

Abstract
Many noncoding regions of genomes appear to be essential to genome function. Conservation of large numbers of noncoding sequences has been reported repeatedly among mammals but not thus far among birds and reptiles. By searching genomes of chicken (Gallus gallus), zebra finch (Taeniopygia guttata), and green anole (Anolis carolinensis), we quantified the conservation among birds and reptiles and across amniotes of long, conserved noncoding sequences (LCNS), which we define as sequences ≥500 bp in length and exhibiting ≥95% similarity between species. We found 4,294 LCNS shared between chicken and zebra finch and 574 LCNS shared by the two birds and Anolis. The percent of genomes comprised by LCNS in the two birds (0.0024%) is notably higher than the percent in mammals (<0.0003% to <0.001%), differences that we show may be explained in part by differences in genome-wide substitution rates. We reconstruct a large number of LCNS for the amniote ancestor (ca. 8,630) and hypothesize differential loss and substantial turnover of these sites in descendent lineages. By contrast, we estimated a small role for recruitment of LCNS via acquisition of novel functions over time. Across amniotes, LCNS are significantly enriched with transcription factor binding sites for many developmental genes, and 2.9% of LCNS shared between the two birds show evidence of expression in brain expressed sequence tag databases. These results show that the rate of retention of LCNS from the amniote ancestor differs between mammals and Reptilia (including birds) and that this may reflect differing roles and constraints in gene regulation.
Keywords: dosage compensation, Blast, regulatory element, reptile, transcription factor binding site
Introduction
The age of comparative genome analysis is upon us, allowing comparisons of both coding and noncoding sequences across closely and distantly related species. One important area of research has been the identification of conserved noncoding elements (CNEs), many of which have been identified in recent years (Nowak 1994; Dermitzakis et al. 2003; Margulies et al. 2003; Sandelin et al. 2004b; de la Calle-Mustienes et al. 2005; Siepel et al. 2005; Derti et al. 2006; Drake et al. 2006; Gardiner et al. 2006; Venkatesh et al. 2006; Sakuraba et al. 2008). CNEs vary in terms of percent similarity, sequence length, and species in which they have been found and have been of interest because of the perceived contradiction between their evolutionary longevity and their apparent lack of function. Human ultraconserved elements (UCEs), for example, are longer than 200 bp with 100% identity with other mammals and they are more highly conserved than coding regions (Katzman et al. 2007), suggesting an important functional role preserved by stabilizing selection. Although these sequences are noncoding, some have been found to overlap protein-coding elements. Most genes overlapped by UCEs are involved in RNA processing (Bejerano et al. 2004). UCEs are not commonly found in segmental duplications but those that are tend to overlap exons (Derti et al. 2006). Long conserved noncoding sequences (LCNS) are yet another class of CNE. LCNS were defined by Sakuraba et al. (2008) as sequences that are >500 bp long and >95% similar between two or more species, a definition we use here (fig. 1). Rather than focusing on a defined type of conserved element, Meader et al. (2010) estimated the total number of constrained bases in eukaryotic genomes and found that between 6.5% and 10% of the human genome is constrained.
FIG. 1.—

Conservation of long noncoding sequences. Four examples of LCNS shared by human, mouse, dog, chicken, zebra finch, and Anolis (lengths: [A] 708 bp; [B] 879 bp; [C] 1,902 bp; [D] 509 bp). Sequences are mapped to the human genome assembly (February 2009 [GRCh37/hg19] at http://www.genome.ucsc.edu). In each figure, the window is a magnification of the region marked by a red bar, and the black box in the LCNS track indicates the position of the conserved sequence.
In addition to stabilizing selection, another hypothesis for the conservation of CNEs is that they simply reflect genomic regions of low mutation rate. Multiple studies, however, suggest that this is not the case. Analysis of HapMap data shows an allele frequency spectrum that is skewed toward rare variants suggesting selective constraint rather than reduced mutation rates (Drake et al. 2006). Mutagenesis studies in mice have also found equivalent mutation capacity between LCNS and other regions of the genome (Sakuraba et al. 2008). LCNS, therefore, do not appear to be mutational cold spots, at least in those mammalian systems tested. However, researchers have known for many years that generation time, metabolic rate, and other physiological mechanisms can influence mutation rate. For this reason, lower LCNS abundance might be expected in lineages such as rodents that have shorter generation times and higher mutation rates (Wu and Li 1985; Martin and Palumbi 1993).
The genomic location of CNEs may also provide clues about their possible function. For example, noncoding sequences are conserved in the neighborhood of the SIM2 gene interval on human chromosome 21 and near the vertebrate Iroquois gene cluster on human chromosome 16 (de la Calle-Mustienes et al. 2005), suggesting a regulatory role (Frazer et al. 2004). In vertebrates, CNEs are found near or within 3′ untranslated regions of regulatory genes, and they seem to enrich RNA secondary structure (Siepel et al. 2005).
Functional studies of conserved sequences have begun to reveal a role in gene regulation. Human–rodent UCEs were found to be developmental enhancers (Visel et al. 2008). Several noncoding sequences appear to regulate gene expression. However, mice that have had UCEs deleted from the genome did not exhibit notable abnormalities. These regions were adjacent to Dmrt1-3, Pax6, Rcn1, and other genes, but their deletion did not appear to affect the function of the adjacent genes (Ahituv et al. 2007). Likewise, mice with point mutations in their LCNS exhibited no clear phenotypic abnormalities (Sakuraba et al. 2008).
As a result of new releases of bird and reptile genome databases, we were able to compare for the first time LCNS shared by mammals, birds, and a nonavian reptile. In this manuscript, we will refer to nonavian reptiles as “reptiles” and reserve Reptilia to describe both reptiles and birds to simplify discussion of the mammalian and reptilian branches of the amniote tree. A mammal–reptile comparison is novel and worthwhile for the characterization of reptile- and mammal-specific rates of genome evolution. Since the lineages leading to chicken and zebra finch and the lineages leading to human and mouse diverged at roughly the same time (respectively, around 81 and 76 MYA; Benton and Donoghue 2007), the number of LCNS shared by chicken and zebra finch and the number shared by human and mouse should be roughly equal, assuming similar rates of reptilian and mammalian LCNS evolution. Also, avian genomes are smaller than those of other amniotes (Hillier et al. 2004; Organ et al. 2008), and if relative numbers of LCNS do not correlate with genome size or timing of divergence, then their presence may suggest differences in functionality or different genomic dynamics among the groups. Finally, if LCNS play a regulatory or other functional role, sequences conserved across amniotes will be of particular interest for functional studies. Toward this goal, we here investigate the frequency, phylogenetic distribution, and possible regulatory role of LCNS in amniotes.
Materials and Methods
Identification of Relevant Sequences
Supplementary table S1 (Supplementary Material online) identifies and describes the assemblies from which genomic sequences were collected. Whole-genome sequences for human (Homo sapiens), mouse (Mus musculus), dog (Canis familiaris), cow (Bos taurus), chicken (Gallus gallus), green anole (Anolis carolinensis), and zebra finch (Taeniopygia guttata) were collected from the Ensembl database (http://www.ensembl.org/). Reptile and bird genome sequences were collected from release 56 (released September 2009) and mammal genome sequences were collected from release 57 (released March 2010). Interspersed repeats and low-complexity regions were detected with RepeatMasker (Smit et al. 2004) by the Ensembl team and masked. Following Sakuraba et al. (2008), we defined LCNS as regions spanning at least 500 bp in which sequences from two species share at least 95% identity and we extend this definition to encompass multispecies comparisons. From the whole-genome assemblies, we masked all exons.
Blast Strategy and Parsing
To compare among the three groups of amniotes (mammals, reptiles, and birds), we used Blast+ 2.2.22 (Zhang et al. 2000). For each species, masked molecular data were transformed into a Blast database. To detect LCNS between two species (1 and 2), we reciprocally compared species 1 sequences with the species 2 database and species 2 sequences with the species 1 database. Both sets of results were compared to detect LCNS. Due to our masking protocol, identified LCNS contained neither exons nor assembly gaps. The search parameter for BlastN was “e-value = 1 × 10−30.” We did not use the post-processing identity filter from the Blast package because it discards whole sequences that are not 95% identical to query instead of looking for a partial extract of the hit with a hyper-conserved core with at least 95% identity.
The results from the Blast analyses were parsed using Python (ver 2.6.1—http://www.python.org/) and Biopython (ver 1.53—http://biopython.org/). We first selected the high-scoring sequence pairs (HSP) in which sequences from both query and database are at least 500 bp long with at least 95% identity and in unmasked portions of the genome. Python scripts investigated the remaining HSP to detect extracts that matched our search criteria. For each LCNS shared by two species, we recorded the length of the alignment and its percent identity. In each two-species LCNS data set, we identified possible duplications by looking for overlap between LCNS.
To detect LCNS shared by three species, each two-species LCNS was queried in the genome of a third species. Therefore, a normal three-species LCNS corresponds to a set of LCNS shared by three species, but the search leading to each LCNS differs in the species (1, 2, or 3) from which the query was initiated. For example, three-species LCNS were classified as a three-species LCNS for species 1 if they were found by using species 1 as a query. By this method, we could identify the differences among trios of queried species and distinguish LCNS shared by either two or three species.
Comparison of Reptilian and Mammalian LCNS
The sequences of mouse and dog LCNS shared with human were compared with masked genome databases of Anolis, chicken, and zebra finch. The bird and reptile genomes were queried by mammalian LCNS to find the two-species and three-species LCNS using the same procedure described above. Multispecies matches were searched among mammals and birds and also among mammals, birds, and Anolis.
LCNS shared by Reptilia and mammals were determined by a different process. The mammalian sequences were mapped in the chicken genome by Blast (e-value of 1 × 10−15). For each reptilian LCNS, the chicken sequence was compared with the results from the mammal/chicken Blast analyses. Any part of this chicken sequence that overlapped with the mapped mammalian sequence was characterized as an amniote LCNS, provided that it was at least 500 bp. We enumerated distinct sets of LCNS for various ancestors in the amniote tree by counting the total number of distinct LCNS among different subsets of extant species in our data set.
Identification of Possible Coding Sequences in Reptile and Bird LCNS
Annotation of the available reptile and bird genomes is currently incomplete. In zebra finch, for example, the Ensembl gene set comprises 17,475 of an expected ∼20,000 genes (Warren et al. 2010). To determine if reptile and bird LCNS actually correspond to unannotated genes, they were compared with the human gene set. FASTA sequences from all human exons were obtained from the Ensembl database using the BioMart tool (Haider et al. 2009). A Blast database was created with these sequences. FASTA sequences from all LCNS were compared with the human exon database with BlastN (parameter: e-value = 1 × 10−15). All LCNS in which one of its sequences had a Blast hit with the human exon database were flagged as possible coding sequences.
Comparison of LCNS to Whole Genomes
To test the hypothesis that LCNS abundance scaled with other measures of genome length, the numbers and total lengths of LCNS were regressed against mean whole-genome sizes of the species in which the LCNS is shared (Olmo 1976; Tiersch et al. 1989; Peterson et al. 1994; Vinogradov 1998; Gregory 2005; Johnston et al. 2007; Pigozzi 2008). Whole-genome sizes were considered in terms of C-value, diploid number of chromosomes, and total sequence length (Gb).
Expression of LCNS
The zebra finch has been intensively studied with respect to patterns of gene expression in the brain. Extensive gene expression data are therefore available and afford the opportunity to test for expression of LCNS. Identified LCNS were compared with brain expressed sequence tags (ESTs) from zebra finch (Replogle et al. 2008). The set of ESTs includes 17,214 nonredundant products that have been spotted on a cDNA microrray (Replogle et al. 2008) and subsequently used in a series of studies of gene expression (Dong et al. 2009; London et al. 2009; Tomaszycki et al. 2009). Surprisingly, many of these transcripts have been identified as intergenic, in which case they lie between genes, or intronic, meaning they are located between exons (Warren et al. 2010). Therefore, these data also provide access to the expressed noncoding portion of the genome. Warren et al. (2010) mapped ESTs in the zebra finch genome using GMAP software (Wu and Watanabe 2005). Coordinates of these mapped ESTs were compared with the sequence coordinates of LCNS. In cases in which sequence from a zebra finch LCNS overlapped a song transcript in the zebra finch genome, the LCNS have been annotated with the name of the song transcript. Noncoding ESTs have also been shown to be differentially regulated (up- or downregulated) in the brain in response to a behavioral stimulus, bird song (Dong et al. 2009). Comparison of these data sets identified expressed LCNS that are regulated in response to song.
Analysis of Transcription Factor Binding Sites
Each LCNS set was tested for associations with cis-regulatory motifs. The zebra finch genome was scanned in nonoverlapping windows of length 500 bp for 104 vertebrate-related motifs collected from the JASPAR database, which is a repository for transcription factor binding sites (Sandelin et al. 2004a). Each window was scored for each motif using Stubb, a Hidden Markov Model–based scoring method for motif clustering (Alaux et al. 2009). Stubb scores a fixed-length (500 bp) window for the presence of one or more weak or strong matches to the motif. It has been demonstrated that scoring short regions rather than individual sites better mirrors the thermodynamic nature of the protein–DNA interaction and adds statistical power. Stubb was previously used for analysis of human (Sinha et al. 2008), honeybee (Sinha et al. 2006), fruit fly (Sinha et al. 2004), and wasp (Kim et al. 2010) genomes, among others. For each JASPAR motif, a set of “motif target windows” was defined in the genome by selecting the 1% top Stubb-scoring windows. The hypergeometric P value was calculated for the enrichment for motif target windows in each LCNS set. Results show each motif and its associated P value of enrichment in ascending order. For a negative control, all enrichment tests were repeated with a randomly generated LCNS set. For each LCNS in the original set, a length matched noncoding sequence was selected randomly from the zebra finch genome.
Evolutionary Patterns of LCNS Retention and Loss
LCNS abundances shared between species were compared with the rate of silent substitution to test whether patterns of LCNS evolution are related to global substitution rates. The number of LCNS between pairs of taxa was compared with the number of synonymous nucleotide substitutions (dS) in protein-coding regions across the genome. We calculated pairwise dS values between all one-to-one and apparent one-to-one orthologs as annotated by Ensembl for the species pairs of human–mouse, human–dog, human–chicken, mouse–chicken, chicken–Anolis, zebra finch–Anolis, and chicken–zebra finch. We aligned each pair of orthologs using TranslatorX (Abascal et al. 2010), which uses protein alignments as a guide for nucleotide alignment with the MUSCLE algorithm (Edgar 2004). Values of dS for each orthologous pair were then determined using the Nei and Gojobori method (Nei and Gojobori 1986), as implemented in the codeml program of the PAML 4.4 package (Yang 2007). We then used the average dS values as a measure of divergence between species pairs. To avoid issues of excessive divergence resulting in saturation, we restricted the average divergence calculation to those genes with dS <2, following the example of Axelsson et al. (2008).
We also investigated the evolutionary dynamics of whole LCNS counts in a phylogenetic framework. We used the inverse of the number of shared LCNS (LCNS−1) as representative of the degree of LCNS divergence among species. For example, chicken and zebra finch share 4,294 LCNS, the inverse of which is 0.000233, and is an order of magnitude smaller than the LCNS−1 of 0.0017 for chicken and Anolis, which share 587 LCNS. We used pairwise LCNS−1 values, with a frog (Xenopus tropicalis) as the outgroup, to construct both topology-constrained and topology-unconstrained phylogenies using the BioNJ method in PAUP (Swofford 2003). In addition, we explored trees in which negative branch lengths were allowed and disallowed. We also scaled branch lengths by time (Benton and Donoghue 2007) to estimate the rate of retention of shared LCNS in amniotes. Molecular clock tests for these trees were performed using the programs Kitsch and Fitch in PHYLIP (Felsenstein 2010). Comparison of the deviations of the sum of squares between the branch lengths in the distance matrix and the matrix of branch lengths in the optimized tree provides a test of the molecular clock for these types of data (Felsenstein 1984).
Results
LCNS Landscape Across Amniotes
Despite a broadly similar divergence time, far more LCNS are shared between chicken and zebra finch (4,294) than between human and mouse (1,236). Even though mouse and human share a more recent common ancestor, human and dog show a larger number of LCNS (4,570) than do human and mouse. The number of LCNS shared by human and dog is similar to that between zebra finch and chicken (table 1). Because of the unusual pattern of conservation among human, mouse, and dog, we also examined LCNS shared by human and cow. Human–cow LCNS abundance (3,191) more closely resembled the number shared between human and dog than between human and mouse. Five hundred and seventy-four LCNS were found in Reptilia (shared by chicken, zebra finch, and Anolis). Of 574 reptilian LCNS, 486 are shared by all three species with the remainder shared by only two (supplementary table S2, Supplementary Material online). These two-species LCNS are also present in the third species but fail to meet the search criteria of >95% conservation across >500 bp.
Table 1.
LCNS Count and Proportion Shared between Pairs of Amniotesa
| Chicken | Zebra Finch | Anolis | Mouse | Dog | Human | Xenopus | |
| Chicken | 3.51 | 0.42 | 0.20 | 0.34 | 0.27 | 0.06 | |
| Zebra finch | 4,294 | 0.40 | 0.19 | 0.33 | 0.26 | 0.05 | |
| Anolis | 587 | 565 | 0.18 | 0.15 | 0.12 | 0.03 | |
| Mouse | 361 | 348 | 361 | 0.47 | 0.42 | 0.02 | |
| Dog | 676 | 652 | 331 | 1,206 | 1.48 | 0.03 | |
| Human | 636 | 610 | 310 | 1,236 | 4,570 | 0.05 | |
| Xenopus | 118 | 99 | 76 | 56 | 82 | 151 |
The top diagonal in the matrix presents LCNS count per megabase of the pair’s mean genome size. The bottom diagonal in the matrix presents count of LCNS per species pair. In addition to pairs, LCNS were also found to be shared among human, dog, chicken, and zebra finch (1,664); human, mouse, chicken, and zebra finch (780); chicken, zebra finch, and Anolis (574); chicken, zebra finch, human, mouse, and dog (471); and across amniotes (Anolis, chicken, zebra finch, human, mouse, and dog [338]).
Twenty-five putative LCNS duplications were identified among the reptilian LCNS. Seventeen of these duplications map to zebra finch “Un” chromosome and eight map to the Anolis assembly (supplementary table S2, Supplementary Material online). Another 250 putative duplications were found among the bird LCNS: 14 duplications were found in chicken and 236 were found in zebra finch. Of the 236 zebra finch duplications, only 46 were assigned to chromosomes. Duplications mapped to the chromosome “Un” should be regarded with some skepticism as they may represent allelic variation or other problems in the assembly. Seven duplications were found in the Anolis assembly from the LCNS shared by Anolis and chicken. Among the 565 LCNS shared by Anolis and zebra finch, 20 duplications were identified (14 in zebra finch and 6 in Anolis). Among the 574 LCNS shared among Reptilia, 4 duplications were found in Anolis and 13 duplications were found in zebra finch. The human–cow LCNS include 79 duplications (75 from cow and 4 from human) yet the human–mouse LCNS include no duplications and human–dog LCNS include 9 duplications from dog and 12 duplications from human (supplementary table S2, Supplementary Material online).
Of the 574 LCNS shared among Reptilia, only 36 (6.28%) have hits with human exons, suggesting that these may, in fact, be coding sequences that have not been properly annotated in reptilian genomes. Six of these are only two-species LCNS and may not be present in chicken (two), zebra finch (three), or Anolis (one). Of the 4,294 LCNS shared by the two birds, only 97 (2.3%) show evidence of expression by comparison with sequenced ESTs. Twenty-eight of these expressed LCNS are differentially regulated in response to song playback treatment (Dong et al. 2009). Seven (1.2%) of the LCNS shared among Reptilia and 123 of uniquely avian LCNS (2.9%) overlap with brain-expressed transcripts found in zebra finch EST databases. Thirty-eight (0.9%) avian LCNS mapped to the chicken Z chromosome, a surprisingly low percentage given that the Z chromosome comprises 7.1% of the chicken genome sequence on Ensembl. Four hundred and eighty-six (84.7%) avian LCNS mapped to chicken macrochromosomes, excluding the Z-linked sequences. The remaining 50 avian LCNS mapped to microchromosomes.
Chicken and zebra finch also share the longest LCNS (2,527 bp) in the data set (fig. 2A). Slight variation is seen among species pairs in the shortest size class (500–600 bp), but LCNS of this size comprise the greatest proportion of LCNS shared by chicken and Anolis, followed by human and mouse. However, chicken and Anolis shared the fewest LCNS of the shortest size class, whereas human and dog shared the most.
FIG. 2.—

Numbers and sizes of LCNS. (A) Size class distributions of LCNS shared by chicken and zebra finch; chicken and Anolis; human and dog; and human and mouse. (B) LCNS comprising whole genomes. For each of six pairs of amniotes, the proportion (left y axis; black bars) and number (right y axis; gray bars) of shared LCNS (>500 bp; >95% similar) are presented per megabase across the whole genome.
Rates of LCNS retention
Linear regressions of LCNS against whole-genome size (C-value) and diploid number of chromosomes (2n) did not demonstrate a discernible correlation (LCNS vs. C-value: R2 = 0.041; LCNS vs. 2n: R2 = 0.206). We refrain from presenting P values because of the well-known problem of phylogenetic correlation between species that requires data transformation to provide independent data points; we are unaware of statistical models that allow the analysis of traits such as LCNS that are by definition shared between species. Comparisons of proportions of genomes composed of LCNS across species demonstrated a more than 2-fold increase in birds as compared with other study taxa (fig. 2B).
The relationship of the paired-taxa measurements of LCNS numbers, divergence time, and dS can reveal patterns of evolutionary rates of LCNS retention between species. Species pairs that diverged more recently share more LCNS, suggesting a relationship between LCNS loss and time among studied species (fig. 3A and B). Elevated LCNS abundance was found in comparison of human–dog to other pairs with respect to dS, indicating that LCNS evolution between human and dog is non-neutral (fig. 3C and D). Similarly, there appears to be greater LCNS conservation between chicken and zebra finch than expected by dS (fig. 3C and D). The trends observed in figure 3A and C relative to figure 3B and D suggest that mouse is an outlier.
FIG. 3.—

Relationship between number of LCNS and divergence time. (A) Counts of LCNS shared between species pairs of varying divergence times. (B) Trend from (A) excluding mouse. (C) Counts of LCNS shared between species with varying mean dS as measured in coding regions. (D) Trend from (C) excluding mouse.
Phylogenetic trees inferred from pairwise LCNS−1 data using Xenopus as an outgroup suggest that the rate of divergence in LCNS number is heterogeneous in amniotes, a result confirmed by molecular clock tests (F = 3.53, degrees of freedom = 5, P = 0.04). Phylogenetic analysis of the raw data results in branch lengths that are relatively long within mammals compared with reptiles and birds (fig. 4A and B). By contrast, rates of LCNS divergence divided by time (LCNS−1) are clearly greater along the branch leading to the birds and Anolis than along the ancestral mammal branch (fig. 4C and D). This tree shows a dramatic increase in the rate of LCNS evolution within the common reptilian ancestor (1.33 × 10−5 LCNS−1/My), but rates of LCNS evolution are an order of magnitude lower within Reptilia. For example, Anolis has the longest rate branch (5.03 × 10−6 LCNS−1/My) within Reptilia. Conversely, the rate of LCNS evolution within the ancestral mammal branch is low (2.83 × 10−6 LCNS−1/My) but increases in the ancestral mouse–human lineage. When constructed without constraining against negative branch lengths, the tree contains negative branches in both Reptilia and mammals, suggesting homoplasy in the extent of retention of LCNS (fig. 4B).
FIG. 4.—

Phylogenetic trends in rates of retention of LCNS. (A) Distance tree based on the pairwise inverse of shared LCNS (a measure of relative LCNS divergence among species). (B) A phylogeny constructed allowing for negative branch lengths. The negative branches suggest homoplasy in LCNS abundance between Reptilia and mammals, excluding mouse. (C) A phylogeny showing branch lengths represented by divergence time in millions of years (Benton and Donoghue 2007). (D) The LCNS−1 tree from panel A, scaled by time from panel C, suggests varying rates of LCNS divergence per million years. This tree suggests an increase in the rate of divergence in LCNS along the reptilian branch before the divergence of lizards (represented by Anolis) and archosaurs (represented by chicken and zebra finch).
Vestiges of LCNS were found across the amniote tree when search parameters were relaxed to identify shorter less similar Blast matches (ca. 100 bp, e-value = 1 × 10−10). Of the 574 LCNS shared among Reptilia, 236 are not found as LCNS in mammals, but with a relaxed search, similar sequences of insufficient length or similarity to be classified as LCNS were identified across human, mouse, and dog (all three: 544; only two: 18 [human and mouse: 1; human and dog: 8; and mouse and dog: 9]; and only one: 9 [mouse: 1 and dog: 8]). Only three LCNS shared among Reptilia did not match any sequences in the three studied mammals. Of the 1,018 mammal LCNS, 680 are not found as LCNS in reptiles or birds. However, a relaxed search found 854 hits across chicken, zebra finch, and Anolis and 123 hits in two of the three studied Reptilia (chicken and zebra finch: 59; chicken and Anolis: 46; and zebra finch and Anolis: 18). Fragments of 16 mammal LCNS were found in only one bird or reptile (chicken: 4; zebra finch: 7; and Anolis: 5), and 25 mammal LCNS did not match any bird or reptile sequence. One can see examples of such vestiges in figure 1.
Summing the unique genome coordinates of all pairwise and three-species LCNS matches enabled us to estimate the number of distinct LCNS for the hypothetical ancestors of the reptile and mammal clades. We found 4,020 and 4,272 LCNS, respectively, for these two ancestors. Summing the distinct LCNS for the ancestors of both Reptilia and mammals and the LCNS shared across all sampled amniotes suggested a total of 8,630 possible sites in the ancestral amniote genome would become future LCNS, once divergence from this ancestor occurred (fig. 5).
FIG. 5.—

Loss of LCNS throughout amniote evolution. The complete series of unique genome coordinates suggests a total of 8,630 LCNS in the amniote ancestor, indicated in large font. The other numbers on various nodes of the tree indicate the number of LCNS lost since the previous node. A simple model of loss of LCNS over time suggests different rates of loss between and within reptilian and mammal lineages. Birds have lost fewer LCNS since their divergence from the reptile ancestor (255 and 272 for chicken and zebra finch, respectively) than Anolis has (3,965). Similarly, human and dog have lost fewer LCNS from the mammalian ancestor than mouse.
Enrichment of Transcription Factor Binding Site Motifs in LCNS
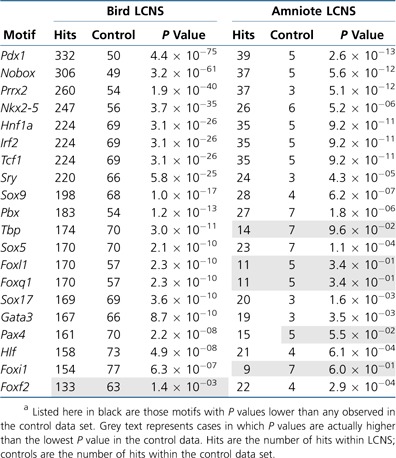
To enhance our understanding of potential function of LCNS, we tested for a statistical overrepresentation of cis-regulatory binding motifs within LCNS. We initially focused on bird (chicken and zebra finch) LCNS and then broadened our scope by adding human, mouse, and dog LCNS for an amniote-wide analysis. As a result of our initial focus on bird LCNS, Anolis was not incorporated into this subset of analyses. In the cases of bird and amniote-wide LCNS, we found strong evidence of regulatory motif enrichment within LCNS (table 2). For each motif that showed enrichment in LCNS, we also compiled lists of all of the LCNS containing that particular motif. Using Ensembl, we then identified the transcripts in the zebra finch genome that were physically closest to each LCNS and conducted a gene ontology (GO) analysis (Wu and Watson 2009). For the majority of LCNS with overrepresented motifs, we found a statistically significant signal for transcription factor activity (GO:0003700), DNA binding (GO:0003677), nucleus (GO:0005634), regulation of transcription, DNA dependent (GO:0006355), positive and negative regulation transcription from RNA polymerase II promoter (GO:0045944, GO:0000122), and sequence-specific DNA binding (GO:004356) among their flanking genes (table 2 and supplementary table S3, Supplementary Material online). This indicates that not only are LCNS enriched for transcription factor binding sites but they also tend to be adjacent to genes with regulatory activity.
Table 2.
Transcription Factor Binding Motifs Overrepresented among LCNSa
 |
Discussion
We have described patterns of sequence conservation and divergence in LCNS among amniotes, with an emphasis on the recently sequenced genomes of zebra finch and Anolis. A simple model of loss over time suggests that Reptilia and mammals have lost similar numbers of LCNS but at different rates (fig. 5). Reptiles and birds have lost LCNS from the amniote ancestor more slowly than mammals. We also find a difference in rate of loss within Reptilia, where Anolis exhibits greater loss of LCNS from the reptile ancestor than birds (fig. 5). A similar and much faster loss is evident in mouse, relative to other mammals, where mouse has lost over five times as many LCNS than other eutherian mammals. This result is perhaps not surprising given the evidence in rodents for high rates of point substitution driven by short generation time and other factors, as well as abundant positive selection in genomes of the Mus species complex (Wu and Li 1985; Halligan et al. 2010).
In addition to simple loss of LCNS depicted in figure 5, differential abundance of LCNS could also be explained, in part, by addition of new LCNS in some lineages. Novel LCNS could come into being through changes to previously nonfunctional sequences or from changes leading to altered function in functional sequences, as suggested by Meader et al. (2010). By this model, some LCNS may have been recruited at different points in amniote history, potentially identified by a decrease in the substitution rate in a lineage in a localized region of the genome over time. Such decreases could be caused by the acquisition of new functions and enrichment for transcription factor binding sites. However, a complete absence of similarity between LCNS of Reptilia versus mammals was found for only 28 of 8,630 hypothesized LCNS in the amniote ancestor. These 28 elements may have taken on a novel regulatory role in one lineage but not the other, causing changes in their function with a novel selective regime and, therefore, substitution rate. In addition to frequent loss as depicted in figure 5, the possibility of infrequent recruitment of novel LCNS over time remains.
Reptiles and birds have retained a landscape of LCNS from the amniote ancestor that is highly distinct compared with mammals. We find a long branch in the extent of shared LCNS per million years in the lineage ancestral to living birds and reptiles followed by much shorter branches in the descendant lineages. The degree of LCNS conservation is explained in large part by divergence time between species. In fact, Meader et al. (2010) also detected more functional sequence shared between mammalian species that had diverged by fewer synonymous substitutions. We support this finding and extend it to reptiles and birds but add that rates of LCNS retention have not been constant within mammals or Reptilia. For example, we have identified a large set of LCNS among birds, and we find many more conserved regions between these two taxa than we found between human and mouse, which diverged approximately at the same time. Phylogenetic analyses of rates of LCNS divergence reveal that the disparity between chicken/zebra finch and human/mouse appears to be due to rapid evolution of the mouse genome, but we also find an effect of slow evolution within the Reptilia, as evidenced by the short branches in that clade (fig. 4D). Overall, these results suggest a strong differential retention of distinct LCNS repertoire in Mammalia and Reptilia as they diverged from the common amniote ancestor.
Finally, whereas Meader et al. (2010) estimated 6.5–10% of the human genome being constrained, our much smaller estimate of the fraction of the genome comprised by LCNS (<0.0003% to <0.001%) is likely due to our choice of focusing only on regions 500 bp or greater, whereas Meader et al. (2010) focused on individual constrained sites in the genome regardless of region size. Meader et al. (2010) also incorporated coding regions into their estimate of total constrained bases, although the discrepancy is not entirely explained by the removal of coding sequences in our analysis, and rather appears to be due to the different units of conservation in the two studies.
One hypothesis for the presence of LCNS is a functional role in gene regulation. We show a strong enrichment for cis-regulatory motifs among avian LCNS and amniote LCNS, a finding that supports a role for LCNS in cis-regulation. Studies of the zebra finch genome and gene expression have indicated the involvement of a large number of noncoding RNAs in transcriptional responses to social stimuli (Dong et al. 2009; Warren et al. 2010). We find, however, that only a small fraction (2.3%) of avian LCNS show evidence of expression in the large zebra finch EST databases. This suggests that if LCNS are playing an important role in gene regulation, this role is largely independent of transcription and that LCNS may instead play a role in binding of cis-acting transcription factors. Even though zebra finch databases consist entirely of brain ESTs, we predict that the patterns we observed will be supported as additional tissues are profiled for gene expression. Although only a small fraction of LCNS show evidence of expression, among these are a small number (28) that are dynamically regulated in response to behavioral stimuli (song). These LCNS therefore warrant further characterization with respect to their role in avian social behavior.
An alternative hypothesis for the existence of LCNS is that they represent mutational cold spots. Because this hypothesis has found no support in mammals, however, it would be surprising if avian LCNS were, in fact, constrained from mutation (Drake et al. 2006; Ahituv et al. 2007; Sakuraba et al. 2008). Also, Shedlock et al. (2007) found evidence for a slowdown in the rate of turnover of oligonucleotide motifs in Reptilia compared with mammals, a result that is reflected in the higher rate of retention of LCNS in Reptilia. The LCNS rate analysis is also consistent with other studies that find accelerated rates of genome evolution in mouse. Mouse LCNS appear to have diverged due to a faster substitution rate than other studied species. Finally, we have identified 338 LCNS that have been conserved across ∼315 Myr of amniote ancestry.
Another fundamental difference among the species analyzed here is in karyotypic organization. The avian karyotype is remarkably conserved despite the diversity of this group (Burt et al. 1999; Burt 2002; Ericson et al. 2002; Hillier et al. 2004), though high rates of chromosomal evolution appear to have occurred at the base of the reptilian tree (Organ et al. 2008). The pattern of karyotype evolution appears similar to the pattern observed here for LCNS, in so far as there was a slowdown in the rate of evolution of both chromosomes and LCNS since the origin of both sets of genomic traits. LCNS may play a role in regulating specific genes. In therian mammals, X-linked gene expression can be affected in females by the inactivation of one X chromosome but dosage compensation is far less prevalent in birds (Melamed and Arnold 2007; Mank 2009; Melamed et al. 2009). The greater proportion of avian genomes composed of LCNS (fig. 2B) may thus indicate an increase in sequence-for-sequence regulation as opposed to global dosage compensation, a mechanism apparently lacking in birds. Finally, if some or all of the 38 Z-linked LCNS found in chicken also map to the chicken W chromosome, then this would distinguish female from male heterogamety because mammalian X-linked LCNS apparently lack Y-linked homologs (Sakuraba et al. 2008).
The number of LCNS unique to nonavian reptiles permits at least an indirect examination of novel roles for gene regulation in reptile genome evolution (Janes et al. 2010). At present, Anolis is the only nonavian reptile for which a genome assembly is available for comparison to avian reptiles and mammals, and additional avian and nonavian reptile genomes will permit the refinement of counts of LCNS in Reptilia. Our conclusions, particularly regarding the loss of LCNS in the Anolis genome, depend of the quality of current genome assemblies. The publicly available Anolis (AnoCar 1.0) genome assembly employed here represents 6.8X coverage with 50% of the sequence carried by scaffolds of at least 2.44 Mb in length (data available at http://genome.ucsc.edu/cgi-bin/hgGateway), indicating that the quality of this Anolis assembly is comparable to assemblies of mammals recently accessed for study of conserved noncoding sequences (Kim and Pritchard 2007). Therefore, our conclusions are unlikely to be affected by the quality of available genome data. A fraction of avian LCNS are related to vocal communication in zebra finch, and a greater proportion of avian genomes is composed of LCNS than is seen in other genomes. Future work should identify functional genomic elements by which the 338 LCNS shared among amniotes interact with transcription factors and measure the effects on gene expression of mutagenized LCNS. Studies of targeted mutagenesis followed by observation of phenotypes will help clarify the roles of LCNS as possible long-range enhancers or as regulatory regions closely linked to coding regions.
Supplementary Material
Supplemental tables S1–S3 are available at Genome Biology and Evolution online (http://www.oxfordjournals.org/our_journals/gbe/).
Acknowledgments
Conversations between Y.G., C.C., and D.E.J. regarding reptile LCNS began at the 13th Evolutionary Biology Meeting at Marseille, and we thank the meeting's organizer, Pierre Pontarotti, for inviting our participation. We thank the Broad Institute Genome Sequencing Platform and Genome Sequencing and Analysis Program, Federica Di Palma, and Kerstin Lindblad-Toh, for making the data for Anolis carolinensis available. Ricardo Godinez provided additional characterization of the Anolis assembly. We thank two anonymous reviewers, Qu Zhang, and Judith Mank, for comments on the manuscript. This work was supported by the National Science Foundation (MCB-0817687 to N. Valenzuela and S.V.E.).
References
- Abascal F, Zardoya R, Telford MJ. TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 2010;38:W7–W13. doi: 10.1093/nar/gkq291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahituv N, et al. Deletion of ultraconserved elements yields viable mice. PLoS Biol. 2007;5:1906–1911. doi: 10.1371/journal.pbio.0050234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alaux C, et al. Honey bee aggression supports a link between gene regulation and behavioral evolution. Proc Natl Acad Sci U S A. 2009;106:15400–15405. doi: 10.1073/pnas.0907043106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelsson E, et al. Natural selection in avian protein-coding genes expressed in brain. Mol Ecol. 2008;17:3008–3017. doi: 10.1111/j.1365-294X.2008.03795.x. [DOI] [PubMed] [Google Scholar]
- Bejerano G, et al. Ultraconserved elements in the human genome. Science. 2004;304:1321–1325. doi: 10.1126/science.1098119. [DOI] [PubMed] [Google Scholar]
- Benton MJ, Donoghue PCJ. Paleontological evidence to date the tree of life. Mol Biol Evol. 2007;24:26–53. doi: 10.1093/molbev/msl150. [DOI] [PubMed] [Google Scholar]
- Burt DW. Origin and evolution of avian microchromosomes. Cytogenet Genome Res. 2002;96:97–112. doi: 10.1159/000063018. [DOI] [PubMed] [Google Scholar]
- Burt DW, et al. The dynamics of chromosome evolution in birds and mammals. Nature. 1999;402:411–413. doi: 10.1038/46555. [DOI] [PubMed] [Google Scholar]
- de la Calle-Mustienes E, et al. A functional survey of the enhancer activity of conserved non-coding sequences from vertebrate Iroquois cluster gene deserts. Genome Res. 2005;15:1061–1072. doi: 10.1101/gr.4004805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dermitzakis ET, et al. Evolutionary discrimination of mammalian conserved non-genic sequences (CNGs) Science. 2003;302:1033–1035. doi: 10.1126/science.1087047. [DOI] [PubMed] [Google Scholar]
- Derti A, Roth FP, Church GM, Wu CT. Mammalian ultraconserved elements are strongly depleted among segmental duplications and copy number variants. Nat Genet. 2006;38:1216–1220. doi: 10.1038/ng1888. [DOI] [PubMed] [Google Scholar]
- Dong S, et al. Discrete molecular states in the brain accompany changing responses to a vocal signal. Proc Natl Acad Sci U S A. 2009;106:11364–11369. doi: 10.1073/pnas.0812998106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake JA, et al. Conserved noncoding sequences are selectively constrained and not mutation cold spots. Nat Genet. 2006;38:223–227. doi: 10.1038/ng1710. [DOI] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ericson PG, et al. A Gondwanan origin of passerine birds supported by DNA sequences of the endemic New Zealand wrens. Proc R Soc Lond B Biol Sci. 2002;269:235–241. doi: 10.1098/rspb.2001.1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. Distance methods for inferring phylogenies: a justification. Evolution. 1984;38:16–24. doi: 10.1111/j.1558-5646.1984.tb00255.x. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. PHYLIP (Phylogeny Inference Package) version 3.69. 2010. Distributed by the author, Seattle (WA): Department of Genetics, University of Washington. [Google Scholar]
- Frazer KA, et al. Noncoding sequences conserved in a limited number of mammals in the SIM2 interval are frequently functional. Genome Res. 2004;14:367–372. doi: 10.1101/gr.1961204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner EJ, Hirons L, Hunter CA, Willett P. Genomic data analysis using DNA structure: an analysis of conserved nongenic sequences and ultraconserved elements. J Chem Inf Model. 2006;46:753–761. doi: 10.1021/ci050384i. [DOI] [PubMed] [Google Scholar]
- Gregory TR. Animal Genome Size Database. Guelph (ON): University of Guelph. 2005. [Google Scholar]
- Haider S, et al. BioMart Central Portal-unified access to biological data. Nucleic Acids Res. 2009;37:W23–W27. doi: 10.1093/nar/gkp265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halligan DL, et al. Evidence for pervasive adaptive protein evolution in wild mice. PLoS Genet. 2010;6:e1000825. doi: 10.1371/journal.pgen.1000825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillier LW, et al. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature. 2004;432:695–716. doi: 10.1038/nature03154. [DOI] [PubMed] [Google Scholar]
- Janes DE, et al. Genome evolution in Reptilia, the sister group of mammals. Annu Rev Genomics Hum Genet. 2010;11:239–264. doi: 10.1146/annurev-genom-082509-141646. [DOI] [PubMed] [Google Scholar]
- Johnston JS, et al. Body lice and head lice (Anoplura: Pediculidae) have the smallest genomes of any hemimetabolous insect reported to date. J Med Entomol. 2007;44:1009–1012. doi: 10.1603/0022-2585(2007)44[1009:blahla]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Katzman S, et al. Human genome ultraconserved elements are ultraselected. Science. 2007;317:915–915. doi: 10.1126/science.1142430. [DOI] [PubMed] [Google Scholar]
- Kim J, et al. Functional characterization of transcription factor motifs using cross-species comparison across large evolutionary distances. PLoS Comput Biol. 2010;6:1–15. doi: 10.1371/journal.pcbi.1000652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SY, Pritchard JK. Adaptive evolution of conserved noncoding elements in mammals. PLoS Genet. 2007;3:e147. doi: 10.1371/journal.pgen.0030147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London SE, Dong S, Replogle K, Clayton DF. Developmental shifts in gene expression in the auditory forebrain during the sensitive period for song learning. Dev Neurobiol. 2009;69:437–450. doi: 10.1002/dneu.20719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mank JE. The W, X, Y and Z of sex-chromosome dosage compensation. Trends Genet. 2009;25:226–233. doi: 10.1016/j.tig.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies EH, et al. Identification and characterization of multi-species conserved sequences. Genome Res. 2003;13:2507–2518. doi: 10.1101/gr.1602203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin AP, Palumbi SR. Body size, metabolic rate, generation time and the molecular clock. Proc Natl Acad Sci U S A. 1993;90:4087–4091. doi: 10.1073/pnas.90.9.4087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meader S, Ponting CP, Lunter G. Massive turnover of functional sequence in human and other mammalian genomes. Genome Res. 2010;20:1335–1343. doi: 10.1101/gr.108795.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melamed E, Arnold AP. Regional differences in dosage compensation on the chicken Z chromosome. Genome Biol. 2007;8:1–10. doi: 10.1186/gb-2007-8-9-r202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melamed E, Elashoff D, Arnold AP. Evaluating dosage compensation on the chicken Z chromosome: should effective dosage compensation eliminate sexual bias? Heredity. 2009;103:357–359. doi: 10.1038/hdy.2009.91. [DOI] [PubMed] [Google Scholar]
- Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986;3:418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- Nowak R. Mining treasures from junk DNA. Science. 1994;263:608–610. doi: 10.1126/science.7508142. [DOI] [PubMed] [Google Scholar]
- Olmo E. Genome size in some reptiles. J Exp Zool. 1976;195:305–310. [Google Scholar]
- Organ CL, Moreno RG, Edwards SV. Three tiers of genome evolution in reptiles. Integr Comp Biol. 2008;48:494–504. doi: 10.1093/icb/icn046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson DG, et al. The relationship between synaptonemal complex length and genome size in four vertebrate classes (Osteichthyes, Reptilia, Aves, Mammalia) Chromosome Res. 1994;2:153–162. doi: 10.1007/BF01553494. [DOI] [PubMed] [Google Scholar]
- Pigozzi MI. Relationship between physical and genetic distances along the zebra finch Z chromosome. Chromosome Res. 2008;16:839–849. doi: 10.1007/s10577-008-1243-5. [DOI] [PubMed] [Google Scholar]
- Replogle K, et al. The Songbird Neurogenomics (SoNG) Initiative: community-based tools and strategies for study of brain gene function and evolution. BMC Genomics. 2008;9:1–20. doi: 10.1186/1471-2164-9-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuraba Y, et al. Identification and characterization of new long conserved noncoding sequences in vertebrates. Mamm Genome. 2008;19:703–712. doi: 10.1007/s00335-008-9152-7. [DOI] [PubMed] [Google Scholar]
- Sandelin A, et al. JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 2004a;32:D91–D94. doi: 10.1093/nar/gkh012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandelin A, et al. Arrays of ultraconserved non-coding regions span the loci of key developmental genes in vertebrate genomes. BMC Genomics. 2004b;5 doi: 10.1186/1471-2164-5-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shedlock AM, et al. Phylogenomics of nonavian reptiles and the structure of the ancestral amniote genome. Proc Natl Acad Sci U S A. 2007;104:2767–2772. doi: 10.1073/pnas.0606204104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S, et al. Cross-species comparison significantly improves genome-wide prediction of cis-regulatory modules in Drosophila. BMC Bioinformatics. 2004;5:1–12. doi: 10.1186/1471-2105-5-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S, et al. Genome scan for cis-regulatory DNA motifs associated with social behavior in honey bees. Proc Natl Acad Sci U S A. 2006;103:16352–16357. doi: 10.1073/pnas.0607448103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S, et al. Systematic functional characterization of cis-regulatory motifs in human core promoters. Genome Res. 2008;18:477–488. doi: 10.1101/gr.6828808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit AFA, Hubley R, Green P. Repeatmasker Open-3.0. 2004. Seattle (WA): Institute for Systems Biology. [Google Scholar]
- Swofford D. PAUP* (Phylogenetic Analysis Using Parsimony *and other methods). Pp. Version 4. 2003. Sunderland (MA): Sinauer Associates, Inc. [Google Scholar]
- Tiersch TR, Chandler RW, Wachtel SS, Elias S. Reference-standards for flow-cytometry and application in comparative studies of nuclear-DNA content. Cytometry. 1989;10:706–710. doi: 10.1002/cyto.990100606. [DOI] [PubMed] [Google Scholar]
- Tomaszycki ML, et al. Sexual differentiation of the zebra finch song system: potential roles for sex chromosome genes. BMC Neurosci. 2009;10:1–14. doi: 10.1186/1471-2202-10-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh B, et al. Ancient noncoding elements conserved in the human genome. Science. 2006;314:1892–1892. doi: 10.1126/science.1130708. [DOI] [PubMed] [Google Scholar]
- Vinogradov AE. Genome size and GC-percent in vertebrates as determined by flow cytometry: the triangular relationship. Cytometry. 1998;31:100–109. doi: 10.1002/(sici)1097-0320(19980201)31:2<100::aid-cyto5>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Visel A, et al. Ultraconservation identifies a small subset of extremely constrained developmental enhancers. Nat Genet. 2008;40:158–160. doi: 10.1038/ng.2007.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren WC, et al. The genome of a songbird. Nature. 2010;464:757–762. doi: 10.1038/nature08819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CI, Li WH. Evidence for higher rates of nucleotide substitution in rodents than in man. Proc Natl Acad Sci U S A. 1985;82:1741–1745. doi: 10.1073/pnas.82.6.1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TD, Watanabe CK. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics. 2005;21:25–29. doi: 10.1093/bioinformatics/bti310. [DOI] [PubMed] [Google Scholar]
- Wu X, Watson M. CORNA: testing gene lists for regulation by microRNAs. Bioinformatics. 2009;25:832–833. doi: 10.1093/bioinformatics/btp059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Schwartz S, Wagner L, Miller W. A greedy algorithm for aligning DNA sequences. J Comput Biol. 2000;7:203–214. doi: 10.1089/10665270050081478. [DOI] [PubMed] [Google Scholar]