

Abstract
Diarrhea caused by enteric infections is a major factor in morbidity and mortality worldwide. An estimated 2–4 billion episodes of infectious diarrhea occur each year and are especially prevalent in infants. This review highlights the cellular and molecular mechanisms underlying diarrhea associated with the three classes of infectious agents, i.e., bacteria, viruses and parasites. Several bacterial pathogens have been chosen as model organisms, including Vibrio cholerae as a classical example of secretory diarrhea, Clostridium difficile and Shigella species as agents of inflammatory diarrhea and selected strains of pathogenic Escherichia coli (E. coli) to discuss the recent advances in alteration of epithelial ion absorption. Many of the recent studies addressing epithelial ion transport and barrier function have been carried out using viruses and parasites. Here, we focus on the rapidly developing field of viral diarrhea including rotavirus, norovirus and astrovirus infections. Finally we discuss Giardia lamblia and Entamoeba histolytica as examples of parasitic diarrhea. Parasites have a greater complexity than the other pathogens and are capable of creating molecules similar to those produced by the host, such as serotonin and PGE2. The underlying mechanisms of infectious diarrhea discussed include alterations in ion transport and tight junctions as well as the virulence factors, which alter these processes either through direct effects or indirectly through inflammation and neurotransmitters.
Key words: infectious diarrhea, ion absorption, enteric pathogens, mechanisms of diarrhea, EPEC
Introduction
At its core, diarrhea is simply an altered movement of ions and water that follows an osmotic gradient. Under normal conditions, the gastro-intestinal tract has tremendous capacity to absorb fluid and electrolytes, where 8–9 liters of fluid are presented to the intestine daily and only 100–200 ml are excreted in the stool. Enteric pathogens, however, can alter this balance towards net secretion, leading to diarrheal disease. The altered movement of ions can occur either through transporters or the lateral spaces between cells, which are regulated by tight junctions (Fig. 1). In this regard, some transporters seem to be tightly coupled with water movement, including sodium-dependent glucose transporter (SGLT1), Na+/H+ exchanger isoform 3 (NHE3) and the apicalCl−/HCO3− exchanger, downregulated in adenoma (DRA). The classical secretory diarrhea caused by cholera toxin (CT) is due to cAMP-dependent activation of the cystic fibrosis transmembrane conductance regulator (CFTR), a Cl-channel (Fig. 1). Alternately, changes in Ca2+ levels increase the activity of the calcium activated chloride channel (CLCA). In some cases, as for CT, the increase in Cl-secretion is paired with a decrease in Na+ absorption. In addition the direct reduction of water transport proteins, such as aquaporins, results in less fluid absorption (Fig. 1).
Figure 1.

An overview of general mechanisms causing diarrhea. At the most basic level, diarrhea is caused by increased secretion or decreased absorption of fluids and electrolytes. Certain ion transport processes are particularly associated with diarrhea. These include CFTR and CLCA, which are chloride channels and the Na+/H+ exchange isoform, NHE3, which is involved in Na+ absorption. Alterations in tight junctions create an important pathway for the movement of both ions and water. DRA is responsible for chloride absorption and is associated with congenital chloride diarrhea. Data on aquaporins is limited but they are expected to contribute to diarrhea when absorption is reduced. SGLT-1 transports sodium and glucose and is tightly coupled with water movement and is the basis for oral rehydration using glucose to enhance sodium absorption.
Enteric pathogens can either directly modulate epithelial ion transport processes and barrier function or do so indirectly through inflammation, neuropeptides or loss of absorptive surface. For example, pathogens such as the intestinal parasite Giardia cause loss of brush border absorptive surface and diffuse shortening of villi. Similarly, enteropathogenic E. coli (EPEC) cause effacement of microvilli, which decreases the surface area for nutrient absorption and causes increased osmolarity of the intestinal contents and malabsorption. However, recent evidence suggests that the rapid onset of diarrhea induced by EPEC could result from direct effects on intestinal epithelial ion transport processes. Several invasive pathogens, including Shigella and Salmonella species, cause an inflammatory diarrhea characterized by fever and polymorphonucleocytes (PMNs) in the stool. PMNs regulate absorption through cytokine secretion but also have a more direct role through the secretion of a precursor to adenosine, a secretagogue that activates CFTR. C. difficle and rotavius infection also work indirectly through modulation of ion transport subsequent to cytokine secretion and activation of enteric nerves via neuropeptides.
This review highlights selected pathogens, which provide both a broad overview of the mechanisms pertaining to ion transport and barrier function that underlie pathophysiology of diarrhea, and presents recent advances regarding specific infectious agents. Major emphasis will be placed on the mechanisms underlying the pathogenesis of bacterial diarrhea, which has been extensively studied in recent years and has served as an important prototype for understanding regulation of intestinal epithelial processes at the cellular and molecular level. However, the mechanisms underlying recent advances in viral and parasitic diarrhea will also be discussed. An increased understanding of these regulatory mechanisms is important to completely define the pathophysiology of infectious diarrhea and explore the potential of novel anti-diarrheal drugs.
Pathophysiological Mechanisms of Infectious Diarrhea
Bacterial diarrhea.
In developing countries, enteric bacterial pathogens and parasites are the leading cause of infectious diarrhea. Although even in the United States, the frequency of bacteria-induced illnesses is considerably high. Enterohemmorhagic E. coli (EHEC) infection causes disease in approximately 75,000 people per year,1 whereas C. difficile remains the major cause of hospital acquired infections.2 Bacterial diarrhea ranges in duration from a few hours for some released toxins to several weeks for active infections of enteroaggregative E. coli. Here we use V. cholerae as an example of secretory diarrhea and then discuss C. difficile-associated diarrhea, which relies on neuropeptides and inflammatory mediators for pathogenesis. In addition, Shigella species are used as an example of inflammatory and invasive diarrhea while E. coli have a variety of strategies, one of which is the reduction in absorption both through a loss of microvilli and through a direct effect on ion transporters. These four groups of pathogens have been selected to explain the major mechanisms involved in diarrhea.
Vibrio cholerae.
Despite being the focus of scientific study since the 1800's, V. cholerae remains a threat today. While people with access to treated water are typically not exposed, areas without adequate chlorination or filtration still suffer from epidemic cholera. V. cholerae have several toxins, which are used to compromise the host, with the most important of these being cholera toxin (CT) itself. CT consists of an A subunit bound to a pentameric ring of B subunits, where the B subunits are responsible for delivery of the A subunit into the cell.3 The A subunit ADP-ribosylates a GTPase, which regulates adenylate cyclase resulting in elevated cAMP production4 (Fig. 2). The production of cAMP activates PKA, which then phosphorylates the regulatory domain of CFTR.5 While CT is the most dangerous part of V. cholorae's arsenal, most of the modern work with CT actually provides insights into understanding key cellular mechanisms. For example, by studying CTs retrograde translocation and the eventual release of the A1 peptide into the host cytoplasm, the details of endogenous retrograde trafficking have been uncovered.6 In addition, movement of CT through the Endoplasmic Reticulum-Associated protein Degradation (ERAD) pathway has shed light on the way cells deal with misfolded proteins in the ER.7 Aside from the knowledge acquired in these studies, the CT B-subunit has shown great promise as an adjuvant in a number of recent vaccine studies. Although there have been additional insights into the transporters affected by CT. In addition to increased Clsecretion, the absorption of Na+ is decreased through a cAMP-dependent mechanism where the activity of both apical sodium transporters, NHE2 and NHE3, is decreased8 (Fig. 2). Together, this leads to an increase in NaCl levels in the intestinal lumen by enhancing secretion or decreasing absorption.
Figure 2.
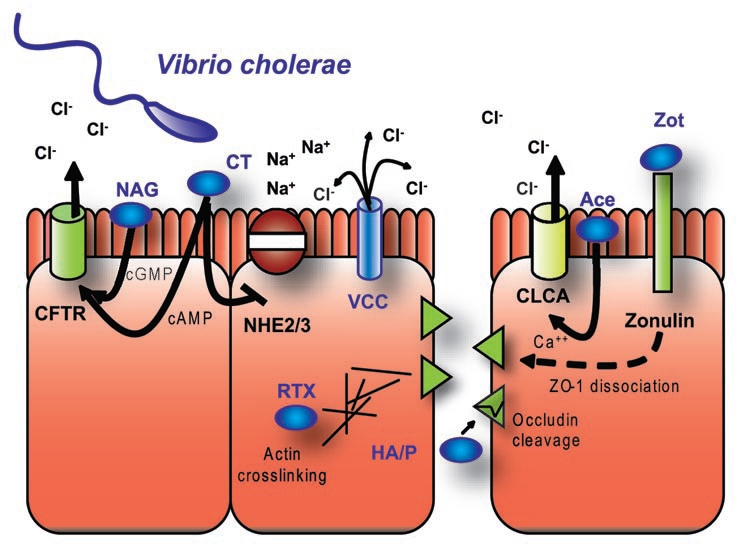
Mechanisms underlying V. cholerae-induced diarrhea. Cholera is characterized by severe watery diarrhea due to changes in ion secretion and absorption. Both CLCA and CFTR-dependent Cl− secretion are activated, the first by Ace and the second by cholera toxin and NAG heat-stabile toxin. Increased cAMP levels also block sodium absorption through NHE2 and NHE3. V. cholerae also creates anion permeable pores through insertion of VCC. In concert with changes in ion transport, paracellular permeability is also decreased. Zot interaction with zonulin causes ZO-1 to dissociate from tight junctions while HA/P cleaves occludin and rTX interferes with the contractile actin ring.
In addition to CT, V. cholerae encodes several other toxins, which modulate ion secretion and perturb barrier function to cause massive diarrhea. The toxins that affect ion secretion directly include accessory cholera toxin (ACE), which stimulates Ca2+ dependent Cl− secretion; NAG-stable toxin, which activates guanylyl cyclase, thus stimulating cGMP production, which leads to PKG-mediated activation of CFTR; and, finally V. cholerae cytolysin (VCC), which creates anion permeable pores9–11 (Fig. 2). One of the phenotypes associated with VCC toxicity has been linked with the newly described phenomenon autophagy.12 The VCC toxin causes large vacuoles to form in host cells in addition to its cytolytic effect on red blood cells. While the mechanism behind this vacuole formation is only beginning to be understood, recent studies have shown that VCC is associated with double membrane vesicles and LC3-II accumulation which are characteristic of autophagy. In addition, mouse embryonic fibroblasts deficient for Atg-5 (which is needed for LC3 conversion) or inhibitors of autophagy including 3-methyladenine block the formation of vacuoles in response to VCC. While vacuole formation appears to be a severe phenotype, it is actually a protective cellular response and its inhibition results in a more than 50% decrease in cell survival after toxin exposure. In this case, vacuole formation appears to be a part of the natural removal of VCC from the cellular membrane, blocking further dysregulated Cl- transport and protecting the cell.
The V. cholerae toxins which alter intestinal barrier function include hemagglutinin/protease or HA/P, RTX and Zot. HA/P is an extracellular protease, which cleaves a tight junction structural protein, occludin, that is known to regulate paracellular permeability (Fig. 2).13 This results in the subsequent loss of the scaffolding protein ZO-1 from the tight junction. RTX cross links actin and induces cell rounding, loss of transepithelial resistance (TER) and an increase in permeability to FITC-dextran 3000.14 Analysis of the crystal structure of RTX cross-linked actin and mass spectrometry showed that residues E270 and K50 form a covalent iso-peptide bond.15 Interestingly, the fungal toxin phalloidin was able to partially restore the ability of actin to polymerize in the presence of the actin cross-linking domain of RTX. Actin and myosin light chain play a critical role in the regulation of tight junctions by forming a belt-like structure around the cell, which can be tightened to increase paracellular flow. In this case, the functionality of the junctional actinomyosin ring is lost due to the sequestration of actin in incorrectly polymerized aggregates. The third toxin, Zot, which also impacts barrier function, serves as both a phage assembly protein and an enterotoxin.16 This is similar to what is seen with Ace, which is also a phage structural protein.17 Many of the virulence factors associated with V. cholerae are part of an integrated prophage called ., which is composed, in part, of Ace and Zot.18 Zot is not functional in its phage associated form but is instead cleaved into an active 12 kDa peptide.16 The Zot peptide binds to an apical receptor for a host protein called zonulin, which regulates permeability exclusively in the small intestine.19 Loss of barrier function occurs only in rabbit ileum but not in colon, consistent with the localization of the zonulin receptor. Recently, a smaller fragment of Zot consisting of only 6 amino acids was shown to be capable of causing the same change in resistance as well as causing the dissociation of ZO-1 from tight junctions.20 However, it should be noted that this work is considered controversial by some. Thus, V. cholerae has three different toxins that promote secretion and three toxins that promote the loss of barrier function and, in combination, this results in a particularly severe diarrhea (Fig. 2).
C. difficile.
C. difficile is the leading cause of nosocomial diarrhea and accounts for almost all cases of pseudomembranous colitis, which is an acute colitis characterized by formation of an adherent inflammatory membrane overlying the site of injury21 (Fig. 3). The recent emergence of novel epidemic strains has caused increased concern in clinical settings because antimicrobial therapy predisposes patients to C. difficile associated diarrhea (CDAD).22 The pathogenic process of C. difficile infection starts with initial colonization followed by the production of two distinct exotoxins, Toxin A and B (TcdA and TcdB), as well as an additional toxin called binary toxin (CDT) which is found in some hypervirulent strains of C. difficile.23 TcdA binds effectively to the apical side of the host cell to glycoprotein gp96, which forms part of the receptor in humans.24 In contrast, TcdB gains access to the basolateral side of the cell after tight junction disruption and binds preferentially to an unidentified receptor25,26 (Fig. 3). TcdA and TcdB are potent cytotoxic enzymes that specifically glucosylate the small GTPase protein Rho, which leads to disruption of cytoskeletal integrity and cytotoxic effects.27 CDT, an actin-specific ADP ribosyltransferase, potentiates the toxicity of TcdA and B and may increase the severity of CDAD.28 While early studies in animals implicate TcdA as the primary factor in the pathogenesis of C. difficile infection, recent studies showed that disruption of the tcdB gene led to a significantly attenuated virulence phenotype in the hamster model suggesting that TcdB might play a key role in disease pathogenesis.29
Figure 3.

Pathogenesis of C. diffcile-associated diarrhea. C. difficile produces toxin A and toxin B (TcdA and TcdB). TcdA binds to the apical side of the cell and, after internalization, causes cytoskeletal modification and disruption of tight junctions. The resulting loss of epithelial barrier function facilitates TcdA and TcdB to cross the epithelium with preferential binding of TcdB to the basolateral cell membrane. Both toxins are cytotoxic and lead to production of proinflammatory cytokines, increase in vascular permeability, recruitment of neutrophils and monocytes, epithelial cell apoptosis and connective tissue degradation, resulting in pseudomembrane formation and diarrhea. Further, the activation and release of various neuropeptides by the toxins stimulates ENS to elicit fluid secretion, causing diarrhea.
Earlier studies showed that TcdA causes direct alterations in barrier function and ion transport. For example, purified toxin A has also been shown to cause net luminal accumulation of sodium, chloride and potassium in rabbit intestine in vivo.30 Ussing chamber studies using isolated mucosal strips from guinea pig ileum demonstrated that TcdA increases transepithelial permeability and decreases electrogenic Na+ absorption while eliciting a Cl-secretory response.31 Rounding up of cells and barrier function disruption corresponding with structural alterations of perijunctional actinomysin ring occurred in human intestinal epithelial T84 cells exposed to TcdA.30,32
Besides the direct effects of the toxins, other mechanisms underlying C. difficile associated diarrhea include inflammation and activation of neuropeptides. The C. difficile toxins initiate an extensive inflammatory cascade that causes increased damage to host tissues resulting in fluid exudation. TcdA causes release of several proinflammatory cytokines such as leukotriene, PGE2, and tumor necrosis factor (TNFα in vivo).33 It also directly activates monocytes to release IL-1 and IL-6,34 and increase neutrophil migration in vitro.35 Other toxin-mediated inflammatory effects include release of reactive oxygen species, activation of mitogen-activated protein kinases and NFκB activation.33 A number of studies suggest that important cellular responses to C. difficile toxins such as p38 MAP kinase activation, mitochondrial damage and IL-8 release occur prior to and independently of Rho glucosylation.33
Another striking feature of C. difficle toxin-associated intestinal responses is activation of enteric nerves and enhanced production or release of neuropeptides including Substance P (SP), calcitonin gene-related peptide (CGRP) and neurotensin,36–38 which are known to elicit Cl-secretion in intestinal epithelial cells. The antagonists of the Substance P receptor, Neurokinin-1 (NK1) and calcitonin gene-related peptide (CGRP), block fluid accumulation and mannitol flux in response to toxin A.33 Also in a NK1-R-/-mouse ileal loop model, TcdA-mediated luminal fluid accumulation was considerably reduced.39 Therefore, unlike cholera toxin and E. coli enterotoxins, which trigger intestinal secretion without intestinal inflammation, the pathophysiology of C. difficile-associated diarrhea involves a necroinflammatory reaction, which activates mast cells, nerves, vascular endothelium, and immune cells in addition to impairment of tight junctions (Fig. 3). Further studies pertaining to detailed analysis of the contribution of the electrogenic versus electroneutral components of ion absorption in the well-established animal models of C. difficile infection would benefit the modeling of disease pathogenesis and help elucidate the mechanisms of C. difficile associated diarrhea.
Shigella species.
There are four major Shigella species that cause diarrheal disease. The most common species in the U.S. and other developed countries is S. sonnei followed by S. flexneri.40 Two other Shigella species, S. dysenteriae and S. boydii, are found very rarely in developed countries and have a generally low infection rate over all, however, S. dysenteriae causes the most life-threatening of all of these infections due to the production of Shiga toxin, which can lead to hemolytic uremic syndrome (HUS). While all four species of Shigella are invasive due to a large virulence plasmid, there is some variation in the plasmid and S. flexneri is the best studied in this regard. The invasion process is complicated and occurs through a trigger mechanism on the basolateral side of epithelial cells after the bacteria have already passed through M-cells and potentially, macrophages.41 In addition, Shigella have actin tails and are capable of cell to cell spread, increasing their ability to colonize the epithelium.42 The type III secreted effector proteins involved in this process are numerous and have been recently and thoroughly reviewed by Schroeder and Hilbi.43
Invasion and inflammatory response. Shigella causes an inflammatory diarrhea and the cellular response to various steps of the invasion process are the primary cause of inflammation (Fig. 4). The destruction of macrophages after emergence from M-cells causes an initial release of IL-1β, which attracts PMNs.44,45 PMNs release a precursor to the secretagogue adenosine, which activates Cl−secretion. This early step in inflammation is exacerbated by the presence of free bacteria on the basolateral side of cells, which allows access to toll-like receptors. Shigella LPS is capable of activating TLR4, although recent studies suggest that it is about 50% less active than LPS from E. coli in activating NFκB due to a reduced level of acetylation.46 Therefore, to some extent, Shigella actively evade TLR4 recognition, while TLR2 is activated by Shigella through lipoproteins.47 In addition, the interaction of Nod1 with shed peptidoglycan from intracellular bacteria also leads to NFκB activation and IL-8 production.48 IL-8 is another pro-inflammatory cytokine, which attracts PMNs. In this case, PMNs cause many of the symptoms of the disease but also lead to its eventual clearance.
Figure 4.

Invasion and inflammation caused by Shigella. Shigella species cross the epithelial barrier through M-cells where they encounter and eliminate macrophages. Binding of lipoprotein to TLR2 results in the production of the chemoattractant IL-1β. After translocation through M-cells LPS can bind to basolateral TLR4 which causes the production of IL-6 and IL-8. This effect is somewhat diminished due to the acetylation of LPS in Shigella. IL-8 is a potent chemoattractant for PMNs and is also produced due to activation of intracellular Nod1 by peptidoglycan. PMNs are the primary destructive force in Shigella infection. PMNs cause Cl− secretion through generation of a precursor to the secretagogue adenosine and can also cause ulceration of the epithelium, which results in a decrease in the absorptive surface but also maximizes permeability and allows easy access of gut flora to the basolateral surface of cells further driving inflammation.
Shigella are very closely related to E. coli and yet the host response is considerably different due to their ability to cross the epithelium and access basolateral toll-like receptors in addition to their ability to invade cells, where there are Nod proteins. Because of this, Shigella species have evolved a number of proteins, which counteract the host immune response. One method is preventative with mechanisms such as the alteration in LPS acetylation, which reduces TLR4 activation.46 In addition, Shigella along with several other types of bacteria have been shown to have a recycling mechanism for their peptidoglycan, which is designed to prevent its release and interaction with Nod1. There are two permeases, AmpG and MppA, which transport free peptidoglycan from the periplasm to the bacterial cytoplasm.48 Mice infected with an MppA mutant were able to clear an otherwise lethal dose of S. flexneri.48 Masking the molecular patterns is not an entirely effective strategy, therefore, Shigella have additional strategies that block signal transduction after pathogen recognition.
Type III secreted effector proteins modulate inflammation. Shigella have a type III secretion system, which injects a number of effector proteins required for initial invasion, escape from the vacuole and movement within the cell. Because shigellosis is primarily due to inflammation, bacterial proteins that modulate the host immune response can differentially modulate the host diarrheal response as well. There are three effector proteins that meet this criteria: OspF, OspG and IpaH (Fig. 5). The first, OspF, has a unique way of inhibiting MAP kinase signaling. It is an entirely new type of enzyme which does not exist in mammalian cells called a phospho-threonine lyase.49 OspF is capable of dephosphorylating threonine residues in a way that not only removes the phosphate group but also the oxygen atom, which is normally part of the amino acid, as well as a hydrogen atom from the adjacent carbon. In doing so it creates a carbon-carbon double bond within the backbone of the amino acid. In this form there is no hydroxyl group capable of being phosphorylated, thus the process is essentially irreversible. Several MAP kinases are affected by OspF, including Erk1/2, p38 and JNK.49 These kinases activate transcription factors which control the production of proinflammatory cytokines including IL-1, IL-6 and TNFα. MAP kinases are also targeted by IpaH which is a ubiquitin ligase.50 This was discovered using a yeast model where the yeast target was Ste7, a MAPKK. Ste7 is actively targeted for proteasomal degradation after being ubiquitinated by IpaH, thus impairing further MAP kinase activation and transcription of proinflammatory cytokines. In contrast, OspG actively interferes with the ubiquitination of phospho-IκBα, preventing its degradation.51 IκBα normally binds to NFκB and prevents its translocation to the nucleus. Under normal conditions IκBα phosphorylation leads to its ubiquitination and subsequent degradation allowing NFκB translocation and transcriptional activation. OspG is autophosphorylated, leading to the association with ubiquitin conjugating enzymes such as UbcH5 and UbcH7.51 While it is clear that OspG effectively blocks phospho-IκBα ubiquitination and degradation, it is unclear whether IκBα is a specific target. The ability of OspG to interact with multiple ubiquitin-conjugating enzymes suggests that there may be additional targets, but the presence of IpaH suggests that at least some ubiquitin-mediated degradation is left intact. OspF, IpaH and OspG interfere with both the MAP kinase cascade and NFκB translocation, blocking many of the critical pathways required for transcriptional activation of host cytokines.
Figure 5.
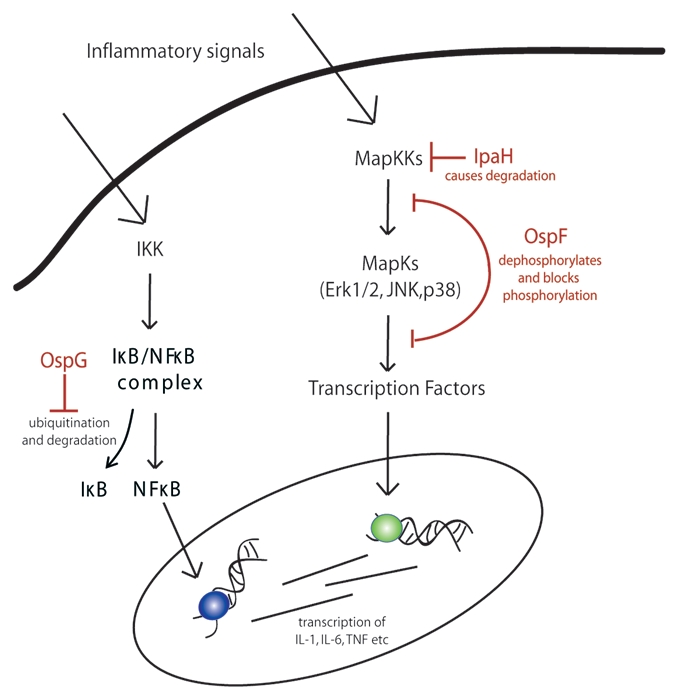
Modulation of pro-inflammatory signaling by Shigella T3SS effectors. Several type III secreted effector proteins have been shown to modulate the host immune response during Shigella infection. The first is OspG which interferes with the ubiquitination of phosphorylated IκB, preventing its degradation, thereby effectively blocking NFκB translocation into the nucleus. In contrast, IpaH actually induces ubiquitination and degradation of MapKKs, preventing further cascade activation. Finally, OspF has a unique way of cleaving the phosphate group and a few extra atoms from MapKs including Erk1/2, JNK and p38 which prevents further phosphorylation.
PMN activation can also lead to a more generalized loss of absorptive function through the destruction of the epithelial layer. In the case of Shigella, this actually enhances bacterial invasion by allowing greater access to the basolateral surface of cells.52 However, it has been recently shown that Shigella actively opposes this loss of the epithelium through the interaction of OspE with the host protein integrin-linked kinase (ILK).53 This interaction slows down turn over of focal adhesions, although the mechanism is not due to increased kinase activity but, instead, increased membrane association of ILK. While this mechanism prevents the initial loss of cells, it also prevents migration in wound healing assays so tissue which is already damaged will not heal. Studies of rectally infected guinea pigs suggest that OspE helps promote greater colonization, more severe diarrhea, inflammation and hemorrhaging. Maintaining epithelial integrity in this manner is somewhat unusual considering the disruption of cellular architecture caused by other pathogens; however, because Shigella colonize the epithelium intracellularly loss of cells which are already colonized could be detrimental to the bacteria.
Alterations in ion transport and barrier function. While diarrhea caused by Shigella is primarily due to host inflammatory processes and many Shigella effector proteins actively affect inflammation, there are additional factors that directly alter ion transport and tight junctions. Viable S. flexneri, in contrast to bacterial supernatant, heat killed bacteria or purified LPS is capable of decreasing transepithelial resistance.54 A number of tight junction proteins including claudin-1, ZO-1, ZO-2 and occludin are altered by the presence of Shigella; however a specific effector protein responsible for these changes has yet to be identified. Shigella also possess three enterotoxins known as SigA, SepA and Pic, which are serine protease autotransporters or SPATES. As the name suggests, the primary function of SPATES is proteolytic. The autotransporter portion of the name refers to the exit mechanism from the bacterial outer membrane through a β-barrel pore formed by the C-terminus of the protein itself. While Pic has been associated with the cleavage of mucin and is believed to play a role in colonization, only SigA plays a clear role in fluid accumulation.55 These proteases are also associated with various cytotoxic effects and induce cell rounding. The actual mechanism involved in SigA-mediated fluid increases in rabbit illeal loops has not been determined. A homologous protein in enteroaggregative E. coli called Pet has been shown to degrade spectrin, a cytoskeletal component, and cause changes in short circuit current in addition to cell lifting. So, while Shigella does produce toxins which alter fluid movement, and proteins which alter tight junction regulation, they are considered less important than inflammation in causing diarrhea.
Escherichia coli.
E. coli is the most abundant facultative anaerobe of the human colonic flora and typically colonizes the gastrointestinal tract within few hours after birth.56 Usually, commensal E. coli interact with the host in a mutually beneficial way; however, some strains of E. coli acquire virulence attributes that can cause a broad spectrum of disease. Several different classes of pathogenic E. coli strains responsible for diarrheal outbreaks have been recognized. These include enteropathogenic E. coli (EPEC), enterohemorrhagic E. coli (EHEC), enterotoxigenic E. coli (ETEC), enetroaggregative E. coli (EAEC), enteroinvasive E. coli and diffusely adherent E. coli (DAEC). This article focuses on diarrheagenic E. coli strains afflicting humans, in particular EPEC, which has been studied extensively in recent years, to gain understanding of the mechanisms underlying the pathophysiology of early diarrhea. A brief introduction regarding ETEC infection is provided primarily for comparison with these more modern studies.
Enterotoxigenic E. coli.
ETEC causes toxigenic secretory diarrhea characterized by massive intestinal fluid secretion. The key virulence attributes of ETEC include adherence to epithelial cell surfaces by colonization factors and elaboration of heat labile (LT) and heat stable (STa) enterotoxins. Some strains of ETEC may also express Entero-aggregative heat stable toxin 1 (EAST1). Similar to cholera toxin, heat labile toxins elicit increases in Cl−secretion via activation of cAMP. STa, however, is known to evoke secretion and diarrhea by elevation of intracellular cGMP. After binding to its receptor, guanylyl cyclase C (GC-C), STa through cGMP dependent pathways is known to stimulate CFTR translocation to the surface of villus enterocytes causing its activation.57 There is net Cl−, HCO3−, and water secretion as well as inhibition of Na+/H+ exchange in jejunal enterocytes by STa.58 Other studies suggested that STa is only anti-absorptive and does not stimulate Cl− secretion.59 Although GC-C has been shown to be the primary receptor involved in STa mediated secretory response, recent studies have shown that a non-GC-C receptor exists in the mouse proximal intestine and functionally stimulates STa-induced duodenal bicarbonate secretion through a mechanism independent of CFTR.60 The CFTR-independent mechanism is further supported by studies demonstrating stimulation in HCO3− secretion in the duodenum of CF patients61 and CFTR knock-out mice.62 Whether this mechanism is relevant in normal individuals or occurs as an adaptive response due to the loss of CFTR needs to be investigated.60 Further elucidation of alternative bicarbonate secretory mechanisms modulated by STa utilizing DRA and PAT-1 knock-out mice would also be of interest.
LT has two serogroups, LTI and LTII, that do not cross react immunologically. LTI is expressed by strains of E. coli that are pathogenic for both animals and humans, whereas LTII is rarely found in human isolates and has not been associated with disease. Both LTI and LTII closely resemble CT structurally and functionally. LTI is composed of an enzymatic A subunit and a pentameric B subunit that binds strongly to ganglioside GM1. After binding to the membrane, LTI is endocytosed and undergoes trafficking through the trans-Golgi network (TGN).63 LT targets adenylate cyclase leading to increased cAMP, activation of PKA leading to increased phosphorylation and activation of CFTR. The resulting stimulation in Cl-secretion is a classical example of how LT and CT cause diarrhea. LT has been shown to decrease H+/PEPT co-transporter through a cAMP dependent pathway in Caco-2 cells.64 Thus, cAMP mediates the hypersecretory effects of LT resulting in decreased absorption and increased secretion of fluids and electrolytes.
Enteropathogenic E. coli (EPEC).
EPEC is a major cause of persistent, watery diarrhea in infants, often accompanied by low-grade fever and vomiting. The pathogenic mechanisms of EPEC remained elusive for many years because, in contrast to prototypic enteric bacterial pathogens, EPEC does not produce classical enterotoxins such as LT in order to influence host cell pathways. In addition, EPEC is typically regarded as non-invasive in comparison to bacteria such as Shigella and Salmonella, although a limited number of the bacteria are internalized. However, EPEC does encode a T3SS and produces a characteristic attaching and effacing (A/E) lesion which is marked by effacement of microvilli on the epithelial surface at the site of bacterial attachment.65 In addition, there is an accumulation of cytoskeletal proteins beneath adherent microcolonies leading to actin cup or pedestal formation, depending on cell type. This profound change in intestinal epithelial cells induced by A/E lesions contributes to the diarrheal phenotype due to loss of overall absorptive surface.66 However, diarrhea occurs as quickly as 3–4 h after the ingestion of the pathogen, suggesting that mechanisms other than malabsorption are at work.66,67 Progress is being made in unraveling the basis of EPEC pathogenesis at the molecular level and the mechanism(s) by which EPEC alters host epithelial responses are beginning to unfold. Studies over the past decade suggest that EPEC-induced diarrhea is a multi-factorial process and involves several factors including disrupted paracellular permeability and disturbances in ion transport. Recent advances suggest that EPEC has a direct effect on ion transport processes notably those related to solute, electrolyte, serotonergic and short chain fatty acid transporters. The following section will review potential mechanisms underlying EPEC-associated diarrhea.
Alterations in Na+ and Cl− transport. Investigation of the direct effects of EPEC on intestinal ion transport suggest that decreased intestinal NaCl absorption underlies the pathophysiology of the early onset of diarrhea rather than an increase in Cl−secretion.68–70 Earlier studies utilizing human intestinal epithelial Caco-2 cells showed that EPEC infection resulted in a small and transient increase in Cl−-dependent short circuit current (Isc), a measure of charged ion movement.71 In contrast, in T84 cells, a colonic crypt cell line widely used to study apical Cl− secretion, there was no increase in Cl− secretion but rather an inhibition of agonist-induced secretory responses.70 Although secretion is minimally affected, EPEC alter Na+ and Cl− absorption in both Caco-2 and T-84 cells.69 While net Na+ uptake mediated by Na+/H+ exchangers (NHE) is actually increased by EPEC in Caco-2 cells, the activity of NHE isoforms is differentially modulated.69 In the intestine, both NHE2 and NHE3 isoforms are apically localized, while expression of NHE1 is restricted to the basolateral membranes. Notably, EPEC infection of intestinal epithelial cells decreases the activity of NHE3, the major Na+-absorbing isoform in the small intestine. In contrast, NHE1 and NHE2 isoforms are stimulated in response to infection with EPEC, which is thought to be a compensatory host response to alteration in NHE3 activity.69 However, NHE2 knock-out mice do not have the same intestinal absorption defects or a diarrheal phenotype as NHE3 knock-out mouse mice.72,73 Additionally, NHE2 is unable to prevent diarrhea in NHE3 knock-out mice.74 This suggests that while absorption of Na+ ions is important, it is not directly coupled with water movement in the case of NHE2. Studies utilizing EPEC mutants revealed that NHE3 inhibition is dependent on an intact T3SS III and occurs via the effector molecule EspF.75 EPEC-induced inhibition of NHE3 activity also requires NHE regulatory factors (NHERF) that are important for regulation of NHE3 function and surface expression.76 The inhibitory effect of EPEC on NHE3 is enhanced when PS120 cells are cotransfected with the scaffolding/regulatory proteins NHERF1 and NHERF2.75 In addition to NHE3, EPEC has been shown to rapidly inactivate sodium-D-glucose transporter (SGLT1), the major contributor of fluid uptake in the small intestine.77 Due to the critical role of SGLT1 in oral rehydration therapy, further research into the mechanism of this process would be of great benefit (Fig. 6).
Figure 6.

EPEC-induced early diarrhea is multifactorial. EPEC infection of human epithelial cells decreases epithelial ion absorption to cause diarrhea. The type III secretion system of EPEC releases E. coli secreted proteins (Esps) into the host cytosol. EspF inhibits the function of Na+/H+ exchange isoform 3, whereas EspG, via disruption of microtubules, decreases surface levels of DRA leading to a decrease in apical Cl−/OH−(HCO3−) exchange activity. The resulting inhibition of coupled, electroneutral NaCl absorption in the infected intestine is thought to contribute to diarrhea. EPEC, via an unknown effector molecule, decreases butyrate absorption by decreasing the plasma membrane expression of monocarboxylate transporter 1. EPEC-induced activation of protein tyrosine phosphatases (PTPases) decreases the function of serotonin transporter (SERT) and increases 5-HT availability, which can further affect ion absorption and motility to cause diarrhea. Furthermore, SGLT-1 inhibition by EPEC is thought to promote fluid accumulation causing early diarrhea.
Cl−-HCO3−(OH−) exchangers function in concert with the Na+/H+ exchangers in mediating coupled electroneutral NaCl absorption in ileal and colonic epithelial cells. Parallel to EPEC mediated inhibition of NHE3, a decrease in apical Cl−/OH− exchange activity was demonstrated in Caco-2 and T84 cells.68 Further studies to delineate the mechanisms underlying the decrease in Cl−/OH− exchange revealed an important role for the T3SS. Mutations in either the effector protein espG or its para-log espG2 partially attenuated the inhibitory effects of EPEC, whereas the double mutant completely abolished the inhibition of Cl−/OH− exchange activity.68 Since EspG and EspG2 play an important role in disrupting the host microtubule network,78 these studies suggested that EPEC-induced inhibition of Cl−/HCO3−(OH−) exchange activity is dependent on the disruption of microtubules. This was further confirmed by the observation that colchicine and nocodazole, microtubule-disrupting agents, decreased Cl−/OH− exchange activity in Caco-2 cells.68 Among the candidate genes for luminal human intestinal Cl−/HCO3− exchangers are two members of the SLC26 gene family: SLC26A3, or DRA, and SLC26A6, or PAT-1 (putative anion transporter-1). DRA is predominantly expressed in the colon and plays a major role in the apical Cl−/HCO3− exchange process based upon its implication in congenital chloride diarrhea (CLD).79 CLD is a rare genetic disorder characterized by voluminous diarrhea, massive loss of Cl−in stool and metabolic alkalosis. In addition, in contrast to PAT-1 knock-out mice, DRA knock-out mice develop a robust diarrheal phenotype as well as serum electrolyte imbalances.80,81 Both biochemical and immunofluorescence studies to assess surface expression of apical anion exchangers demonstrated reduced levels of DRA (but not PAT-1) on the apical plasma membrane in response to EPEC infection. This reduction in plasma membrane levels of DRA was EspG/EspG2 dependent.68 An important role of DRA in the pathophysiology of diarrhea was further confirmed in vivo. A marked redistribution of DRA from apical to subapical compartments was demonstrated in EPEC-infected mouse colon.68 Furthermore, mice challenged with C. rodentium, the mouse equivalent of EPEC, showed ∼10 fold downregulation of DRA and fatal fluid loss.82,83 A decrease in DRA and NHE3 activities impairs luminal NaCl absorption and underlies the pathophysiology of EPEC induced early diarrhea (Fig. 6).
Water transport. Aside from modulating electrolyte transport, C. rodentium has been shown to directly impact water transport. During C. rodentium infection, water channels called aquaporins 2 and 3 (AQP2 and AQP3) redistribute from cell membranes to the cytoplasm partly due to EspF and EspG.84 This altered AQP localization correlates with increased fluid levels in internal stool although the mice do not exhibit diarrhea per se. This loss of water absorption is thought to be a contributing factor to diarrhea during EPEC infection. In addition, genome-wide transcriptional array studies utilizing C. rodentium infected mouse colon showed a drastic downregulation of AQP8 mRNA83 (Fig. 6).
Modulation of serotonin and butyrate transporters. EPEC is also known to alter other epithelial processes that indirectly influence electrolyte transport, epithelial integrity and immune responses in intestinal epithelial cells. 5-HT, a neurotransmitter and hormone of the GI tract, affects several physiological processes including absorption and secretion of fluids and electrolytes via serotonin receptors.85 5-HT is internalized through the highly selective Na+ and Cl−-coupled serotonin transporter, SERT, which facilitates 5-HT degradation by intracellular 5-HT catabolizing enzymes. Therefore, SERT regulates 5-HT content and availability in the gut lumen. Enterotoxins such as CT are known to cause the release of serotonin in the small bowel which supports the secretory mechanism causing diarrhea.86 Recent studies demonstrated that EPEC infection of Caco-2 cells decreases SERT function.87 Protein tyrosine phosphatases (PTPases) were involved in inhibiting SERT in Caco-2 cells infected with EPEC. Decreased SERT expression and mucosal 5-HT content also occurred in EPEC-infected murine small intestine.87 Future identification of the EPEC effector molecule(s) that activates PTPases and mediates inhibitory effects on SERT will be of importance in developing new targets to modulate the serotonergic system in treatment of infectious diarrheal diseases.
Another potential conributor to EPEC-induced diarrhea is the modulation of butyrate absorption mediated by monocarboxylate transporter (MCT1) in the intestine.88 Butyrate, the major short chain fatty acid, provides energy for colonocytes and is known to stimulate electrolyte absorption.88 Butyrate transport in epithelial cells is reduced by EPEC infection via a TTSS dependent mechanism and occurs via decreasing the levels of MCT1 on plasma membrane88 (Fig. 6).
Disruption of tight junctions and immune responses to EPEC. Several lines of evidence demonstrate that EPEC infection of intestinal epithelial cells disrupts the tight junction barrier due to activation of myosin light chain kinase (MLCK) and dephosphorylation of occludin.89,90 These effects are dependant on a functional T3SS and several effector proteins including EspF, NleA and Map. The effect of NleA on tight junction integrity occurs via inhibition of host cell protein trafficking through COPII-dependent pathways, while the mechanism of EspF activity remains unknown.90 The decrease in TER seen with EPEC infection also occurs in vivo in ileal and colonic mucosa.91 This particular phenotype is EspF-dependent and correlates with the redistribution of occludin, replicating in vitro data.91 Disruption of tight junctions increases paracellular permeability allowing electrochemical gradients to reach an equilibrium and has been shown to have a synergistic effect with altered ion transport in causing diarrhea.
Initiation of the inflammatory response is another effect of EPEC infection. EPEC is known to activate NFκB, which stimulates the transcription of IL-8, a prototypical cytokine that recruits PMNs to the site of infection.92 EPEC-induced activation of NFκB also upregulates expression of the galanin-1 receptor, which results in increased Cl− secretion and fluid accumulation within the lumen after activation by its ligand, galanin-1.66 Although inflammation is certainly the net effect of EPEC infection, the disease is not as inflammatory as other bacterial pathogens such as Shigella and recent studies have demonstrated that EPEC effector proteins dampen the inflammatory response somewhat.93 The ability of EPEC to suppress the degree or severity of the net inflammatory response was suggested to be essential for the survival of these noninvasive bacteria in the host.93
Collectively, these observations suggest that EPEC-induced diarrhea is a multifactorial process with perturbation of electrolyte, solute and water transport contributing to the development of early onset diarrhea induced by EPEC infection (Fig. 6). A prominent conclusion that emerged from these studies is the use of distinct effector proteins, suggesting selective effects of EPEC on ion transporters. For example, EPEC infection of host intestinal epithelial cells differentially modulates NHE2 and NHE3 activities as well causes a decrease in plasma membrane levels of DRA but not PAT-1.68,69 Also, EspG which is involved in disruption of the host microtubule network, is involved in perturbation of Cl− and water transport but does not modulate other apical membrane transporters such as Na+ or butyrate transporters.68,75,84,88
Enterohemorrhagic E. coli.
EHEC is another major intestinal pathogen that is a subset of Shiga toxin producing E. coli (STEC). EHEC adheres to epithelial cells, expresses a T3SS and causes A/E lesions much like EPEC.63 Unlike EPEC, infection with EHEC can cause more severe symptoms including bloody diarrhea and life-threatening conditions such as hemolytic uremic syndrome (HUS) and thrombotic thrombocytopenic pupura. These symptoms are largely attributable to the production of Shiga toxins (Stx1 and/or Stx2), both of which are composed of one 32 kDa A subunit bound non-covalently to the pentameric ring of identical B subunits. The A subunit possesses N-glycosidase activity that catalyzes depurination of a single adenine residue of 60S ribosomes, rendering them inactive.94 The B subunits mediate the binding of toxin to the globotriaosylceramide (Gb3) glycolipid receptor present on the plasma membrane of certain eukaryotic cells.95 Although EHEC infections often originate from contaminated beef, EHEC colonization of ruminants is actually asymptomatic because of differences in Gb3 receptor distribution. CD77/Gb3 is present on kidney glomerular cells in humans but not in cattle, making HUS a serious complication only for humans. Interestingly, human intestinal epithelial cells do not even express GB3 receptors,94,96 however, a novel binding site for Stx has been described at the base of small intestinal crypts of leiberkuhn in paneth cells.97
With respect to mechanisms of diarrhea, both Stx1 and Stx2 elicit luminal fluid accumulation in the intestine.98 In this regard, purified Stx1 inoculated into adult rabbit ileum induces fluid accumulation and is associated with apoptosis in villus absorptive cells.99 Similarly, Stx2 holotoxin evokes fluid accumulation in vivo and inhibits net absorptive water transport across human colon in vitro.98 Recent studies utilizing T84 intestinal cells and a rabbit model of EHEC infection demonstrate a novel mechanism of Stx-mediated diarrhea. Stx1 infection decreases the secretion of galectin-3, a β-galactoside-specific lectin, resulting in mistargeting of several important brush border proteins.100 Particularly, trafficking of villin, NHE2 and dipeptidyl peptidase is impaired, which is expected to alter epithelial absorption, leading to diarrhea.100 Interestingly, decreased galectin-3 levels have also been described in Crohn's disease.101 Whether EHEC virulence factors encoded by T3SS directly affect epithelial ion transport processes and contribute to diarrheal phenotype of this pathogen, as in the case of EPEC, requires further investigation.
Viral diarrhea. Viruses including norovirus, sapovirus, adenovirus, rotavirus and astroviruses are responsible for 30–40% of acute episodes of diarrhea in the US.40,102 Compared to bacterial infections, many viral infections are mild and self-limited. Norovirus infection affects people of all ages and accounts for ∼40% of non-bacterial diarrheal outbreaks in the US.103 In contrast, rotavirus causes life-threatening gastroenteritis in children, accounting for 35% of the hospitalized cases of diarrhea in the US.104 In the following section, we discuss three viral species that cause diarrhea, including rotavirus, which has the only known viral enterotoxin, as well as norovirus and astrovirus which are only beginning to be understood.
Rotavirus.
Rotavirus is the leading cause of life-threatening diarrheal diseases among young children. Research over the past several years has provided important insights into mechanism of viral pathogenesis and led to successful development of live, attenuated vaccines for gastroenteritis.105 Rotavirus primarily infects small intestinal villus cells and can cause watery diarrhea without any significant intestinal inflammation. The double stranded RNA genome of rotavirus encodes for 6 structural proteins that form virus particles (Vps) and 6 non-structural proteins (NSPs).106 NSP4 is the first described virus-encoded enterotoxin and has been suggested to play a critical role in fluid and electrolyte secretion.107 Interestingly, NSP4 is absent in the mature infective virion and is synthesized in infected villus enterocytes (Fig. 7). NSP4 and virus particles are released through the apical membrane of polarized epithelial cells by a non-classical secretory pathway.105 However, NSP4 is also released from the basolateral side of infected enterocytes, although the role of basolaterally-released NSP4 in diarrhea is not clearly understood108 (Fig. 7). The virology and pathogenesis of rotavirus has been extensively reviewed recently.105
Figure 7.

Mechanisms of rotavirus-mediated diarrhea. Rotavirus infection of enterocytes leads to virus entry, formation of viroplasms (VI) and release of the virus and its toxin, NSP4. Intracellular NSP4 (iNSP4) induces an increase in intracellular Ca2+ primarily through release from er and a PLC-independent mechanism. NSP4 released from the apical side increases intracellular calcium levels through a receptor-mediated PLC-dependent mechanism. The increase in calcium by NSP4 disrupts microvillus cytoskeleton as well as barrier function, leading to an increase in the paracellular flow of water and electrolytes, causing diarrhea. The NSP4 induced increase in Ca2+ levels can also increase Cl− secretion directly through a CFTR-independent mechanism and can cause release of amines, peptides, cytokines and reactive oxygen species, which can stimulate the enteric nervous system indirectly to increase Cl− secretion. The basolateral release of NSP4 may also stimulate ENS. Maldigestion of carbohydrates due to a decrease in surface levels of sucrase-isomaltase and decreased function of SGLT-1 appears to be a major mechanism underlying diarrhea caused by rotavirus infection. eNSP4 is extracellular NSP4.
In contrast to classical secretory diarrhea, the viral enterotoxin, NSP4, induces diarrhea subsequent to maldigestion of carbohydrates concomitant with decreased water absorption, increased Ca2+ mobilization and a relatively mild Cl− secretory component (Fig. 7). Maldigestion of carbohydrates has been suggested as a major mechanism underlying the pathophysiology of rotavirus-induced diarrhea. Rotavirus infection of Caco-2 cells decreases sucrose-isomaltase activity and apical expression in the absence of enterocyte destruction, suggesting the involvement of trafficking mechanisms.109 Similarly, infection of young rabbits or mice with rotavirus decreases disaccharidase activity.110,111 In addition, NSP4 applied exogenously is known to induce Ca2+ release from intracellular stores and plasmalemmal Ca2+ influx through a phospholipase C-dependent mechanism.105 This NSP4-mediated Ca2+ mobilization can support diarrhea by influencing Ca2+-dependent epithelial processes such as ion transport, barrier function or cytoskeletal regulation. Indeed, rotavirus has been demonstrated to increase paracellular permeability in Caco-2 cells.112 In addition, NSP4-mediated Ca2+ mobilization may trigger the release of amines/peptides as well as the release of cytokines, prostaglandins and reactive oxygen species, which can alone or collectively activate the enteric nervous system (ENS).113 This was further confirmed with studies demonstrating that net rotavirus-mediated fluid transport was inhibited by treatment of mice with drugs that affect ENS function.114 Further, clinical studies in hospitalized, rotavirus-infected children show that an enkephalinase inhibitor reduces diarrhea duration.115 Intracellular NSP4, however, is also known to increase intracellular calcium levels through a PLC-independent mechanism.116 Utilizing NSP4-EGFP expression in HEK 293 cells, recent studies demonstrate that intracellular NSP4 causes actin reorganization in a calcium-dependent manner through decreased phosphorylation of the actin remodeling protein cofilin.117 Modulation of subcortical actin dynamics and dysregulation of cofilin influences membrane trafficking events and ion transport processes.118
The Cl− secretory component underlying the pathogenesis of rotavirus-associated diarrhea is complex, comprised of both pro- and anti-secretory components.119 Unlike the purely secretory diarrhea caused by CT, rotavirus infection only moderately increases luminal Cl−concentration.119 An increase in luminal Cl−concentrations could be a consequence of decreased absorption and/or increased secretion. Early studies demonstrated that NSP4 can cause diarrhea in young mice, which is associated with Ca2+ mobilization and potentiation of cAMP-dependent fluid secretion.107 Interestingly, in CFTR-deficient mouse pups, NSP4 continues to result in diarrhea ruling out the involvement of CFTR in fluid accumulation.120 Unexpectedly, rotavirus infection of rabbits actually stimulates Cl− absorption in intestinal brush border membrane isolated from villus cells and does not alter Cl− secretory responses in crypt cells.119 However, the net Cl− secretory response is weak, suggesting that NSP4 exerts both secretory and anti-secretory actions to limit overall Cl− secretion.119 More in-depth studies are required to delineate the cellular mechanisms underlying rotavirus associated Cl− secretory responses. The potential role of apical Cl−/HCO3− exchangers, CLC chloride channels and key signaling events in the pathogenesis of rotavirus infection would be of utmost interest.
Norovirus.
Noroviruses, previously known as Norwalk-like viruses, are a member of the Caliciviridae family.40 They are one of a subset of viruses, which cause viral gastroenteritis, more commonly known as the stomach flu. This illness is typified by short term vomiting and diarrhea, both of which are infectious. The stomach flu is caused by a large number of viruses from several different families and includes sapoviruses, which are also members of the Caliciviridae family, a subtype of adenovirus, as well as astrovirus. In contrast to rotavirus infection, none of these viruses are particularly well-studied and infection rarely results in hospitalization, except in immune compromised individuals. A typical norovirus infection lasts anywhere from 1–3 days and not all infected individuals develop symptoms. Transmission is both foodborne and person-to-person via infectious GI fluids.
Research has been somewhat limited with regard to the molecular basis of norovirus induced diarrhea, since the virus has only recently been grown in tissue culture and earlier studies relied on transfection of viral genes into host cells. Several viral factors can interfere with basic cellular functions without causing cell death. One of these is the 3C-like proteinase (3CLpro), which is similar to a previously described enzyme in rhinovirus called 3Cpro. 3Cpro interferes with eukaryotic translation by cleaving poly(A)-binding protein (PABP).121 Since both ion transporters and tight junction proteins have intermediate turnover times of 12–18 hours on average, inhibition of host translation could limit the abundance of these important regulators of intestinal homeostasis. Another mechanism which has the potential to interfere with host proteins involves the nonstructural protein p48 of Norwalk virus. P48 binds to a cellular protein known as VAMP associated protein or VAP-A, which associates with v-SNARES, regulators of vesicle trafficking.122 As with the inhibition of translation, inhibition of protein trafficking is likely to interfere with cellular processes critical for maintaining absorptive function as well as barrier function.
While cell culture models have been impossible until recently and animal models are limited, a recent study was carried out using human intestinal biopsies from patients suffering from norovirus infection.123 Initial observations showed that, in infected patients, villus length was decreased by 25%, while crypt length remained unchanged, reducing the overall absorptive surface of the epithelium (Fig. 8). Miniature Ussing chambers (0.049 cm2) used to determine resistance, flux and short circuit current on biopsies samples, showed an increase in Isc in patients infected with norovirus, which was dependent on active Cl− secretion, consistent with CFTR activation. SGLT1 activity was also increased suggesting that electrogenic Na+ absorption is not impaired. In addition to changes in Cl− secretion, there was a marked decrease in transepithelial resistance that corresponded with a decrease in the levels of occludin as well as claudins 4 and 5. Because this work was carried out in human tissue, little is known about the mechanism which underlies these changes, however, recently developed norovirus cell culture models should prove useful.
Figure 8.
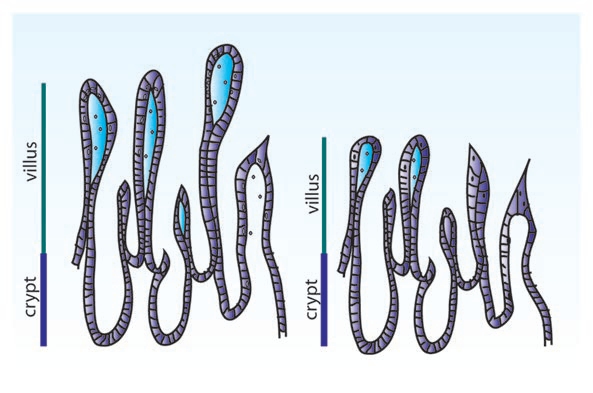
Shortening of villi associated with viral infections. Viral infections including astrovirus and norovirus cause villus blunting or shortening of the villi. There is a decrease in the number of cells making up the villi, reducing the overall absorptive surface. Intestinal epithelial cells are generated from stem cells within the crypts and move toward the villus tip, where they are ultimately shed. Blunting can be caused by reduced cell proliferation, although in the case of norovirus it is associated with an apical villus infection, resulting in increased cell death. Villus blunting is also the cause of diarrhea associated with Celiac disease, which is autoimmune rather than pathogen caused.
Astrovirus.
Astroviruses, like noroviruses, are another cause of the stomach flu. Astrovirus infection includes both diarrhea and vomiting and spreads in a similar manner to the previously described viruses. Little is known about the pathology of astrovirus infection as hospitalization is unlikely. One report is available describing the pathology of an immune-suppressed 4 year old male who showed villus shortening, which is consistent with what is seen in turkey models124 (Fig. 8). In addition, in both these human biopsy specimens and in animal models, there is relatively little inflammation, suggesting that this is not an immune-mediated diarrhea.125 However, there is some evidence that pro-inflammatory pathways are activated in the cell. In fact, activation of ERK1/2 is absolutely required for viral replication.126 UV-inactivated astrovirus is also capable of activating ERK1/2 with a similar response at 15 minutes post infection followed by inactivation at 30 minutes.126 The reduction in both viral protein and RNA levels compared to controls is most consistent with the case of HIV, which requires ERK activation for entry, however, the actual mechanism of ERK activation on adenovirus entry and replication is not yet known. The activation of ERK in these cells is moderate and more is known about the inactivation of the innate immune response due complement inhibition. The astrovirus coat protein is capable of binding to the classical complement component C1q, blocking the subsequent cascade of complement cleavage and membrane attack complex formation.127
Astrovirus infection causes diarrhea with mild inflammation and has been shown to affect barrier function. Moser et al.128 demonstrated a decrease in TER in Caco-2 cells infected with astrovirus. Interestingly, there was also a loss of TER in cells which were treated with UV-inactivated astrovirus, which is incapable of replication, or with virus-like particles, which lack RNA. This suggests that the entry step is responsible for a loss of TER and is consistent with the fact that other viruses are known to target tight junctions during entry. However, the cellular receptor for astrovirus remains unknown. The authors also suggest that there is a disruption of occludin in infected cells, however, cellular morphology is changed substantially after viral infection and is consistent with cells undergoing the early stages of apoptosis. The alteration of occludin could represent an early apoptotic event, although there is a loss in barrier function which is not influenced by early apoptotic events.
The association of astrovirus with cell death in humans is in question due to the lack of patient samples and the variability seen in animal models, specifically turkeys and pigs. In vitro, however, cell death has been seen in Caco-2 cells starting at 36 hours post-infection.129 This apoptosis is blocked by the caspase inhibitor z-VAD-fmk. Interestingly, this viral activation of the caspase system is critical for the production of infectious virions. A precursor of the capsid protein called VP90 has a caspase cleavage site, which is clipped as a step in generating an intermediate capsid protein, VP70. As such, the caspase inhibitor z-VAD-fmk substantially inhibits viral processing to VP70. Virions can be formed by VP90, although they are not stable or infectious and in host cells the cleavage of VP90 to VP70 is critical for release from the cell. While cell death is not particularly associated with astrovirus infection, the characteristic blunting of villus tips could be explained through a loss of cells. This villus blunting along with changes in barrier function are the primary means through which astrovirus causes diarrhea, although changes in ion transport have not yet been studied.
Parasitic diarrhea.
Diarrhea caused by parasites is unlike that of either bacterial or viral infections. For example, Giardia has a slow onset of diarrhea and can be present for months, while most bacterial and viral infections are limited to 1–2 weeks.40 In addition, parasites are eukaryotic, which makes them larger and more complex than either viruses or bacteria and also more difficult to eradicate due to their similarity to the host. In fact, it is not unusual for parasites to make their own versions of host proteins: For example serotonin and PGE2 are both made by Entamoeba histolytica. The advanced mechanisms used by the two important protozoan parasites, Giardia lamblia and E. histolytica, to cause diarrhea will be discussed below.
Giardia lamblia.
Giardia lamblia (syn G. duodenalis and G. intestinalis) is the most common parasite of the human small bowel and causes waterborne diarrheal disease worldwide. Giardia is a non-invasive organism and trophozoites, the active form of the parasite, colonize the upper small intestine by adhering to the apical surface of the epithelium.130 Symptomatic infection with Giardia causes acute or chronic diarrhea, dehydration, abdominal pain and malabsorption leading to malnutrition and weight loss. Also, this parasitic infection can trigger exacerbation of IBS131 and may cause development of post-infectious functional gastrointestinal disorders.132 Several studies utilizing a variety of cell systems, animal models and recent studies in humans with chronic infections have provided important insights into the pathophysiology of giardiasis. An important finding emanating from these studies is that malabsorption, secretion of electrolytes and impairment of tight junctions may underlie the luminal fluid accumulation during infection. Infections with Giardia species are known to cause a diffuse shortening of the microvillus brush border via activated T lymphocytes, accompanied by the reorganization of F-actin and ZO-1 in enterocytes through MLC phosphorylation.133 Enterocyte apoptotic pathways are also induced by G. lamblia via caspase-3 activation, which may also adversely affect epithelial tight junctional integrity.134 Previous studies from both in vitro and in vivo animal models demonstrated decreased absorption of glucose and Na+ and reduced disaccharidase activity due to the loss of epithelial absorptive surface area.135,136 Also, G. lamblia-infected mouse intestine demonstrated secretion of Na+ and Cl− that was protein kinase C dependent.137 However, the role of Giardia virulence factors, such as proteinases and lectins, in modulating secretory responses needs to be fully determined.
The studies from in vitro and animal models were recently confirmed with observations from human subjects infected with Giardia.138 It was established that G. lamblia infection is comprised of active electrogenic anion secretion, impaired barrier dysfunction and malabsorption. Analysis of tight junction proteins in membrane extracts from duodenal biopsy samples of infected patients showed a decrease in the epithelial tight junction protein pool primarily caused by reduction in mucosal surface area.138 However, after mucosal surface area correction, only claudin 1 expression was differentially downregulated in chronic giardiasis.138 Despite the decreased mucosal surface area, an increase in basal Isc reflecting increased Cl−/HCO3(OH−)secretion was observed in giardiasis.138 Importantly, the mucosal surface reduction in human biopsy samples is predominantly loss of villus surface while crypts, an important site of active anion secretion, are not affected. Additionally, apoptosis and a dramatic reduction in villus surface leading to impaired Na+-dependent glucose absorption was observed in the duodenum of patients chronically infected with G. lamblia.133,138 Detailed investigation of the signaling mechanisms and virulence factors underlying epithelial dysfunction following infection will be of importance to understand the pathophysiology of giardiasis as well as development of post-infectious IBS.
Entamoeba histolytica.
E. histolytica is a single cell protozoan which causes amebiasis. Pathologic effects are due to both parasite virulence factors and the host response to the parasite. In fact, only about 10% of people infected with E. histolytica actually develop symptoms.40 In those who present with invasive disease, there is an initial ulceration of the intestinal epithelium due to the expression of several cysteine proteases, which degrade the extracellular matrix, and due to amoebapores which cause cell lysis139,140 (Fig. 9). This results in cell detachment, reducing the absorptive surface of the intestine. In addition, the cysteine pro-teases cause degradation of several complement factors including C3, C3a and C5a, as well as immunoglobulins, thus blunting the innate immune response. While E. histolytica infection is inflammatory, the parasite is resistant to neutrophils due both to complement degradation and a C59-like complement-inactivating adhesion on the surface of the parasite.141 In addition, PMNs can be killed or alternately, neutralized through inhibition of cathepsin G, a serine protease, thus decreasing the activity of PMNs in areas of infection.142,143 In addition, macrophages are activated due to stimulation of endosomal TLR-9 by parasite DNA as well as activation of TLR2 and 4 by lipopeptidophos-phoglycan.144,145 These inflammatory cells are thought to enlarge the initial flask-shaped ulcerations which are characteristic of E. histolytica infection. Therefore, a combination of parasite mediated destruction and subsequent inflammation causes the ulceration of the epithelium which reduces absorption and disrupts barrier function.
Figure 9.

Epithelial destruction and inflammation caused by E. histolytica. E. histolytica induces a flask-shaped ulcer usually spanning several villi rather than several cells as shown here. The epithelial layer is destroyed both due to pathogen factors, such as the pore forming amoebapores and extracellular matrix cleavage by cysteine proteases, as well as host inflammatory factors. E. histolytica releases PGE2 and causes a host PGE2 response that results in colonic Cl− secretion, IL-8 secretion and PMN recruitment. While PMNs can be neutralized and phagocytosed by the parasite and complement is degraded by cysteine proteases, the infection is still highly inflammatory. Macrophages have been shown to be activated by parasite DNA through TLR9 or alternately through lipophoshpopeptidoglycan activation of TLR 2 or 4. In addition to PGE2, E. histolytica also secretes serotonin, which is capable of activating Ca+ dependent Cl− secretion in the small intestine.
In addition to these morphological changes there are also direct effects on barrier function. Because of their ability to induce cell damage and make apparent holes in monolayers, work with E. histolytica and TER must be done early in infection. Leroy et al. found there was a drop in resistance as early as 1 hour post infection, while monlayers began losing cells between 3 and 6 hours post infection.146 This decrease in TER was associated with an increase in mannitol flux and can not be replicated with soluble factors or sonicated trophozoites. Infection does not alter levels of occuldin or its localization but does cause a decrease in levels of ZO-1 without obvious dissociation from the tight junction.146 While secreted cysteine proteases seem a likely cause of this drop in TER, typical protease inhibitors such as E-64 or aprotinin did not block the drop in resistance. However, inactivation of cysteine proteases has been shown to block the later drop in resistance associated with hole formation in monolayers.147
In addition to changes in tight junction activity, there are also changes in ion transport. Analysis of amebic lysates suggests that there are two distinct actions on Cl− secretion: there is a Ca2+ dependent process, which is active in ileum and a cAMP-dependent process, which is active in colon (Fig. 9). The Ca2+-dependent response is only active on the serosal surface, suggesting that these mechanisms are not activated until the epithelial layer is breached.148 HPLC analysis and immunodetection found that amebic lysates actually contain low levels of serotonin and pathogenic strains contain twice the level of non-pathogenic strains. Serotonin is not the only host protein made by E. histolytica; recent studies show that prostaglandin E2 (PGE2) is also made when the amoebae are supplied with arachidonic acid (AA)149 (Fig. 9). They have their own cyclooxygenase (COX)-like enzyme, which is capable of converting AA to PGE2 but is not inhibited by indomethacin as are human COX enzymes.149 However, this appears to be only a small part of the PGE2 response because indomethacin is capable of blocking PGE2 production in human intestinal xenografts in SCID mice.150 The effects of PGE2 are multifold with an increase in both production of IL-8, a potent chemoattractant, and an increase in Cl− secretion.150,151 Secretory effects in mouse colon are attributed to PGE2 synthesis with only a minor Ca2+-dependent component (25%).151 Pretreatment of cells with PGE2 causes desensitization to amebic lysates and the COX inhibitors indomethacin and piroxicam block the rise in Isc almost entirely.151 This mechanism is dependent on the production of cAMP and can be blocked with piroxicam, suggesting a CFTR-dependent mechanism in contrast to the Ca2+-dependent mechanism found in small intestine151 (Fig. 9). Both mechanisms are active on the basolateral side of cells, suggesting that this is not an early event but instead depends on initial infiltration of the epithelial layer by the parasite prior to activation. The overall picture of E. histolytica infection depends both on parasite components, including cysteine proteases, amoebapores, serotonin and PGE2 as well as the host inflammatory response with no single component providing a complete answer.
Conclusion
The elucidation of the mechanisms underlying infectious diarrhea has progressed remarkably over the last decade and will continue to advance. Pathogen-specific virulence factors and signaling cascades affect wide-range of cellular functions such as ion secretion, absorption, barrier function and membrane trafficking events that elicit luminal fluid accumulation causing diarrhea (summarized in Table 1). This is further evident from the huge versatility in mechanisms utilized by pathogenic strains of E. coli, thus contributing to the complexity of pathogenesis. Remarkably, studies in recent years have progressed to define E. coli-secreted proteins (Esps) that specifically decrease ion and water absorption, in contrast to toxin producing strains, which stimulate secretion. Furthermore, activation of the mucosal immune and enteric nervous system as well as cytoskeletal arrangements and loss of absorptive surface seems to augment the outcome of infection (Table 1). Although, malabsorption seems to be a prominent factor in rotavirus and Giardia infections, much progress needs to be made on the molecular mechanisms and virulence factors underlying ion transport modulations associated with these infections. Further investigation into how pathogenic strains of E. coli modulate host physiological responses in vivo to cause diarrhea is critical to completely define the disease. Similarly, our understanding of the pathogenic mechanisms of noroviruses and astroviruses is inadequate and needs extensive research. Unraveling the pathophysiological mechanisms of these pathogens may help identify novel therapeutic targets not only for diarrhea associated with enteric infections but also for a variety of other gastrointestinal diarrheal disorders.
Table 1.
An overview of the data presented in this review assessing the relative contribution of each phenotype to diarrhea for each pathogen presented
| Pathogen | Type | Symptoms | Duration | Inflammation | Increased secretion | Tight junctions | Decreased absorption | Loss of cells* |
| Vibrio cholerae | gram (−) | profuse watery diarrhea | 3–4 days | + | +++ | +++ | +++ | − |
| Clostridium difficile | gram (+) | diarrhea + fever | 3 days+ | +++ | ++ | +++ | + | ++ |
| Shigella species | gram (-) | diarrhea + fever | 5–7 days | +++ | + | +++ | ? | + |
| Escherichia coli (EPEC) | gram (−) | diarrhea | 1–3 weeks | + | − | +++ | +++ | − |
| Rotavirus | dsRNA | vomiting + diarrhea + fever | 3–8 days | − | + | ++ | +++ | + |
| Norovirus | (+) RNA | vomiting + diarrhea | 1–2 days | + | ++ | +++ | ? | +++ |
| Astrovirus | (+) RNA | vomiting + diarrhea + fever | 1–4 days | + | ? | ++ | ? | ++ |
| Giardia | protozoan | diarrhea | 2–6 weeks | − | + | +++ | ++ | +++ |
| Entamoeba histolytica | protozoan | diarrhea | weeks to months | ++ | ++ | ++ | ? | +++ |
Loss of cells includes shortening of villi.
Acknowledgements
We would like to thank Prof. Pradeep Dudeja, Dr. Waddah Alrefai, Andrew W. Weflen, Athanasia Koutsouris and Dr. Sei Yoshida for critical reading of the manuscript.
Footnotes
Previously published online: www.landesbioscience.com/journals/gutmicrobes/article/11036
References
- 1.Slutsker L, Ries AA, Greene KD, Wells JG, Hutwagner L, Griffin PM. Escherichia coli O157:H7 diarrhea in the United States: clinical and epidemiologic features. Ann Intern Med. 1997;126:505–513. doi: 10.7326/0003-4819-126-7-199704010-00002. [DOI] [PubMed] [Google Scholar]
- 2.Kyne L, Hamel MB, Polavaram R, Kelly CP. Health care costs and mortality associated with nosocomial diarrhea due to Clostridium difficile. Clin Infect Dis. 2002;34:346–353. doi: 10.1086/338260. [DOI] [PubMed] [Google Scholar]
- 3.Zhang RG, Scott DL, Westbrook ML, Nance S, Spangler BD, Shipley GG, Westbrook EM. The three-dimensional crystal structure of cholera toxin. J Mol Biol. 1995;251:563–573. doi: 10.1006/jmbi.1995.0456. [DOI] [PubMed] [Google Scholar]
- 4.Cassell JT, Neil A. Dayton 1893–1977. A personal tribute. Am J Ment Defic. 1977;82:227–228. [PubMed] [Google Scholar]
- 5.Cheng SH, Rich DP, Marshall J, Gregory RJ, Welsh MJ, Smith AE. Phosphorylation of the R domain by cAMP-dependent protein kinase regulates the CFTR chloride channel. Cell. 1991;66:1027–1036. doi: 10.1016/0092-8674(91)90446-6. [DOI] [PubMed] [Google Scholar]
- 6.Lencer WI, Constable C, Moe S, Jobling MG, Webb HM, Ruston S, et al. Targeting of cholera toxin and Escherichia coli heat labile toxin in polarized epithelia: role of COOH-terminal KDEL. J Cell Biol. 1995;131:951–962. doi: 10.1083/jcb.131.4.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lord JM, Roberts LM, Lencer WI. Entry of protein toxins into mammalian cells by crossing the endoplasmic reticulum membrane: co-opting basic mechanisms of endoplasmic reticulum-associated degradation. Curr Top Microbiol Immunol. 2005;300:149–168. doi: 10.1007/3-540-28007-3_7. [DOI] [PubMed] [Google Scholar]
- 8.Subramanya SB, Rajendran VM, Srinivasan P, Nanda Kumar NS, Ramakrishna BS, Binder HJ. Differential regulation of cholera toxin-inhibited Na-H exchange isoforms by butyrate in rat ileum. Am J Physiol Gastrointest Liver Physiol. 2007;293:857–863. doi: 10.1152/ajpgi.00462.2006. [DOI] [PubMed] [Google Scholar]
- 9.Trucksis M, Conn TL, Wasserman SS, Sears CL. Vibrio cholerae ACE stimulates Ca(2+)-dependent Cl(-)/HCO(3)(-) secretion in T84 cells in vitro. Am J Physiol Cell Physiol. 2000;279:567–577. doi: 10.1152/ajpcell.2000.279.3.C567. [DOI] [PubMed] [Google Scholar]
- 10.Krasilnikov OV, Muratkhodjaev JN, Zitzer AO. The mode of action of Vibrio cholerae cytolysin. The influences on both erythrocytes and planar lipid bilayers. Biochim Biophys Acta. 1992;1111:7–16. doi: 10.1016/0005-2736(92)90268-q. [DOI] [PubMed] [Google Scholar]
- 11.Visweswariah SS, Shanthi G, Balganesh TS. Interaction of heat-stable enterotoxins with human colonic (T84) cells: modulation of the activation of guanylyl cyclase. Microb Pathog. 1992;12:209–218. doi: 10.1016/0882-4010(92)90055-s. [DOI] [PubMed] [Google Scholar]
- 12.Gutierrez MG, Saka HA, Chinen I, Zoppino FC, Yoshimori T, Bocco JL, Colombo MI. Protective role of autophagy against Vibrio cholerae cytolysin, a pore-forming toxin from V. cholerae. Proc Natl Acad Sci USA. 2007;104:1829–1834. doi: 10.1073/pnas.0601437104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu Z, Nybom P, Magnusson KE. Distinct effects of Vibrio cholerae haemagglutinin/protease on the structure and localization of the tight junction-associated proteins occludin and ZO-1. Cell Microbiol. 2000;2:11–17. doi: 10.1046/j.1462-5822.2000.00025.x. [DOI] [PubMed] [Google Scholar]
- 14.Fullner KJ, Lencer WI, Mekalanos JJ. Vibrio cholerae-induced cellular responses of polarized T84 intestinal epithelial cells are dependent on production of cholera toxin and the RTX toxin. Infect Immun. 2001;69:6310–6317. doi: 10.1128/IAI.69.10.6310-6317.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kudryashov DS, Cordero CL, Reisler E, Satchell KJ. Characterization of the enzymatic activity of the actin cross-linking domain from the Vibrio cholerae MARTX Vc toxin. J Biol Chem. 2008;283:445–452. doi: 10.1074/jbc.M703910200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uzzau S, Cappuccinelli P, Fasano A. Expression of Vibrio cholerae zonula occludens toxin and analysis of its subcellular localization. Microb Pathog. 1999;27:377–385. doi: 10.1006/mpat.1999.0312. [DOI] [PubMed] [Google Scholar]
- 17.Davis BM, Waldor MK. Filamentous phages linked to virulence of Vibrio cholerae. Curr Opin Microbiol. 2003;6:35–42. doi: 10.1016/s1369-5274(02)00005-x. [DOI] [PubMed] [Google Scholar]
- 18.Waldor MK, Mekalanos JJ. Lysogenic conversion by a filamentous phage encoding cholera toxin. Science. 1996;272:1910–1914. doi: 10.1126/science.272.5270.1910. [DOI] [PubMed] [Google Scholar]
- 19.Wang W, Uzzau S, Goldblum SE, Fasano A. Human zonulin, a potential modulator of intestinal tight junctions. J Cell Sci. 2000;113:4435–4440. doi: 10.1242/jcs.113.24.4435. [DOI] [PubMed] [Google Scholar]
- 20.Gopalakrishnan S, Pandey N, Tamiz AP, Vere J, Carrasco R, Somerville R, et al. Mechanism of action of ZOT-derived peptide AT-1002, a tight junction regulator and absorption enhancer. Int J Pharm. 2009;365:121–130. doi: 10.1016/j.ijpharm.2008.08.047. [DOI] [PubMed] [Google Scholar]
- 21.Bartlett JG, Gorbach SL. Pseudomembranous enterocolitis (antibiotic-related colitis) Adv Intern Med. 1977;22:455–476. [PubMed] [Google Scholar]
- 22.Deneve C, Janoir C, Poilane I, Fantinato C, Collignon A. New trends in Clostridium difficile virulence and pathogenesis. Int J Antimicrob Agents. 2009;33:24–28. doi: 10.1016/S0924-8579(09)70012-3. [DOI] [PubMed] [Google Scholar]
- 23.Perelle S, Gibert M, Bourlioux P, Corthier G, Popoff MR. Production of a complete binary toxin (actin-specific ADP-ribosyltransferase) by Clostridium difficile CD196. Infect Immun. 1997;65:1402–1407. doi: 10.1128/iai.65.4.1402-1407.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Na X, Kim H, Moyer MP, Pothoulakis C, LaMont JT. gp96 is a human colonocyte plasma membrane binding protein for Clostridium difficile toxin A. Infect Immun. 2008;76:2862–2871. doi: 10.1128/IAI.00326-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rupnik M, Wilcox MH, Gerding DN. Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat Rev Microbiol. 2009;7:526–536. doi: 10.1038/nrmicro2164. [DOI] [PubMed] [Google Scholar]
- 26.Thelestam M, Chaves-Olarte E. Cytotoxic effects of the Clostridium difficile toxins. Curr Top Microbiol Immunol. 2000;250:85–96. doi: 10.1007/978-3-662-06272-2_4. [DOI] [PubMed] [Google Scholar]
- 27.Thelestam M. Bacterial protein toxins—general conclusions. Int J Med Microbiol. 2000;290:503–506. [PubMed] [Google Scholar]
- 28.Barbut F, Decre D, Lalande V, Burghoffer B, Noussair L, Gigandon A, et al. Clinical features of Clostridium difficile-associated diarrhoea due to binary toxin (actin-specific ADP-ribosyltransferase)-producing strains. J Med Microbiol. 2005;54:181–185. doi: 10.1099/jmm.0.45804-0. [DOI] [PubMed] [Google Scholar]
- 29.Lyras D, O'Connor JR, Howarth PM, Sambol SP, Carter GP, Phumoonna T, et al. Toxin B is essential for virulence of Clostridium difficile. Nature. 2009;458:1176–1179. doi: 10.1038/nature07822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lima AA, Lyerly DM, Wilkins TD, Innes DJ, Guerrant RL. Effects of Clostridium difficile toxins A and B in rabbit small and large intestine in vivo and on cultured cells in vitro. Infect Immun. 1988;56:582–588. doi: 10.1128/iai.56.3.582-588.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moore R, Pothoulakis C, LaMont JT, Carlson S, Madara JL. C. difficile toxin A increases intestinal permeability and induces Cl− secretion. Am J Physiol. 1990;259:165–172. doi: 10.1152/ajpgi.1990.259.2.G165. [DOI] [PubMed] [Google Scholar]
- 32.Hecht G, Pothoulakis C, LaMont JT, Madara JL. Clostridium difficile toxin A perturbs cytoskeletal structure and tight junction permeability of cultured human intestinal epithelial monolayers. J Clin Invest. 1988;82:1516–1524. doi: 10.1172/JCI113760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pothoulakis C, Lamont JT. Microbes and microbial toxins: paradigms for microbial-mucosal interactions II. The integrated response of the intestine to Clostridium difficile toxins. Am J Physiol Gastrointest Liver Physiol. 2001;280:178–183. doi: 10.1152/ajpgi.2001.280.2.G178. [DOI] [PubMed] [Google Scholar]
- 34.Flegel WA, Muller F, Daubener W, Fischer HG, Hadding U, Northoff H. Cytokine response by human monocytes to Clostridium difficile toxin A and toxin B. Infect Immun. 1991;59:3659–3666. doi: 10.1128/iai.59.10.3659-3666.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kelly CP, Becker S, Linevsky JK, Joshi MA, O'Keane JC, Dickey BF, et al. Neutrophil recruitment in Clostridium difficile toxin A enteritis in the rabbit. J Clin Invest. 1994;93:1257–1265. doi: 10.1172/JCI117080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Keates AC, Castagliuolo I, Qiu B, Nikulasson S, Sengupta A, Pothoulakis C. CGRP upregulation in dorsal root ganglia and ileal mucosa during Clostridium difficile toxin A-induced enteritis. Am J Physiol. 1998;274:196–202. doi: 10.1152/ajpgi.1998.274.1.G196. [DOI] [PubMed] [Google Scholar]
- 37.Pothoulakis C, Castagliuolo I, Leeman SE, Wang CC, Li H, Hoffman BJ, Mezey E. Substance P receptor expression in intestinal epithelium in Clostridium difficile toxin A enteritis in rats. Am J Physiol. 1998;275:68–75. doi: 10.1152/ajpgi.1998.275.1.G68. [DOI] [PubMed] [Google Scholar]
- 38.Castagliuolo I, Wang CC, Valenick L, Pasha A, Nikulasson S, Carraway RE, Pothoulakis C. Neurotensin is a proinflammatory neuropeptide in colonic inflammation. J Clin Invest. 1999;103:843–849. doi: 10.1172/JCI4217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Castagliuolo I, Riegler M, Pasha A, Nikulasson S, Lu B, Gerard C, et al. Neurokinin-1 (NK-1) receptor is required in Clostridium difficile-induced enteritis. J Clin Invest. 1998;101:1547–1550. doi: 10.1172/JCI2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Centers for Disease Control and Prevention. www.cdc.gov.
- 41.Wassef JS, Keren DF, Mailloux JL. Role of M cells in initial antigen uptake and in ulcer formation in the rabbit intestinal loop model of shigellosis. Infect Immun. 1989;57:858–863. doi: 10.1128/iai.57.3.858-863.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Prevost MC, Lesourd M, Arpin M, Vernel F, Mounier J, Hellio R, Sansonetti PJ. Unipolar reorganization of F-actin layer at bacterial division and bundling of actin filaments by plastin correlate with movement of Shigella flexneri within HeLa cells. Infect Immun. 1992;60:4088–4099. doi: 10.1128/iai.60.10.4088-4099.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schroeder GN, Hilbi H. Molecular pathogenesis of Shigella spp.: controlling host cell signaling, invasion and death by type III secretion. Clin Microbiol Rev. 2008;21:134–156. doi: 10.1128/CMR.00032-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yee RB, Buffenmyer CL. Infection of Cultured Mouse Macrophages with Shigella flexneri. Infect Immun. 1970;1:459–463. doi: 10.1128/iai.1.5.459-463.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sansonetti PJ, Phalipon A, Arondel J, Thirumalai K, Banerjee S, Akira S, et al. Caspase-1 activation of IL-1beta and IL-18 are essential for Shigella flexneri-induced inflammation. Immunity. 2000;12:581–590. doi: 10.1016/s1074-7613(00)80209-5. [DOI] [PubMed] [Google Scholar]
- 46.Rallabhandi P, Awomoyi A, Thomas KE, Phalipon A, Fujimoto Y, Fukase K, et al. Differential activation of human TLR4 by Escherichia coli and Shigella flexneri 2a lipopolysaccharide: combined effects of lipid A acylation state and TLR4 polymorphisms on signaling. J Immunol. 2008;180:1139–1147. doi: 10.4049/jimmunol.180.2.1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aliprantis AO, Weiss DS, Radolf JD, Zychlinsky A. Release of Toll-like receptor-2-activating bacterial lipoproteins in Shigella flexneri culture supernatants. Infect Immun. 2001;69:6248–6255. doi: 10.1128/IAI.69.10.6248-6255.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nigro G, Fazio LL, Martino MC, Rossi G, Tattoli I, Liparoti V, et al. Muramylpeptide shedding modulates cell sensing of Shigella flexneri. Cell Microbiol. 2008;10:682–695. doi: 10.1111/j.1462-5822.2007.01075.x. [DOI] [PubMed] [Google Scholar]
- 49.Li H, Xu H, Zhou Y, Zhang J, Long C, Li S, et al. The phosphothreonine lyase activity of a bacterial type III effector family. Science. 2007;315:1000–1003. doi: 10.1126/science.1138960. [DOI] [PubMed] [Google Scholar]
- 50.Rohde JR, Breitkreutz A, Chenal A, Sansonetti PJ, Parsot C. Type III secretion effectors of the IpaH family are E3 ubiquitin ligases. Cell Host Microbe. 2007;1:77–83. doi: 10.1016/j.chom.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 51.Kim DW, Lenzen G, Page AL, Legrain P, Sansonetti PJ, Parsot C. The Shigella flexneri effector OspG interferes with innate immune responses by targeting ubiquitin-conjugating enzymes. Proc Natl Acad Sci USA. 2005;102:14046–14051. doi: 10.1073/pnas.0504466102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sansonetti PJ, Arondel J, Huerre M, Harada A, Matsushima K. Interleukin-8 controls bacterial transepithelial translocation at the cost of epithelial destruction in experimental shigellosis. Infect Immun. 1999;67:1471–1480. doi: 10.1128/iai.67.3.1471-1480.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim M, Ogawa M, Fujita Y, Yoshikawa Y, Nagai T, Koyama T, et al. Bacteria hijack integrin-linked kinase to stabilize focal adhesions and block cell detachment. Nature. 2009;459:578–582. doi: 10.1038/nature07952. [DOI] [PubMed] [Google Scholar]
- 54.Sakaguchi T, Kohler H, Gu X, McCormick BA, Reinecker HC. Shigella flexneri regulates tight junction-associated proteins in human intestinal epithelial cells. Cell Microbiol. 2002;4:367–381. doi: 10.1046/j.1462-5822.2002.00197.x. [DOI] [PubMed] [Google Scholar]
- 55.Al-Hasani K, Henderson IR, Sakellaris H, Rajakumar K, Grant T, Nataro JP, et al. The sigA gene which is borne on the she pathogenicity island of Shigella flexneri 2a encodes an exported cytopathic protease involved in intestinal fluid accumulation. Infect Immun. 2000;68:2457–2463. doi: 10.1128/iai.68.5.2457-2463.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kaper JB, Nataro JP, Mobley HL. Pathogenic Escherichia coli. Nat Rev Microbiol. 2004;2:123–140. doi: 10.1038/nrmicro818. [DOI] [PubMed] [Google Scholar]
- 57.Golin-Bisello F, Bradbury N, Ameen N. STa and cGMP stimulate CFTR translocation to the surface of villus enterocytes in rat jejunum and is regulated by protein kinase G. Am J Physiol Cell Physiol. 2005;289:708–716. doi: 10.1152/ajpcell.00544.2004. [DOI] [PubMed] [Google Scholar]
- 58.Vaandrager AB. Structure and function of the heat-stable enterotoxin receptor/guanylyl cyclase C. Mol Cell Biochem. 2002;230:73–83. [PubMed] [Google Scholar]
- 59.Lucas ML. Enterocyte chloride and water secretion into the small intestine after enterotoxin challenge: unifying hypothesis or intellectual dead end? J Physiol Biochem. 2008;64:69–88. doi: 10.1007/BF03168236. [DOI] [PubMed] [Google Scholar]
- 60.Sellers ZM, Mann E, Smith A, Ko KH, Giannella R, Cohen MB, et al. Heat-stable enterotoxin of Escherichia coli (STa) can stimulate duodenal HCO3(-) secretion via a novel GC-C-and CFTR-independent pathway. Faseb J. 2008;22:1306–1316. doi: 10.1096/fj.06-7540com. [DOI] [PubMed] [Google Scholar]
- 61.Pratha VS, Hogan DL, Martensson BA, Bernard J, Zhou R, Isenberg JI. Identification of transport abnormalities in duodenal mucosa and duodenal enterocytes from patients with cystic fibrosis. Gastroenterology. 2000;118:1051–1060. doi: 10.1016/s0016-5085(00)70358-1. [DOI] [PubMed] [Google Scholar]
- 62.Sellers ZM, Childs D, Chow JY, Smith AJ, Hogan DL, Isenberg JI, et al. Heat-stable enterotoxin of Escherichia coli stimulates a non-CFTR-mediated duodenal bicarbonate secretory pathway. Am J Physiol Gastrointest Liver Physiol. 2005;288:654–663. doi: 10.1152/ajpgi.00386.2004. [DOI] [PubMed] [Google Scholar]
- 63.Nataro JP, Kaper JB. Diarrheagenic Escherichia coli. Clin Microbiol Rev. 1998;11:142–201. doi: 10.1128/cmr.11.1.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Muller U, Brandsch M, Prasad PD, Fei YJ, Ganapathy V, Leibach FH. Inhibition of the H+/peptide cotransporter in the human intestinal cell line Caco-2 by cyclic AMP. Biochem Biophys Res Commun. 1996;218:461–465. doi: 10.1006/bbrc.1996.0082. [DOI] [PubMed] [Google Scholar]
- 65.Kenny B, Abe A, Stein M, Finlay BB. Enteropathogenic Escherichia coli protein secretion is induced in response to conditions similar to those in the gastrointestinal tract. Infect Immun. 1997;65:2606–2612. doi: 10.1128/iai.65.7.2606-2612.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hecht G. Microbes and microbial toxins: paradigms for microbial-mucosal interactions VII. Enteropathogenic Escherichia coli: physiological alterations from an extracellular position. Am J Physiol Gastrointest Liver Physiol. 2001;281:1–7. doi: 10.1152/ajpgi.2001.281.1.G1. [DOI] [PubMed] [Google Scholar]
- 67.Donnenberg MS, Tacket CO, James SP, Losonsky G, Nataro JP, Wasserman SS, et al. Role of the eaeA gene in experimental enteropathogenic Escherichia coli infection. J Clin Invest. 1993;92:1412–1417. doi: 10.1172/JCI116717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gill RK, Borthakur A, Hodges K, Turner JR, Clayburgh DR, Saksena S, et al. Mechanism underlying inhibition of intestinal apical Cl/OH exchange following infection with enteropathogenic E. coli. J Clin Invest. 2007;117:428–437. doi: 10.1172/JCI29625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hecht G, Hodges K, Gill RK, Kear F, Tyagi S, Malakooti J, et al. Differential regulation of Na+/H+ exchange isoform activities by enteropathogenic E. coli in human intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2004;287:370–378. doi: 10.1152/ajpgi.00432.2003. [DOI] [PubMed] [Google Scholar]
- 70.Hecht G, Koutsouris A. Enteropathogenic E. coli attenuates secretagogue-induced net intestinal ion transport but not Cl− secretion. Am J Physiol. 1999;276:781–788. doi: 10.1152/ajpgi.1999.276.3.G781. [DOI] [PubMed] [Google Scholar]
- 71.Collington GK, Booth IW, Knutton S. Rapid modulation of electrolyte transport in Caco-2 cell mono-layers by enteropathogenic Escherichia coli (EPEC) infection. Gut. 1998;42:200–207. doi: 10.1136/gut.42.2.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schultheis PJ, Clarke LL, Meneton P, Miller ML, Soleimani M, Gawenis LR, et al. Renal and intestinal absorptive defects in mice lacking the NHE3 Na+/H+ exchanger. Nat Genet. 1998;19:282–285. doi: 10.1038/969. [DOI] [PubMed] [Google Scholar]
- 73.Schultheis PJ, Clarke LL, Meneton P, Harline M, Boivin GP, Stemmermann G, et al. Targeted disruption of the murine Na+/H+ exchanger isoform 2 gene causes reduced viability of gastric parietal cells and loss of net acid secretion. J Clin Invest. 1998;101:1243–1253. doi: 10.1172/JCI1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ledoussal C, Woo AL, Miller ML, Shull GE. Loss of the NHE2 Na(+)/H(+) exchanger has no apparent effect on diarrheal state of NHE3-deficient mice. Am J Physiol Gastrointest Liver Physiol. 2001;281:1385–1396. doi: 10.1152/ajpgi.2001.281.6.G1385. [DOI] [PubMed] [Google Scholar]
- 75.Hodges K, Alto NM, Ramaswamy K, Dudeja PK, Hecht G. The enteropathogenic Escherichia coli effector protein EspF decreases sodium hydrogen exchanger 3 activity. Cell Microbiol. 2008;10:1735–1745. doi: 10.1111/j.1462-5822.2008.01163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cha B, Donowitz M. The epithelial brush border Na+/H+ exchanger NHE3 associates with the actin cytoskeleton by binding to ezrin directly and via PDZ domain-containing Na+/H+ exchanger regulatory factor (NHERF) proteins. Clin Exp Pharmacol Physiol. 2008;35:863–871. doi: 10.1111/j.1440-1681.2008.04931.x. [DOI] [PubMed] [Google Scholar]
- 77.Dean P, Maresca M, Schuller S, Phillips AD, Kenny B. Potent diarrheagenic mechanism mediated by the cooperative action of three enteropathogenic Escherichia coli-injected effector proteins. Proc Natl Acad Sci USA. 2006;103:1876–1881. doi: 10.1073/pnas.0509451103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tomson FL, Viswanathan VK, Kanack KJ, Kanteti RP, Straub KV, Menet M, et al. Enteropathogenic Escherichia coli EspG disrupts microtubules and in conjunction with Orf3 enhances perturbation of the tight junction barrier. Mol Microbiol. 2005;56:447–464. doi: 10.1111/j.1365-2958.2005.04571.x. [DOI] [PubMed] [Google Scholar]
- 79.Makela S, Kere J, Holmberg C, Hoglund P. SLC26A3 mutations in congenital chloride diarrhea. Hum Mutat. 2002;20:425–438. doi: 10.1002/humu.10139. [DOI] [PubMed] [Google Scholar]
- 80.Schweinfest CW, Spyropoulos DD, Henderson KW, Kim JH, Chapman JM, Barone S, et al. slc26a3 (dra)-deficient mice display chloride-losing diarrhea, enhanced colonic proliferation, and distinct upregulation of ion transporters in the colon. J Biol Chem. 2006;281:37962–37971. doi: 10.1074/jbc.M607527200. [DOI] [PubMed] [Google Scholar]
- 81.Seidler U, Rottinghaus I, Hillesheim J, Chen M, Riederer B, Krabbenhoft A, et al. Sodium and chloride absorptive defects in the small intestine in Slc26a6 null mice. Pflugers Arch. 2008;455:757–766. doi: 10.1007/s00424-007-0318-z. [DOI] [PubMed] [Google Scholar]
- 82.Borenshtein D, Schlieper KA, Rickman BH, Chapman JM, Schweinfest CW, Fox JG, Schauer DB. Decreased expression of colonic Slc26a3 and carbonic anhydrase iv as a cause of fatal infectious diarrhea in mice. Infect Immun. 2009;77:3639–3650. doi: 10.1128/IAI.00225-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Borenshtein D, Fry RC, Groff EB, Nambiar PR, Carey VJ, Fox JG, Schauer DB. Diarrhea as a cause of mortality in a mouse model of infectious colitis. Genome Biol. 2008;9:122. doi: 10.1186/gb-2008-9-8-r122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Guttman JA, Samji FN, Li Y, Deng W, Lin A, Finlay BB. Aquaporins contribute to diarrhoea caused by attaching and effacing bacterial pathogens. Cell Microbiol. 2007;9:131–141. doi: 10.1111/j.1462-5822.2006.00773.x. [DOI] [PubMed] [Google Scholar]
- 85.Gill RK, Saksena S, Tyagi S, Alrefai WA, Malakooti J, Sarwar Z, et al. Serotonin inhibits Na+/H+ exchange activity via 5-HT4 receptors and activation of PKC alpha in human intestinal epithelial cells. Gastroenterology. 2005;128:962–974. doi: 10.1053/j.gastro.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 86.Eklund S, Brunsson I, Jodal M, Lundgren O. Changes in cyclic 3′5′-adenosine monophosphate tissue concentration and net fluid transport in the cat's small intestine elicited by choleratoxin, arachidonic acid, vasoactive intestinal polypeptide and 5-hydroxytryptamine. Acta Physiol Scand. 1987;129:115–125. doi: 10.1111/j.1748-1716.1987.tb08046.x. [DOI] [PubMed] [Google Scholar]
- 87.Esmaili A, Nazir SF, Borthakur A, Yu D, Turner JR, Saksena S, et al. Enteropathogenic E. coli Infection Inhibits Intestinal Serotonin Transporter (SERT) Function and Expression. Gastroenterology. 2009 doi: 10.1053/j.gastro.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Borthakur A, Gill RK, Hodges K, Ramaswamy K, Hecht G, Dudeja PK. Enteropathogenic Escherichia coli inhibits butyrate uptake in Caco-2 cells by altering the apical membrane MCT1 level. Am J Physiol Gastrointest Liver Physiol. 2006;290:30–35. doi: 10.1152/ajpgi.00302.2005. [DOI] [PubMed] [Google Scholar]
- 89.Manjarrez-Hernandez HA, Baldwin TJ, Williams PH, Haigh R, Knutton S, Aitken A. Phosphorylation of myosin light chain at distinct sites and its association with the cytoskeleton during enteropathogenic Escherichia coli infection. Infect Immun. 1996;64:2368–2370. doi: 10.1128/iai.64.6.2368-2370.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Thanabalasuriar A, Koutsouris A, Weflen A, Mimee M, Hecht G, Gruenheid S. The bacterial virulence factor NleA is required for the disruption of intestinal tight junctions by enteropathogenic Escherichia coli. Cell Microbiol. 2009 doi: 10.1111/j.1462-5822.2009.01376.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shifflett DE, Clayburgh DR, Koutsouris A, Turner JR, Hecht GA. Enteropathogenic E. coli disrupts tight junction barrier function and structure in vivo. Lab Invest. 2005;85:1308–1324. doi: 10.1038/labinvest.3700330. [DOI] [PubMed] [Google Scholar]
- 92.Savkovic SD, Koutsouris A, Hecht G. PKCzeta participates in activation of inflammatory response induced by enteropathogenic E. coli. Am J Physiol Cell Physiol. 2003;285:512–521. doi: 10.1152/ajpcell.00444.2002. [DOI] [PubMed] [Google Scholar]
- 93.Sharma R, Tesfay S, Tomson FL, Kanteti RP, Viswanathan VK, Hecht G. Balance of bacterial pro- and anti-inflammatory mediators dictates net effect of enteropathogenic Escherichia coli on intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2006;290:685–694. doi: 10.1152/ajpgi.00404.2005. [DOI] [PubMed] [Google Scholar]
- 94.Obrig TG, Moran TP, Brown JE. The mode of action of Shiga toxin on peptide elongation of eukaryotic protein synthesis. Biochem J. 1987;244:287–294. doi: 10.1042/bj2440287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cohen A, Hannigan GE, Williams BR, Lingwood CA. Roles of globotriosyl- and galabiosylceramide in verotoxin binding and high affinity interferon receptor. J Biol Chem. 1987;262:17088–17091. [PubMed] [Google Scholar]
- 96.Hauf N, Chakraborty T. Suppression of NFkappaB activation and proinflammatory cytokine expression by Shiga toxin-producing Escherichia coli. J Immunol. 2003;170:2074–2082. doi: 10.4049/jimmunol.170.4.2074. [DOI] [PubMed] [Google Scholar]
- 97.Schuller S, Heuschkel R, Torrente F, Kaper JB, Phillips AD. Shiga toxin binding in normal and inflamed human intestinal mucosa. Microbes Infect. 2007;9:35–39. doi: 10.1016/j.micinf.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 98.Creydt VP, Miyakawa MF, Martin F, Zotta E, Silberstein C, Ibarra C. The Shiga toxin 2 B subunit inhibits net fluid absorption in human colon and elicits fluid accumulation in rat colon loops. Braz J Med Biol Res. 2004;37:799–808. doi: 10.1590/s0100-879x2004000600004. [DOI] [PubMed] [Google Scholar]
- 99.Ferreira AJ, Elias WP, Jr, Pelayo JS, Giraldi R, Pedroso MZ, Scaletsky IC. Culture supernatant of Shiga toxin-producing Escherichia coli strains provoke fluid accumulation in rabbit ileal loops. FEMS Immunol Med Microbiol. 1997;19:285–288. doi: 10.1111/j.1574-695X.1997.tb01098.x. [DOI] [PubMed] [Google Scholar]
- 100.Laiko M, Murtazina R, Malyukova I, Zhu C, Boedeker EC, Gutsal O, et al. Shiga toxin 1 interaction with enterocytes causes apical protein mistargeting through the depletion of intracellular galectin-3. Exp Cell Res. 2009 doi: 10.1016/j.yexcr.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Muller S, Schaffer T, Flogerzi B, Fleetwood A, Weimann R, Schoepfer AM, Seibold F. Galectin-3 modulates T cell activity and is reduced in the inflamed intestinal epithelium in IBD. Inflamm Bowel Dis. 2006;12:588–597. doi: 10.1097/01.MIB.0000225341.37226.7c. [DOI] [PubMed] [Google Scholar]
- 102.Musher DM, Musher BL. Contagious acute gastrointestinal infections. N Engl J Med. 2004;351:2417–2427. doi: 10.1056/NEJMra041837. [DOI] [PubMed] [Google Scholar]
- 103.Ciarlet M, Estes MK. Rotavirus and calicivirus infections of the gastrointestinal tract. Curr Opin Gastroenterol. 2001;17:10–16. doi: 10.1097/00001574-200101000-00003. [DOI] [PubMed] [Google Scholar]
- 104.Rodriguez WJ, Kim HW, Brandt CD, Schwartz RH, Gardner MK, Jeffries B, et al. Longitudinal study of rotavirus infection and gastroenteritis in families served by a pediatric medical practice: clinical and epidemiologic observations. Pediatr Infect Dis J. 1987;6:170–176. doi: 10.1097/00006454-198702000-00006. [DOI] [PubMed] [Google Scholar]
- 105.Greenberg HB, Estes MK. Rotaviruses: from pathogenesis to vaccination. Gastroenterology. 2009;136:1939–1951. doi: 10.1053/j.gastro.2009.02.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pesavento JB, Crawford SE, Estes MK, Prasad BV. Rotavirus proteins: structure and assembly. Curr Top Microbiol Immunol. 2006;309:189–219. doi: 10.1007/3-540-30773-7_7. [DOI] [PubMed] [Google Scholar]
- 107.Ball JM, Tian P, Zeng CQ, Morris AP, Estes MK. Age-dependent diarrhea induced by a rotaviral non-structural glycoprotein. Science. 1996;272:101–104. doi: 10.1126/science.272.5258.101. [DOI] [PubMed] [Google Scholar]
- 108.Boshuizen JA, Rossen JW, Sitaram CK, Kimenai FF, Simons-Oosterhuis Y, Laffeber C, et al. Rotavirus enterotoxin NSP4 binds to the extracellular matrix proteins laminin-beta3 and fibronectin. J Virol. 2004;78:10045–10053. doi: 10.1128/JVI.78.18.10045-10053.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Jourdan N, Brunet JP, Sapin C, Blais A, Cotte-Laffitte J, Forestier F, et al. Rotavirus infection reduces sucrase-isomaltase expression in human intestinal epithelial cells by perturbing protein targeting and organization of microvillar cytoskeleton. J Virol. 1998;72:7228–7236. doi: 10.1128/jvi.72.9.7228-7236.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Halaihel N, Lievin V, Alvarado F, Vasseur M. Rotavirus infection impairs intestinal brush-border membrane Na(+)-solute cotransport activities in young rabbits. Am J Physiol Gastrointest Liver Physiol. 2000;279:587–596. doi: 10.1152/ajpgi.2000.279.3.G587. [DOI] [PubMed] [Google Scholar]
- 111.Collins J, Candy DC, Starkey WG, Spencer AJ, Osborne MP, Stephen J. Disaccharidase activities in small intestine of rotavirus-infected suckling mice: a histochemical study. J Pediatr Gastroenterol Nutr. 1990;11:395–403. doi: 10.1097/00005176-199010000-00020. [DOI] [PubMed] [Google Scholar]
- 112.Dickman KG, Hempson SJ, Anderson J, Lippe S, Zhao L, Burakoff R, Shaw RD. Rotavirus alters paracellular permeability and energy metabolism in Caco-2 cells. Am J Physiol Gastrointest Liver Physiol. 2000;279:757–766. doi: 10.1152/ajpgi.2000.279.4.G757. [DOI] [PubMed] [Google Scholar]
- 113.Lorrot M, Vasseur M. Physiopathology of Rotavirus diarrhea. Arch Pediatr. 2007;14:145–151. doi: 10.1016/s0929-693x(07)80018-2. [DOI] [PubMed] [Google Scholar]
- 114.Morris AP, Estes MK. Microbes and microbial toxins: paradigms for microbial-mucosal interactions VIII. Pathological consequences of rotavirus infection and its enterotoxin. Am J Physiol Gastrointest Liver Physiol. 2001;281:303–310. doi: 10.1152/ajpgi.2001.281.2.G303. [DOI] [PubMed] [Google Scholar]
- 115.Salazar-Lindo E, Santisteban-Ponce J, Chea-Woo E, Gutierrez M. Racecadotril in the treatment of acute watery diarrhea in children. N Engl J Med. 2000;343:463–467. doi: 10.1056/NEJM200008173430703. [DOI] [PubMed] [Google Scholar]
- 116.Berkova Z, Morris AP, Estes MK. Cytoplasmic calcium measurement in rotavirus enterotoxin-enhanced green fluorescent protein (NSP4-EGFP) expressing cells loaded with Fura-2. Cell Calcium. 2003;34:55–68. doi: 10.1016/s0143-4160(03)00022-8. [DOI] [PubMed] [Google Scholar]
- 117.Berkova Z, Crawford SE, Blutt SE, Morris AP, Estes MK. Expression of rotavirus NSP4 alters the actin network organization through the actin remodeling protein cofilin. J Virol. 2007;81:3545–3553. doi: 10.1128/JVI.01080-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.DesMarais V, Macaluso F, Condeelis J, Bailly M. Synergistic interaction between the Arp2/3 complex and cofilin drives stimulated lamellipod extension. J Cell Sci. 2004;117:3499–3510. doi: 10.1242/jcs.01211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lorrot M, Vasseur M. How do the rotavirus NSP4 and bacterial enterotoxins lead differently to diarrhea? Virol J. 2007;4:31. doi: 10.1186/1743-422X-4-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Morris AP, Scott JK, Ball JM, Zeng CQ, O'Neal WK, Estes MK. NSP4 elicits age-dependent diarrhea and Ca(2+)mediated I(-) influx into intestinal crypts of CF mice. Am J Physiol. 1999;277:431–444. doi: 10.1152/ajpgi.1999.277.2.G431. [DOI] [PubMed] [Google Scholar]
- 121.Kuyumcu-Martinez M, Belliot G, Sosnovtsev SV, Chang KO, Green KY, Lloyd RE. Calicivirus 3C-like proteinase inhibits cellular translation by cleavage of poly(A)-binding protein. J Virol. 2004;78:8172–8182. doi: 10.1128/JVI.78.15.8172-8182.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ettayebi K, Hardy ME. Norwalk virus nonstructural protein p48 forms a complex with the SNARE regulator VAP-A and prevents cell surface expression of vesicular stomatitis virus G protein. J Virol. 2003;77:11790–11797. doi: 10.1128/JVI.77.21.11790-11797.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Troeger H, Loddenkemper C, Schneider T, Schreier E, Epple HJ, Zeitz M, et al. Structural and functional changes of the duodenum in human norovirus infection. Gut. 2009;58:1070–1077. doi: 10.1136/gut.2008.160150. [DOI] [PubMed] [Google Scholar]
- 124.Sebire NJ, Malone M, Shah N, Anderson G, Gaspar HB, Cubitt WD. Pathology of astrovirus associated diarrhoea in a paediatric bone marrow transplant recipient. J Clin Pathol. 2004;57:1001–1003. doi: 10.1136/jcp.2004.017178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Koci MD, Moser LA, Kelley LA, Larsen D, Brown CC, Schultz-Cherry S. Astrovirus induces diarrhea in the absence of inflammation and cell death. J Virol. 2003;77:11798–11808. doi: 10.1128/JVI.77.21.11798-11808.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Moser LA, Schultz-Cherry S. Suppression of astrovirus replication by an ERK1/2 inhibitor. J Virol. 2008;82:7475–7482. doi: 10.1128/JVI.02193-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bonaparte RS, Hair PS, Banthia D, Marshall DM, Cunnion KM, Krishna NK. Human astrovirus coat protein inhibits serum complement activation via C1, the first component of the classical pathway. J Virol. 2008;82:817–827. doi: 10.1128/JVI.01847-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Moser LA, Carter M, Schultz-Cherry S. Astrovirus increases epithelial barrier permeability independently of viral replication. J Virol. 2007;81:11937–11945. doi: 10.1128/JVI.00942-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Mendez E, Salas-Ocampo E, Arias CF. Caspases mediate processing of the capsid precursor and cell release of human astroviruses. J Virol. 2004;78:8601–8608. doi: 10.1128/JVI.78.16.8601-8608.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Muller N, von Allmen N. Recent insights into the mucosal reactions associated with Giardia lamblia infections. Int J Parasitol. 2005;35:1339–1347. doi: 10.1016/j.ijpara.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 131.D'Anchino M, Orlando D, De Feudis L. Giardia lamblia infections become clinically evident by eliciting symptoms of irritable bowel syndrome. J Infect. 2002;45:169–172. doi: 10.1016/s0163-4453(02)91038-8. [DOI] [PubMed] [Google Scholar]
- 132.Hanevik K, Dizdar V, Langeland N, Hausken T. Development of functional gastrointestinal disorders after Giardia lamblia infection. BMC Gastroenterol. 2009;9:27. doi: 10.1186/1471-230X-9-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Buret AG. Mechanisms of epithelial dysfunction in giardiasis. Gut. 2007;56:316–317. doi: 10.1136/gut.2006.107771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Chin AC, Teoh DA, Scott KG, Meddings JB, Macnaughton WK, Buret AG. Strain-dependent induction of enterocyte apoptosis by Giardia lamblia disrupts epithelial barrier function in a caspase-3-dependent manner. Infect Immun. 2002;70:3673–3680. doi: 10.1128/IAI.70.7.3673-3680.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Buret A, Hardin JA, Olson ME, Gall DG. Pathophysiology of small intestinal malabsorption in gerbils infected with Giardia lamblia. Gastroenterology. 1992;103:506–513. doi: 10.1016/0016-5085(92)90840-u. [DOI] [PubMed] [Google Scholar]
- 136.Cevallos A, Carnaby S, James M, Farthing JG. Small intestinal injury in a neonatal rat model of giardiasis is strain dependent. Gastroenterology. 1995;109:766–773. doi: 10.1016/0016-5085(95)90383-6. [DOI] [PubMed] [Google Scholar]
- 137.Gorowara S, Ganguly NK, Mahajan RC, Walia BN. Study on the mechanism of Giardia lamblia induced diarrhoea in mice. Biochim Biophys Acta. 1992;1138:122–126. doi: 10.1016/0925-4439(92)90051-n. [DOI] [PubMed] [Google Scholar]
- 138.Troeger H, Epple HJ, Schneider T, Wahnschaffe U, Ullrich R, Burchard GD, et al. Effect of chronic Giardia lamblia infection on epithelial transport and barrier function in human duodenum. Gut. 2007;56:328–335. doi: 10.1136/gut.2006.100198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bracha R, Nuchamowitz Y, Mirelman D. Transcriptional silencing of an amoebapore gene in Entamoeba histolytica: molecular analysis and effect on pathogenicity. Eukaryot Cell. 2003;2:295–305. doi: 10.1128/EC.2.2.295-305.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Que X, Reed SL. Cysteine proteinases and the pathogenesis of amebiasis. Clin Microbiol Rev. 2000;13:196–206. doi: 10.1128/cmr.13.2.196-206.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Braga LL, Ninomiya H, McCoy JJ, Eacker S, Wiedmer T, Pham C, et al. Inhibition of the complement membrane attack complex by the galactose-specific adhesion of Entamoeba histolytica. J Clin Invest. 1992;90:1131–1137. doi: 10.1172/JCI115931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Takeuchi A, Phillips BP. Electron microscope studies of experimental Entamoeba histolytica infection in the guinea pig II. Early cellular and vascular changes accompanying invasion of the lamina propria. Virchows Arch B Cell Pathol. 1976;20:1–13. doi: 10.1007/BF02890322. [DOI] [PubMed] [Google Scholar]
- 143.Riahi Y, Siman-Tov R, Ankri S. Molecular cloning, expression and characterization of a serine proteinase inhibitor gene from Entamoeba histolytica. Mol Biochem Parasitol. 2004;133:153–162. doi: 10.1016/j.molbiopara.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 144.Maldonado-Bernal C, Kirschning CJ, Rosenstein Y, Rocha LM, Rios-Sarabia N, Espinosa-Cantellano M, et al. The innate immune response to Entamoeba histolytica lipopeptidophosphoglycan is mediated by toll-like receptors 2 and 4. Parasite Immunol. 2005;27:127–137. doi: 10.1111/j.1365-3024.2005.00754.x. [DOI] [PubMed] [Google Scholar]
- 145.Ivory CP, Prystajecky M, Jobin C, Chadee K. Toll-like receptor 9-dependent macrophage activation by Entamoeba histolytica DNA. Infect Immun. 2008;76:289–297. doi: 10.1128/IAI.01217-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Leroy A, Lauwaet T, De Bruyne G, Cornelissen M, Mareel M. Entamoeba histolytica disturbs the tight junction complex in human enteric T84 cell layers. FASEB J. 2000;14:1139–1146. doi: 10.1096/fasebj.14.9.1139. [DOI] [PubMed] [Google Scholar]
- 147.Lauwaet T, Oliveira MJ, Callewaert B, De Bruyne G, Mareel M, Leroy A. Proteinase inhibitors TPCK and TLCK prevent Entamoeba histolytica induced disturbance of tight junctions and microvilli in enteric cell layers in vitro. Int J Parasitol. 2004;34:785–794. doi: 10.1016/j.ijpara.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 148.McGowan K, Kane A, Asarkof N, Wicks J, Guerina V, Kellum J, et al. Entamoeba histolytica causes intestinal secretion: role of serotonin. Science. 1983;221:762–764. doi: 10.1126/science.6308760. [DOI] [PubMed] [Google Scholar]
- 149.Dey I, Keller K, Belley A, Chadee K. Identification and characterization of a cyclooxygenase-like enzyme from Entamoeba histolytica. Proc Natl Acad Sci USA. 2003;100:13561–13566. doi: 10.1073/pnas.1835863100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Stenson WF, Zhang Z, Riehl T, Stanley SL., Jr Amebic infection in the human colon induces cyclooxygenase-2. Infect Immun. 2001;69:3382–3388. doi: 10.1128/IAI.69.5.3382-3388.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.McGowan K, Piver G, Stoff JS, Donowitz M. Role of prostaglandins and calcium in the effects of Entamoeba histolytica on colonic electrolyte transport. Gastroenterology. 1990;98:873–880. doi: 10.1016/0016-5085(90)90010-x. [DOI] [PubMed] [Google Scholar]