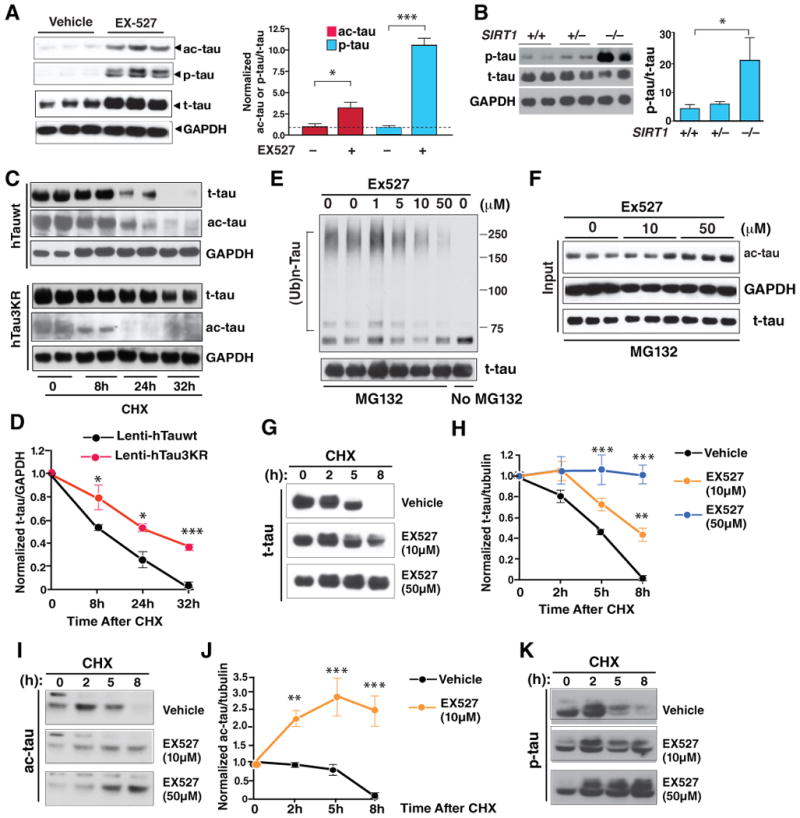
(A) Inhibiting SIRT1 with EX527 (50 μM) elevated ac-tau and p-tau in rat primary neurons (DIV=10). Left: Representative western blots. p-tau was detected with AT8. Right: Levels of ac-tau/t-tau or p-tau/t-tau in vehicle-treated cells were set as 1. n=6 independent treatments. ***, P<0.001; *, P<0.05 (paired t test).
(B) Deletion of SIRT1 elevated AT8-positive p-tau in the brain. n=3–4 mice/genotype. *, P<0.05 (SIRT1+/+ vs. SIRT1−/−) (one-way ANOVA and Tukey-Kramer posthoc test).
(C and D) Tau3KR was more stable than wildtype tau in primary neurons. Cells were infected with Lenti-hTauwt or Lenti-hTau3KR and treated with CHX for 8–32 h 4 days after infection (DIV=9). (C) Representative western blot of 2 experiments showing ac-tau, t-tau, and GAPDH. (D) The turnover of t-tau was slower in cells expressing Tau3KR. t-tau/GAPDH levels in cells harvested at time 0 were set as 1. n=3–5 from 2 experiments. *, P=0.04 (8 h), P=0.015 (24 h); ***, P<0.0001 (32 h) (unpaired t test for each time-point).
(E and F) SIRT1 inhibitor EX527 (1–50 μM) suppressed tau ubiquitination and elevated ac-tau in a dose-dependent manner. Blots are representative of 3 experiments. See Figure S4, which shows that resveratrol-enhanced tau ubiquitination depended on SIRT1's deacetylase activity.
(G–K) The SIRT1 inhibitor EX527 increases the half-life of tau in rat primary neurons (DIV=8) in a dose-dependent manner. Neurons were treated with CHX for 0–8 h in the presence or absence of EX527 (10–50 μM). Representative western blots of 3 experiments showing the turnover of t-tau (G), ac-tau (I), or p-tau (K) in neurons with or without EX527. (H and J) The turnover of t-tau (H) or ac-tau (J) was markedly slowed by treatment of EX527. Levels of t-tau/tubulin or ac-tau/tubulin in cells harvested at time 0 were set as 1. n=3. **, P<0.01; ***, P<0.001 (two-way ANOVA, EX527-treated vs. vehicle-treated). See Figure S5 for comparison of the half-life of ac-tau vs. t-tau. Values are means ± SEM (A–B, D, G, I).
How might elevated tau acetylation lead to higher levels of p-tau? Acetylation of lysines can preclude its ubiquitination and stabilize proteins that are normally degraded by the UPS, including p53 (Ito et al., 2002), Runx3 (Jin et al., 2004), β-catenin (Ge et al., 2009), and other regulatory factors (Caron et al., 2005). Since tau is ubiquitinated and the degradation of tau, especially p-tau, involves the proteasome-mediated pathway (Petrucelli et al., 2004; Tan et al., 2008), we hypothesized that acetylation precludes tau ubiquitination and suppresses its degradation.
To test this hypothesis, we assessed the involvement of acetylated lysines in regulating protein turnover. We compared the turnover rates of human wildtype tau (hTauwt) and human tau3KR (hTau3KR). Primary cortical neurons were infected with Lenti-hTauwt and Lenti-hTau3KR and treated with CHX (Figure 6C). Infection of Lenti-hTau3KR resulted in much weaker Ab708-positive signal than that of Lenti-hTauwt, providing further support that Ab708 recognizes acetylation of lysines 163, 174, and 180. Mutating these three lysines to arginines significantly increased the half-life of tau, possibly by permanently blocking ubiquitination at the three sites (Figure 6D). These results support the notion that the acetylated lysines can be ubiquitinated.
To directly test if enhancing acetylation can block ubiquitination, we transfected HEK293T cells with expression plasmids encoding tau and hemagglutinin (HA)-tagged ubiquitin and then treated with EX527 to inhibit SIRT1 and with MG132 to block the proteasome-mediated degradation. Ubiquitinated tau was immunoprecipitated with an anti-FLAG antibody and detected with an anti-HA antibody. EX527 prevented polyubiquitination of tau in a dose-dependent manner, indicating that tau ubiquitination is suppressed by enhanced acetylation (Figure 6E). EX527 also elevated ac-tau levels as expected (Figure 6F). In contrast, SIRT1-mediated deacetylation appears to enhance tau ubquitination. Treatment with resveratrol, which may be indirectly involved in activating SIRT1, significantly increased tau ubiquitination in cells transfected with wildtype SIRT1, but not those with H363Y mutant (Figure S4).
We then directly examined whether enhancing acetylation of tau slows the turnover of endogenous tau. Primary neurons were treated with EX527 to enhance tau acetylation and with cycloheximide (CHX) to inhibit translation of new proteins. Endogenous rat tau in primary neurons had a half-life of around 5 h. Inhibiting SIRT1 with EX527 slowed tau turnover and increased the half-life of t-tau in a dose-dependent manner (Figure 6G, 6H). Consistent with this notion, ac-tau appears to be degraded slower than that of t-tau. In primary neurons, CHX markedly reduced t-tau levels after 5 h, whereas ac-tau levels were only slightly reduced after 8 h (Figure S5). Moreover, inhibition of SIRT1 with 10 μM of EX527 blocked the turnover of ac-tau, leading its accumulation (Figure 6I, 6J). Higher dose of EX527 (50 μM) resulted in more pronounced accumulation of ac-tau (Figure 6I). Treatment with EX527 also blocked the degradation of AT8-positive p-tau in a dose-dependent manner (Figure 6K).
Elevation of Tau Acetylation in Pathological Conditions
Since degradation of tau was slowed by its acetylation, we hypothesized that acetylation is a critical early event that contributes to accumulation of p-tau that is normally degraded via the proteasome-mediated pathway (Petrucelli et al., 2004; Tan et al., 2008). In primary neurons, treatment with low levels of amyloid β (Aβ) oligomers, a key pathogen in AD, increased levels of ac-tau in a dose-dependent manner (Figure 7A). Higher levels of ac-tau were observed in primary neurons expressing human tau carrying FTD-linked mutation (hTauP301L) than those expressing similar levels of hTauwt (Figure 7B). These findings suggest that tau acetylation is elevated by stress, such as Aβ accumulation, or FTD-linked mutations.
Figure 7. Tau Acetylation Is Elevated under Pathological Conditions.
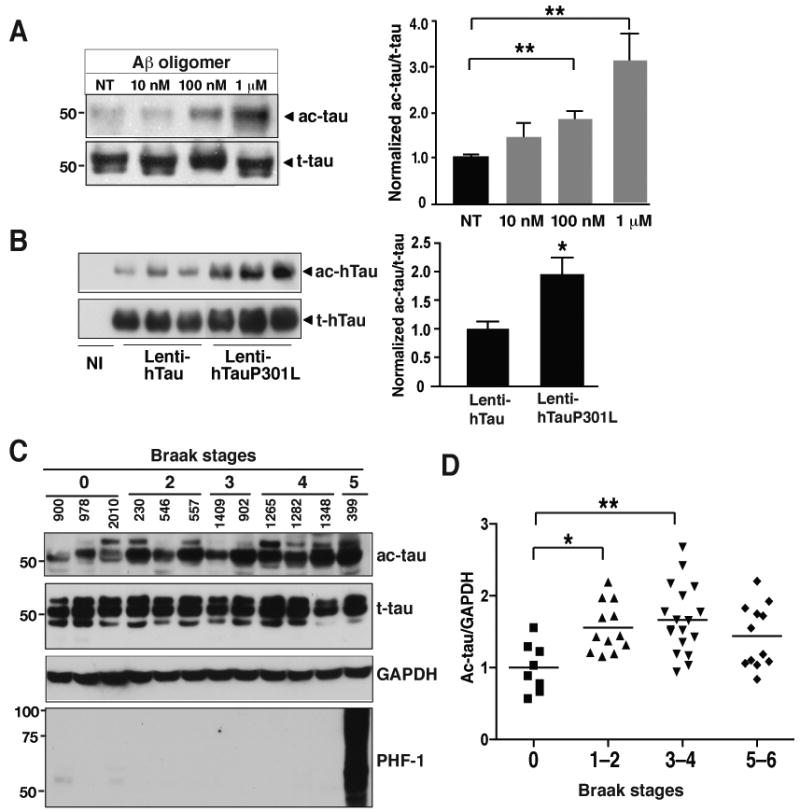
(A) Tau acetylation was increased by low levels of Aβ oligomers in primary cortical neurons (DIV=11). n=5 from 3 experiments. **, P=0.003 (one-way ANOVA and Tukey-Kramer posthoc test).
(B) Tau acetylation was associated with familial MAPT mutations in primary neurons (DIV=13). Ac-tau/t-tau levels in neurons infected with Lenti-hTauwt were set as 1. n=9 from three experiments. *, P=0.013 (unpaired t test).
(C) Representative western blots showing levels of ac-tau, t-tau, and hyperphosphorylated tau in human brains (Bm-22, superior temporal gyrus) at different Braak stages (0–5).
(D) Ac-tau levels were elevated in patients with mild (Braak stages 1–2) to moderate (Braak stages 3–4) levels of tau pathology. n=8–18 cases/Braak range. *, P<0.05; **, P<0.01, one-way ANOVA Tukey-Kramer posthoc analyses. See Table-S2 for the patient information. Values are means±SEM (A, B, D).
We next examined tau acetylation in the frontal cortex of patients with different degrees of tau pathology (Braak and Braak, 1991) (Table-S2). Patients at Braak stages 1–2 or 3–4 had significantly higher levels of ac-tau in the soluble fraction of the brain lysates than patients at Braak stage 0 (Figure 7C). Ab708 can recognize various human tau isoforms in transgenic mice overexpressing human tau (Figure 1D). However, unlike Tau 5, which detects all isoforms, Ab708 appears to detect some isoforms preferentially in AD brains (Figure 7C). Hyperphosphorylated tau detected with PHF-1 (Figure 1D) or AT8 (data not shown) was observed only in patients at stages 5–6, consistent with lack of significant NFTs in the frontal cortex of patients at earlier Braak stages (Braak and Braak, 1991). Thus, our findings support the notion that enhanced tau acetylation precedes hyperphosphorylation of tau and NFT formation. However, in patients at Braak stages 5–6, especially those at stage 6, with NFTs in the frontal cortex, levels of ac-tau were slightly lower than patients at mild to moderate stages. This end-stage reduction might be explained by severe loss of neurons, or sequestration of ac-tau in the NFTs, thus remaining in the insoluble fractions of the lysates.
Reducing Tau Acetylation Eliminates p-Tau Induced by FTD-linked Mutation
To test the hypothesis that tau acetylation contributes to p-tau accumulation, we determined if inhibiting tau acetylation eliminates p-tau and protects against tauopathy. Inhibiting p300 in primary neurons with the small molecule C646 eliminated ac-tau without affecting t-tau levels (Figure 8A). Strikingly, pathogenic tau phosphorylated at serine 202, detected with AT8 antibody, was also abolished within 2 h treatment with C646. Its inactive analog C37 had no effects (Figure 8B). These results suggest that deacetylation preferentially enhances degradation of p-tau, consistent with the observation that p-tau species are selectively degraded via the UPS pathway (Dickey et al., 2007; Dickey et al., 2006). In primary neurons expressing hTauP301L, a cellular model of tauopathy, AT8-positive p-tau was also diminished by C646 treatment (Figure 8C). Reducing tau improves cognitive function in mouse models of AD and FTDP-17 (Roberson et al., 2007; Santacruz et al., 2005) and protects against excitotoxicity (Roberson et al., 2007). Our results implicate modulating lysine acetylation as a new therapeutic strategy to reduce levels of tau, especially pathogenic forms of p-tau, in neurodegenerative tauopathies.
Figure 8. Reducing Tau Acetylation Eliminates p-Tau.
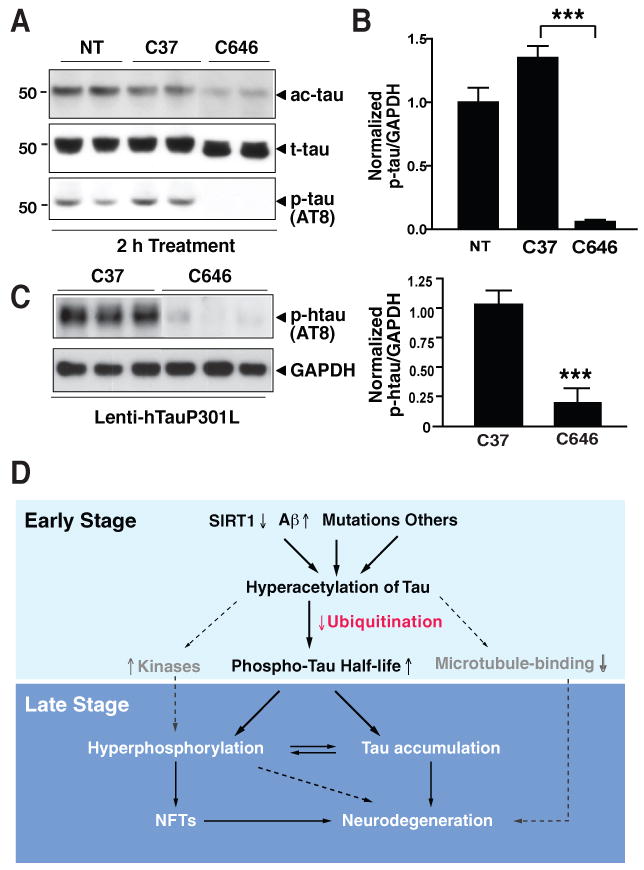
(A) C646 (20 μM) eliminated ac-tau and AT8-positive p-tau within 2 h in primary cortical neurons (DIV=9). Representative western blot of two experiments.
(B) C646 (20 μM) eliminated p-tau. Levels of p-tau/GAPDH in non-treated cells were set as 1. n=4. ***, P<0.0001 (unpaired t test).
(C) C646 (20 μM) eliminated AT8-positive p-tau in primary neurons expressing hTauP301L (DIV=12). Left, Representative western blot of two experiments. Right, Levels of p-tau/GAPDH in cells treated with control compound (C37) were set as 1. n=7. ***, P=0.0001 (unpaired t test).
(D) Hypothetical model of how tau acetylation may contribute to tau-mediated neurodegeneration. Dashed lines and factors in grey indicate pathways not yet tested. Values are means±SEM (B–C).
Discussion
Using two antibodies specific for ac-tau that we developed, we found that tau is acetylated and that acetylation of tau contributes to the accumulation of p-tau. In primary neurons, tau acetylation was elevated by an FTDP-17-linked mutation and by low levels of oligomeric Aβ. Tau acetylation was enhanced in patients at early and moderate Braak stages of tau pathology, before NFTs form. Moreover, we demonstrated that SIRT1 deacetylates tau and that acetyltransferase p300 mediates its acetylation. SIRT1 deficiency elevated ac-tau in vivo. Inhibition of SIRT1 blocked tau polyubiquitination and tau turnover, resulting in p-tau accumulation. In contrast, deacetylating tau by inhibiting p300 with a highly specific small-molecule inhibitor effectively eliminated p-tau.
Our ac-tau-specific antibodies Ab708 or 9AB did not recognize nonacetylated recombinant tau, allowing us to specifically detect ac-tau in vivo. The mutagenesis study showed that Ab708 recognizes the acetylated lysines at positions 163, 174 and 180, and possibly other acetylated lysines. Mass spectrometry detected p300-induced tau acetylation at 23 lysines. However, p300 likely acetylates other lysines in the 13% of the tau sequence we did not analyze. Importantly, the sites acetylated in vivo probably would differ from those mapped under cell-free conditions. In addition, depending on neuronal subtypes and cellular conditions, different sets of lysines could be acetylated. Systematic mass spectrometry will be needed to map the acetylation sites of tau under different pathophysiological conditions.
p300 induced tau acetylation both in vitro and in vivo. Exactly how p300 acetylates tau in vivo remains to be determined. Although p300 is mainly a nuclear protein, it can be cytosolic, acting as an E4 ubiquitin ligase for p53 (Shi et al., 2009). Since small amount of tau also may be present in the nucleus in some cell types (Wang et al., 1993), p300 might acetylate tau directly. However, p300 could also induce tau acetylation indirectly by unknown mechanisms. Inhibition of p300 with siRNA or the small molecule C646 reduced ac-tau levels significantly. C646 induced a much stronger decetylation effects than p300 siRNA, perhaps because C646 may also inhibit CBP, which shares 90% homology with p300 in the acetyltransferase domain (Liu et al., 2008). Due to the functional redundancy of CBP and p300, inhibition of both by C646 is likely to result in much stronger effects than inhibition of p300 only with siRNA.
Our study suggests that SIRT1 interacts with tau in vivo and directly deacetylates tau under cell-free conditions. Inhibition of SIRT1 with siRNA or the small molecule EX527 increased tau acetylation, and deleting SIRT1 elevated ac-tau in mouse brains. However, whether SIRT1 directly deacetylates tau in vivo remains to be established. SIRT1 shuttles between the nucleus and cytosol, and its subcellular localization is regulated by pathophysiological stimuli. SIRT1 is localized in the cytoplasm of embryonic and adult neural precursor cells, but transiently translocated in the nucleus in response to differentiation stimuli, resulting in reduction of cytosolic SIRT1 activities during differentiation (Hisahara et al., 2008). We found that levels of ac-tau were increased in primary neurons during maturation and correlated negatively with SIRT1 levels. These findings support the model that increased tau acetylation during neural maturation could be mediated by a reduction in cytosolic SIRT1 activity. The subcellular localization of SIRT1 is also modulated by stress and apoptosis (Greiss et al., 2008; Jin et al., 2007), providing potential mechanisms by which SIRT1 regulates tau acetylation and tauopathy during neuronal injury.
In human brains, elevated tau acetylation preceded the accumulation of NFTs, supporting the model that tau acetylation is an early event in tau-mediated neurodegeneration (Figure 8D). Our findings also suggest that tau acetylation is increased by stress due to Aβ accumulation or by mutations associated with tauopathy. However, the exact mechanisms underlying increased tau acetylation are undefined. Some likely mechanisms include deficiency in SIRT1 levels or SIRT1 activities in the cytosol, enhanced p300 levels or p300 activity, or alterations in tau conformation that blocks the access/binding to deacetylases or enhances the access/binding to acetyltransferases. These mechanisms are not mutually exclusive. Inhibition of p300 diminished ac-tau in primary neurons expressing TauP301L, consistent with the notion that P301L-induced hyperacetylation requires active p300. In AD brains, SIRT1 levels are reduced, and this reduction correlates with the amount of tau aggregates (Julien et al., 2009), consistent with a role of SIRT1 deficiency in Aβ-induced tau hyperacetylation. SIRT1 was found to reduce Aβ generation by activating transcription of a gene encoding α-secretase (Donmez et al., 2010). SIRT1 deficiency could also exacerbate the accumulation of Aβ, which could increase tau acetylation and tau phosphorylation even further. Other stress pathways induced during neuronal injury may also contribute to tau hyperacetylation and hyperphosphorylation.
Little is known how acetylation affects the functions of tau. We focused on the role of acetylation in ubiquitin-dependent degradation and effects on p-tau. However, acetylation of tau is likely to play other important roles in the development of tau-mediated neuropathology. We identified at least 13 putatively acetylated lysines in the microtubule-binding domains, raising the possibility that acetylation of tau could affect its ability to bind to and to stabilize microtubules, leading to neuronal dysfunction. The positively charged proline-rich regions are tightly bound to negatively charged microtubule surface (Amos, 2004; Kar et al., 2003). Since acetylation neutralizes charges in the microtubule-binding domain, aberrant acetylation might interfere with the binding of tau to microtubule, leading to tau dysfunction (Figure 8D). These residues appear to be variably acetylated by p300 in vitro, consistent with a dynamic acetylation process that is amenable to modulation in vivo.
The cross talk of tau acetylation with tau ubiquitination or phosphorylation may have implications for tau-mediated neurodegeneration (Figure 8D). Our findings suggest that tau acetylation directly contributes to accumulation of p-tau, a hallmark of tauopathy. Besides affecting p-tau turnover, tau acetylation may also modulate the activities of kinases involved in tau phosphorylation (Figure 8D). How kinase activities are modulated by tau acetylation is unknown. Regardless of the exact mechanisms, SIRT1 deficiency, high Aβ levels, or FTDP-17-linked mutations could lead to hyperacetylation of lysines on p-tau, preventing it from being ubiquitinated and degraded via the UPS pathway. In contrast, inhibition of p300 diminished ac-tau and effectively eliminated p-tau, suggesting that interfering with tau acetylation may be a new approach to reduce tauopathy.
Experimental Procedures
Chemicals and Reagents
C646 was from Chembridge (San Diego, CA). C37 (inactive analog of C646) was synthesized by David Meyers (Johns Hopkins University). We purchased EX527 (Tocris Bioscience, Ellisville, MO), resveratrol (EMD Chemicals, Gibbstown, NJ), MG-132 (Sigma, St. Louis, MO), cycloheximide (Sigma), Aβ42 peptide (rPeptide, Bogart, GA), and recombinant tau (rPeptide). Aβ42 oligomers were prepared as described (Chen et al., 2005).
Primary Antibodies
Two rabbit polyclonal anti-ac-tau antisera were generated against two acetylated peptides of tau (Abgent, San Diego, CA). PHF1 antibody was a kind gift of Peter Davis (Albert Einstein College of Medicine). Other antibodies were obtained and used at the indicated concentrations: Tau 5 (1:5000; Abcam, Cambridge, MA), anti-p300 (1:500; Santa Cruz Biotechnology, Santa Cruz, CA), anti-GAPDH (1:10000; Sigma), anti-tubulin (1:10000; Sigma), anti-FLAG (1:2000; Sigma), AT8 for p-tau (1:500; Thermo Fisher Scientific, Rockford, IL), anti-Sir2 (1:2000; Millipore, Billerica, MA), anti-HA (1:1000; Cell Signaling Technology, Danvers, MA). Secondary antibodies: peroxidase-conjugated goat anti-rabbit and anti-mouse IgGs (1:2000; GE Healthcare, Piscataway, NJ).
Expression Plasmids
For expression in HEK293T cells, cDNAs encoding hTauwt, hTau2KR (K174R, K180R), hTau3KR (K163R, K174R, K180 R), p300, SIRT1, H363Y (SIRT1), SIRT2, HDAC5, HDAC6, and HA-ubiquitin were cloned into pcDNA3.1 vector (Invitrogen). For protein expression in bacteria, hTauwt cDNA was cloned into pGEX4T-1 vector for GST-fusion protein expression. For expression in primary neurons, cDNAs encoding hTauwt, hTau3KR, hTauP301, and cre recombinase were cloned into lentiviral FUGW vectors.
Mice
All procedures involving animals were in compliance with the policies of the Animal Care and Use Committee at the University of California, San Francisco. PS19 mice were obtained from Jackson Laboratory (Bar Harbor, ME). The hT-PAC-N line was a generous gift of Gerard D. Schellenberg (University of Pennsylvania). SIRT-null and SIRT1F/F mice were kindly provided by Fred Alt (Harvard Medical School).
Cell Cultures and Transient Transfections
HEK293T cells and MEFs were grown at 37°C in Dulbecco's modified Eagles medium supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. For overexpression, transfections were performed with Lipofectamine 2000 (Invitrogen). For siRNA oligonucleotide transfection, HEK 293T cells were seeded at 1×105 cells/well on 12-well culture plates. After 12 h, cells were transfected with 10 nM ON-TARGETplus SMARTpool siRNA (Thermo Scientific-Dharmacon, Chicago, IL) with Lipofectamine RNAiMAX transfection reagent (Invitrogen), according to the manufacturer's protocol. SIRT1 siRNA (L-094699-01), SIRT2 siRNA (L-004826-00), HDAC6 siRNA (L-003499-00), p300 siRNA (L-003486-00), and PCAF siRNA (L-005055-00) were used to target specific cellular genes; siControl Non-Targeting siRNA#1 (Dharmacon) was used as a negative control. About 48 h after siRNA transfection, plasmid pcDNA3.1-hTauwt was transfected into the same culture plates. Cells were harvested 24 h later for real-time RT-PCR or western blot analyses.
Primary Neuronal Cultures and Lentiviral Infections
Primary cultures were established from cortices of Sprague-Dawley rat pups (Charles River Laboratories) or SIRT1F/F mice on postnatal day 0 or 1. Purified cells were plated at 160,000 cells/ml in Neurobasal medium supplemented with B27 (Invitrogen) on poly-ornithine coated plates. All treatments were performed at 7–13 DIV in Neurobasal medium supplemented with N2 (Invitrogen) unless noted otherwise.
Lentivirus was generated, purified, and used for infection as described (Chen et al., 2005). Recombinant lentivirus was produced by co-transfection of the shuttle vector (FUGW), two helper plasmids, delta8.9 packaging vector, and VSV-G envelope vector into 293T cells and purified by ultracentrifugation. Viral titers were measured by p24 enzyme-linked immunosorbent assays at the Gladstone-UCSF Laboratory of Clinical Virology.
Homogenization of Cells and Tissues and Western Blot Analyses
Cells or human or mouse brain tissues were lysed in RIPA buffer containing protease inhibitor cocktail (Sigma), 1 mM phenylmethyl sulfonyl fluoride (Sigma), phosphotase inhibitor cocktail (Roche), and HDAC inhibitors, including 5 mM nicotinamide (Sigma) and 1 μM trichostatin A (Sigma). After sonication, lysates from human or mouse brain tissues were centrifuged at 170,000 g at 4°C for 15 min and at 18,000 g at 4°C for 10 min. Supernatants were collected and analyzed by western blot. Bands in immunoblots were visualized by enhanced chemiluminescence (Pierce) and quantified by densitometry and Quantity One 4.0 software (Bio-Rad, Hercules, CA).
In Vitro Acetylation Assays
The reactions were performed as described (Pagans et al., 2005). Briefly, 1 μg of human recombinant tau, 2 nM of acetyl CoA (Sigma), and 1 μl of purified GST-p300 in acetylation buffer (50 mM HEPES, pH 8.0, 10% glycerol, 1 mM dithiothreitol (DTT), and 10 mM Na butyrate) were incubated for 30 min at 30°C with constant shaking. Reactions were stopped by adding 2× LDS sampling buffer (Invitrogen), followed by SDS-PAGE and western blot analyses.
MALDI-TOF Analyses
Samples from in vitro acetylation reactions were run on SDS-polyacrylamide gels and stained with Coomassie Blue. The band at approximately 65 kDa was cut out and sent to Stanford Mass Spectrometry Laboratory for analyses. In-gel digestion was done with Promega MS grade trypsin overnight. Before digestion, the gel slices were cut into approximately 1 mm × 1 mm cubes, reduced with 5 mM DTT and alkylated with acrylamide. Peptides were extracted and dried down using a speed-vac before reconstitution and analysis.
Nano reversed-phase HPLC was done with an Eksigent 2D nanoLC (Eksigent, Dublin, CA) with buffer A consisting of 0.1 % formic acid in water and buffer B consisting of 0.1% formic acid in acetonitrile. A fused silica column self packed with Duragel C18 (Peeke, Redwood City, CA) matrix was used with a linear gradient from 5% B to 40% B over 80 min at a flow rate of 450 nl/min. The nanoHPLC was interfaced with an Advion Nanomate (Ithaca, NY) for nano-electrospray ionization into the mass spectrometer. The mass spectrometer was a LCQ Deca XP Plus (Thermo Scientific), which was set in data dependent acquisition mode to perform MS/MS on the top three most intense ions with a dynamic exclusion setting of two. The DTA files were extracted from the raw data and systematically searched with Mascot. At least two peptides with a probability >95% were needed for the assignment of a protein.
In Vitro Deacetylation Assays
The reactions were modified from established procedures (Pagans et al., 2005). HEK293T cells were transfected with human FLAG-tagged SIRT1 plasmid or mock plasmid with Lipofectamine 2000 (Invitrogen). After 24 h, cells were lysed in lysis buffer (50 mM Tris-HCl, pH 7.5, 0.5 mM EDTA, 0.5 % NP-40, 150 mM NaCl, and protease inhibitor cocktails). After centrifugation at 13,000 rpm at 4°C for 10 min, equal amounts of supernatant proteins were immunoprecipitated with FLAG M2 agarose beads (Sigma) for 3 h at 4°C. Immuoprecipitated beads were washed twice in lysis buffer and once in deacetylation buffer (50 mM Tris-HCl, pH 9.0, 4 mM MgCl2, and 0.2 mM DTT) and incubated with in vitro ac-tau in deacetylation buffer at 30°C for 3 h with constant shaking. Reactions were stopped by adding 2× LDS sampling buffer (Invitrogen) and analyzed by western blot.
In Vivo Ubiquitination Assays
Procedures were modified from a published study (Oh et al., 2009). HEK293T cells were transfected with expression vectors encoding FLAG-tagged human tau and HA-ubiquitin with or without Myc-SIRT1 (wildtype or H363Y mutant). After 2 h of incubation, cells were treated with EX527, resveratrol, or DMSO in Dulbecco's modified Eagle's medium and incubated for 20 h. MG-132 (20 μM) was added and incubated for 4 h. Cells were lysed in ubiquitination buffer (20 mM Tris-HCl, pH 7.5, 0.1 mM EDTA, 0.2% Triton X-100, 150 mM NaCl, and protease inhibitor cocktail). Supernatant proteins were immunoprecipitated with FLAG M2 agarose beads (Sigma) for 3 h at 4°C. Reactions were washed at least three times with ubiquitination buffer and analyzed by SDS-PAGE and western blot with anti-HA antibody (Cell Signaling Technology).
Purification of GST Fusion Proteins and Interaction Assays
Full-length cDNA encoding human tau was subcloned into pGEX-4T-1 bacterial expression vector (Sigma) and transformed in the BL21 (DE3) strain. After induction with 100 μM isopropyl β-D-1-thiogalactopyranoside, bacterial cells were harvested and sonicated in phosphate-buffered saline with 1 mM EDTA, 0.5% Triton X-100, and protease inhibitor cocktail (Sigma). GST-tagged human tau or GST protein was purified with glutathione-agarose beads (GenScript).
In GST pull-down assays, bead-bound forms of purified GST-tau were incubated with lysates from HEK293T cells that were not transfected (for interaction with endogenous SIRT1) or transfected with FLAG-tagged human SIRT1. Beads were washed at least three times with lysis buffer containing Triton X-100 or Nonidet-40 and analyzed by SDS-PAGE and western blot with anti-SIRT1 antibody (Millipore) or anti-FLAG antibody (Sigma).
In coimmunoprecipitation assays, HEK293T cells were transfected with pcDNA3.1-hTau-FLAG. Triton X-100-solubilized lysates were incubated with FLAG M2 agarose beads for 3 h at 4°C. Beads were washed at least three times with lysis buffer containing Triton X-100 (0.5%) and analyzed by SDS-PAGE and western blot with anti-SIRT1 antibody (Millipore) or anti-FLAG antibody (Sigma).
Characterization of C646, a Selective p300 Inhibitor
C646 was identified as one of the putative inhibitors of p300 by a computational docking screen (Bowers et al., 2010). A convenient spectrophotometric assay was performed to validate it as a p300 inhibitor (Kim et al., 2000), followed by a series of secondary assays. In the coupled spectrophotometric assay, the acetyltransferase reaction product CoASH becomes a substrate for alphaketoglutarate dehydrogenase, which converts NAD to NADH, resulting in an increase of UV absorbance at 340 nm (Kim et al., 2000). A radioactive p300 HAT assay was subsequently performed to directly measure the IC50 of C646. The specific inhibition of p300 versus other acetyltransferases by C646 was further examined. These acetyltransferases included serotonin N-acetyltransferase, and the HATs pCAF, GCN5, Rtt109, Sas, and MOZ.
Data analyses
Statistical analyses were conducted with Graphpad Prism. Differences among multiple (≥3) means with one variable were evaluated by one-way ANOVA and the Tukey-Kramer posthoc test. Differences between two means were assessed with the paired or unpaired two-tailed t test. P < 0.05 was considered significant.
Supplementary Material
1. Tables:
Table S1 Putative modification of human tau peptides in the presence of p300; related to Figure 1.
Table S2 Summary of patients information used in the study; related to Figure 7.
2. Figures:
Figure S1 Representative spectrum of MS/MS fragmentation; related to Figure 1.
Figure S2 Alignment of human tau with rat tau and mouse tau; related to Figure 1 and Figure 2
Figure S3 Deleting SIRT1 in MEFs elevated ac-tau; related to Figure 3
Figure S4 Resveratrol (Res)-enhanced tau ubiquitination depended on SIRT1's deacetylase activity; related to Figure 6.
Figure S5 Ac-tau was more stable than t-tau in primary neurons; related to Figure 6
Acknowledgments
We thank Drs. Eric Verdin and Lennart Mucke for insightful discussions, Dr. Fred Alt from Harvard Medical School for SIRT1 null and floxed SIRT1 knockout mice, Drs. Chris Adams and Andrew Guzzetta from Stanford Mass Spectrometry Laboratory for MALDI-TOF analyses, Dr. Katerina Mancevska from New York Brain Bank at Columbia University for human brain samples, Stephen Ordway and Gary Howard for editorial review, and Kelley Nelson for administrative assistance. This work was supported in part by a grant from Whittier Foundation (to L.G.), UCSF ADRC grant AG023501-06 (to W. W. S and E. J. H), grants from NIH and FAMRI foundation (to P.A.C, D. M and C. M), a grant from NIH (to M. O), and NIH/NCRR CO6 RRO18928 (a facility grant to the J. David Gladstone Institutes). L. G. receives research funding from élan Pharmaceuticals. P.A.C is a cofounder and consults for Acylin Inc.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amos LA. Microtubule structure and its stabilisation. Org Biomol Chem. 2004;2:2153–2160. doi: 10.1039/b403634d. [DOI] [PubMed] [Google Scholar]
- Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci. 2007;8:663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- Bowers EM, Yan G, Mukherjee C, Orry A, Wang L, Holbert MA, Crump NT, Hazzalin CA, Liszczak G, Yuan H, et al. Virtual ligand screening of the p300/CBP histone acetyltransferase: identification of a selective small molecule inhibitor. Chem Biol. 2010;17:471–482. doi: 10.1016/j.chembiol.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol Berl. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Cairns NJ, Bigio EH, Mackenzie IR, Neumann M, Lee VM, Hatanpaa KJ, White CL, 3rd, Schneider JA, Grinberg LT, Halliday G, et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 2007;114:5–22. doi: 10.1007/s00401-007-0237-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caron C, Boyault C, Khochbin S. Regulatory cross-talk between lysine acetylation and ubiquitination: role in the control of protein stability. Bioessays. 2005;27:408–415. doi: 10.1002/bies.20210. [DOI] [PubMed] [Google Scholar]
- Chen J, Zhou Y, Mueller-Steiner S, Chen LF, Kwon H, Yi S, Mucke L, Gan L. SIRT1 Protects against Microglia-dependent Amyloid-{beta} Toxicity through Inhibiting NF-{kappa}B Signaling. J Biol Chem. 2005;280:40364–40374. doi: 10.1074/jbc.M509329200. [DOI] [PubMed] [Google Scholar]
- Cheng HL, Mostoslavsky R, Saito S, Manis JP, Gu Y, Patel P, Bronson R, Appella E, Alt FW, Chua KF. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc Natl Acad Sci USA. 2003;100:10794–10799. doi: 10.1073/pnas.1934713100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- Chua KF, Mostoslavsky R, Lombard DB, Pang WW, Saito S, Franco S, Kaushal D, Cheng HL, Fischer MR, Stokes N, et al. Mammalian SIRT1 limits replicative life span in response to chronic genotoxic stress. Cell Metab. 2005;2:67–76. doi: 10.1016/j.cmet.2005.06.007. [DOI] [PubMed] [Google Scholar]
- Cohen DE, Supinski AM, Bonkowski MS, Donmez G, Guarente LP. Neuronal SIRT1 regulates endocrine and behavioral responses to calorie restriction. Genes Dev. 2009;23:2812–2817. doi: 10.1101/gad.1839209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cripps D, Thomas SN, Jeng Y, Yang F, Davies P, Yang AJ. Alzheimer disease-specific conformation of hyperphosphorylated paired helical filament-Tau is polyubiquitinated through Lys-48, Lys-11, and Lys-6 ubiquitin conjugation. J Biol Chem. 2006;281:10825–10838. doi: 10.1074/jbc.M512786200. [DOI] [PubMed] [Google Scholar]
- Dickey CA, Kamal A, Lundgren K, Klosak N, Bailey RM, Dunmore J, Ash P, Shoraka S, Zlatkovic J, Eckman CB, et al. The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J Clin Invest. 2007;117:648–658. doi: 10.1172/JCI29715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickey CA, Yue M, Lin WL, Dickson DW, Dunmore JH, Lee WC, Zehr C, West G, Cao S, Clark AM, et al. Deletion of the ubiquitin ligase CHIP leads to the accumulation, but not the aggregation, of both endogenous phospho- and caspase-3-cleaved tau species. J Neurosci. 2006;26:6985–6996. doi: 10.1523/JNEUROSCI.0746-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donmez G, Wang D, Cohen DE, Guarente L. SIRT1 suppresses beta-amyloid production by activating the alpha-secretase gene ADAM10. Cell. 2010;142:320–332. doi: 10.1016/j.cell.2010.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Gan L, Mucke L. Paths of convergence: sirtuins in aging and neurodegeneration. Neuron. 2008;58:10–14. doi: 10.1016/j.neuron.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge X, Jin Q, Zhang F, Yan T, Zhai Q. PCAF acetylates {beta}-catenin and improves its stability. Mol Biol Cell. 2009;20:419–427. doi: 10.1091/mbc.E08-08-0792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman RH, Smolik S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000;14:1553–1577. [PubMed] [Google Scholar]
- Greiss S, Hall J, Ahmed S, Gartner A. C. elegans SIR-2.1 translocation is linked to a proapoptotic pathway parallel to cep-1/p53 during DNA damage-induced apoptosis. Genes Dev. 2008;22:2831–2842. doi: 10.1101/gad.482608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronroos E, Hellman U, Heldin CH, Ericsson J. Control of Smad7 stability by competition between acetylation and ubiquitination. Mol Cell. 2002;10:483–493. doi: 10.1016/s1097-2765(02)00639-1. [DOI] [PubMed] [Google Scholar]
- Haigis MC, Guarente LP. Mammalian sirtuins--emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006;20:2913–2921. doi: 10.1101/gad.1467506. [DOI] [PubMed] [Google Scholar]
- Hisahara S, Chiba S, Matsumoto H, Tanno M, Yagi H, Shimohama S, Sato M, Horio Y. Histone deacetylase SIRT1 modulates neuronal differentiation by its nuclear translocation. Proc Natl Acad Sci U S A. 2008;105:15599–15604. doi: 10.1073/pnas.0800612105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang XF, Yao TP. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417:455–458. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- Ito A, Kawaguchi Y, Lai CH, Kovacs JJ, Higashimoto Y, Appella E, Yao TP. MDM2-HDAC1-mediated deacetylation of p53 is required for its degradation. Embo J. 2002;21:6236–6245. doi: 10.1093/emboj/cdf616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Q, Yan T, Ge X, Sun C, Shi X, Zhai Q. Cytoplasm-localized SIRT1 enhances apoptosis. J Cell Physiol. 2007;213:88–97. doi: 10.1002/jcp.21091. [DOI] [PubMed] [Google Scholar]
- Jin YH, Jeon EJ, Li QL, Lee YH, Choi JK, Kim WJ, Lee KY, Bae SC. Transforming growth factor-beta stimulates p300-dependent RUNX3 acetylation, which inhibits ubiquitination-mediated degradation. J Biol Chem. 2004;279:29409–29417. doi: 10.1074/jbc.M313120200. [DOI] [PubMed] [Google Scholar]
- Julien C, Tremblay C, Emond V, Lebbadi M, Salem N, Jr, Bennett DA, Calon F. Sirtuin 1 reduction parallels the accumulation of tau in Alzheimer disease. J Neuropathol Exp Neurol. 2009;68:48–58. doi: 10.1097/NEN.0b013e3181922348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar S, Fan J, Smith MJ, Goedert M, Amos LA. Repeat motifs of tau bind to the insides of microtubules in the absence of taxol. EMBO J. 2003;22:70–77. doi: 10.1093/emboj/cdg001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Nguyen MD, Dobbin MM, Fischer A, Sananbenesi F, Rodgers JT, Delalle I, Baur JA, Sui G, Armour SM, et al. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer's disease and amyotrophic lateral sclerosis. Embo J. 2007;26:3169–3179. doi: 10.1038/sj.emboj.7601758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Tanner KG, Denu JM. A continuous, nonradioactive assay for histone acetyltransferases. Anal Biochem. 2000;280:308–314. doi: 10.1006/abio.2000.4546. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. Acetylation: a regulatory modification to rival phosphorylation? Embo J. 2000;19:1176–1179. doi: 10.1093/emboj/19.6.1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Wang L, Zhao K, Thompson PR, Hwang Y, Marmorstein R, Cole PA. The structural basis of protein acetylation by the p300/CBP transcriptional coactivator. Nature. 2008;451:846–850. doi: 10.1038/nature06546. [DOI] [PubMed] [Google Scholar]
- Ludolph AC, Kassubek J, Landwehrmeyer BG, Mandelkow E, Mandelkow EM, Burn DJ, Caparros-Lefebvre D, Frey KA, de Yebenes JG, Gasser T, et al. Tauopathies with parkinsonism: clinical spectrum, neuropathologic basis, biological markers, and treatment options. Eur J Neurol. 2009;16:297–309. doi: 10.1111/j.1468-1331.2008.02513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan P, Korvatska E, Poorkaj P, Evstafjeva Z, Robinson L, Greenup L, Leverenz J, Schellenberg GD, D'Souza I. Tau isoform regulation is region- and cell-specific in mouse brain. J Comp Neurol. 2008;511:788–803. doi: 10.1002/cne.21867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michan S, Sinclair D. Sirtuins in mammals: insights into their biological function. Biochem J. 2007;404:1–13. doi: 10.1042/BJ20070140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napper AD, Hixon J, McDonagh T, Keavey K, Pons JF, Barker J, Yau WT, Amouzegh P, Flegg A, Hamelin E, et al. Discovery of indoles as potent and selective inhibitors of the deacetylase SIRT1. J Med Chem. 2005;48:8045–8054. doi: 10.1021/jm050522v. [DOI] [PubMed] [Google Scholar]
- North BJ, Marshall BL, Borra MT, Denu JM, Verdin E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol Cell. 2003;11:437–444. doi: 10.1016/s1097-2765(03)00038-8. [DOI] [PubMed] [Google Scholar]
- Oh YM, Kwon YE, Kim JM, Bae SJ, Lee BK, Yoo SJ, Chung CH, Deshaies RJ, Seol JH. Chfr is linked to tumour metastasis through the downregulation of HDAC1. Nat Cell Biol. 2009;11:295–302. doi: 10.1038/ncb1837. [DOI] [PubMed] [Google Scholar]
- Ohsawa S, Miura M. Caspase-mediated changes in Sir2alpha during apoptosis. FEBS Lett. 2006;580:5875–5879. doi: 10.1016/j.febslet.2006.09.051. [DOI] [PubMed] [Google Scholar]
- Pagans S, Pedal A, North BJ, Kaehlcke K, Marshall BL, Dorr A, Hetzer-Egger C, Henklein P, Frye R, McBurney MW, et al. SIRT1 regulates HIV transcription via Tat deacetylation. PLoS Biol. 2005;3:e41. doi: 10.1371/journal.pbio.0030041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrucelli L, Dickson D, Kehoe K, Taylor J, Snyder H, Grover A, De Lucia M, McGowan E, Lewis J, Prihar G, et al. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum Mol Genet. 2004;13:703–714. doi: 10.1093/hmg/ddh083. [DOI] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi D, Pop MS, Kulikov R, Love IM, Kung AL, Grossman SR. CBP and p300 are cytoplasmic E4 polyubiquitin ligases for p53. Proc Natl Acad Sci U S A. 2009;106:16275–16280. doi: 10.1073/pnas.0904305106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimura H, Schwartz D, Gygi SP, Kosik KS. CHIP-Hsc70 complex ubiquitinates phosphorylated tau and enhances cell survival. J Biol Chem. 2004;279:4869–4876. doi: 10.1074/jbc.M305838200. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci U S A. 1998;95:7737–7741. doi: 10.1073/pnas.95.13.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan JM, Wong ES, Kirkpatrick DS, Pletnikova O, Ko HS, Tay SP, Ho MW, Troncoso J, Gygi SP, Lee MK, et al. Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum Mol Genet. 2008;17:431–439. doi: 10.1093/hmg/ddm320. [DOI] [PubMed] [Google Scholar]
- Tanno M, Sakamoto J, Miura T, Shimamoto K, Horio Y. Nucleocytoplasmic shuttling of the NAD+-dependent histone deacetylase SIRT1. J Biol Chem. 2007;282:6823–6832. doi: 10.1074/jbc.M609554200. [DOI] [PubMed] [Google Scholar]
- Wang Y, Loomis PA, Zinkowski RP, Binder LI. A novel tau transcript in cultured human neuroblastoma cells expressing nuclear tau. J Cell Biol. 1993;121:257–267. doi: 10.1083/jcb.121.2.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XJ, Seto E. Lysine acetylation: codified crosstalk with other posttranslational modifications. Mol Cell. 2008;31:449–461. doi: 10.1016/j.molcel.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1. Tables:
Table S1 Putative modification of human tau peptides in the presence of p300; related to Figure 1.
Table S2 Summary of patients information used in the study; related to Figure 7.
2. Figures:
Figure S1 Representative spectrum of MS/MS fragmentation; related to Figure 1.
Figure S2 Alignment of human tau with rat tau and mouse tau; related to Figure 1 and Figure 2
Figure S3 Deleting SIRT1 in MEFs elevated ac-tau; related to Figure 3
Figure S4 Resveratrol (Res)-enhanced tau ubiquitination depended on SIRT1's deacetylase activity; related to Figure 6.
Figure S5 Ac-tau was more stable than t-tau in primary neurons; related to Figure 6