

Summary
Developing small-molecule antagonists against protein-protein interactions will provide powerful tools for mechanistic/functional studies and the discovery of new antibacterials. We have developed a reverse yeast three-hybrid approach that allows high-throughput screening for antagonist peptides against essential protein-protein interactions. We have applied our methodology to the essential bacterial helicase-primase interaction in Bacillus stearothermophilus and isolated a unique antagonist peptide. This peptide binds to the primase, thus excluding the helicase and inhibiting an essential interaction in bacterial DNA replication. We provide proof of principle that our reverse yeast three-hybrid method is a powerful “one-step” screen tool for direct high-throughput antagonist peptide selection against any protein-protein interaction detectable by traditional yeast two-hybrid systems. Such peptides will provide useful “leads” for the development of new antibacterials.
Introduction
Many functions in biology involve large macromolecular assemblies. The ability of individual protein components to interact with each other in a subtle and highly specific manner often defines mechanistic and regulatory aspects of the functional requirements of such assemblies. Characterization of the molecular details that underpin specific protein-protein interactions is a considerable challenge. Traditional structural biology, X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy, and microscopy, provides powerful tools for studying such interactions, but technological and experimental limits have made it difficult to apply such approaches in a generic, high-throughput manner. Instead, biochemical techniques, such as phage-display, yeast two-hybrid (Y2H), and mass spectrometry-based technologies, have been developed as more generic methods [1-4]. Such methods have been used extensively to identify protein-protein interaction interfaces, interacting domains, and small ligands that may mediate such interactions.
The discovery of agonist and/or antagonist peptides that regulate protein-protein interactions is of great importance, not only in understanding structural, regulatory, and mechanistic aspects of macromolecular assemblies, but also in designing new drugs employing combinatorial and bioinformatics approaches [5, 6]. The use of peptide antagonists to disrupt protein-protein interactions has been demonstrated in a variety of systems. Examples include peptides that block IL-1a binding to type I IL-1 receptor [7], insulin-like growth factor 1 binding to its regulatory binding protein, IGFBP-1 [8], the angiogenesis factor VEFGF binding to its cell surface receptor, KDR [9, 10], IgG Fc binding to streptococcal protein A [11], and HIV-1 gp120 binding to CD4 [12]. By comparison, the use of such peptides against DNA replication is less well documented.
To overcome mechanistic and topological problems associated with DNA replication, evolution has created large, complex multiprotein assemblies, like the primosome and the replisome. The functions of these assemblies are dependent on delicate collaborations of their component proteins [13-15]. These collaborations are based upon allosteric protein-protein and protein-DNA interactions that ensure the effective progression of the entire assembly through various functional stages in order to complete the effective and faithful replication of the genetic information. Interference with any of these essential interactions will inevitably result in stalling of DNA replication and, ultimately, cell death. Small molecules and/or peptides may act as antagonists to block such interactions, and may represent a new avenue to explore and develop new antibacterial drugs. Indeed, three very recent examples have demonstrated that a peptide can bind to geminin and suppress DNA replication in HCT116 colon cancer cells by disrupting the geminin-Cdt1 interaction [16], whereas a small molecule can mimic the growth-inhibitory effect of phage polypeptides by disrupting the interaction within a prototypic pair, ORF104 and DnaI (the putative helicase loader) in Staphylococcus aureus [17] and also other small molecules can inhibit the interaction of the herpes simplex virus DNA polymerase and its processivity subunit UL42 thus inhibiting viral replication [18].
In this article, we report the discovery, using a reverse yeast three-hybrid (Y3H) approach, of an antagonist peptide against the essential interaction between the main replicative hexameric helicase, DnaB, and the primase, DnaG, in the Gram-positive Bacillus stearothermophilus. This peptide competes with DnaB for binding to DnaG, thus disrupting an essential bacterial protein-protein interaction during DNA replication and making it a powerful “lead compound” for the development of synthetic peptidomimetics as novel broad-spectrum antibiotics against bacterial DNA replication. This antagonist peptide will also be useful in studying the molecular details of the helicase-primase interaction.
Furthermore, although the potential of Y3H in the analysis of small molecule-protein interactions, as well as its applications in drug discovery, has been emphasized before [19], our data presented here provide proof of principle that the use of the Y3H technology in a reverse manner to screen random peptide libraries is a valuable advance over existing methods. Apart from being a direct screen for isolating antagonist peptides against any protein-protein interaction, it also offers a unique flexibility, allowing the targeting of either partner of a binary protein complex. It can also be used to investigate, very rapidly, the effects of mutations on the activity of the antagonist peptide, and can be easily adapted as a high-throughput screen in a manner similar to other Y2H high-throughput screens to amplify its potential as an industrial method for direct antagonist peptide screening. It offers new opportunities in the field of peptide-based drug discovery against any protein-protein interaction that is detectable by the traditional Y2H system.
Results and Discussion
Antagonist Peptides against the Helicase-Primase Interaction
The helicase (DnaB)-primase (DnaG) interaction in B. stearothermophilus is stable and the complex can be reconstituted and detected from the purified proteins [20]. A C-terminal domain of DnaG, known as P16, can substitute for the full-length DnaG in the complex and interacts with a linker region that connects the N- and C-terminal domains of DnaB to mediate this interaction both structurally and functionally [21]. However, the precise structural details of this interaction still remain unresolved. The stoichiometry of the complex is an unresolved issue, with a DnaB6-DnaG3 model being the more likely [20], but with DnaB6-DnaG2 and DnaB6-DnaG1 complexes also detectable [21].
The interactions of both DnaG and P16 with DnaB can also be detected by traditional Y2H. We devised a novel use of the Y3H technology in a reverse manner to screen a commercial MATCHMAKER random peptide library from Clontech in order to discover antagonist peptides against this essential bacterial protein-protein interaction. Such peptides will provide us with potential leads for the development of drugs that will target bacterial DNA replication.
Using the pBridgeBP16 plasmid that carries both GAL4BD-DnaB and GAL4AD-P16 fusions, we proceeded to test whether we could detect the DnaB-P16 interaction. GAL4BD-DnaB is under the control of the constitutive PADH promoter, whereas GAL4AD-P16 was placed under the control of the PMET25 inducible promoter (Figures 1A–1D). In the absence of methionine, the PMET25 promoter is switched on and GAL4AD-P16 is expressed. We found that the DnaB-P16 interaction was detectable in trp−/met− media using the pBridge-BP16 plasmid (data not shown), indicating that we could now proceed to use the pBridgeBP16 plasmid in a reverse Y3H experiment to screen for antagonist peptides using a random peptide library, as shown in Figure 1E.
Figure 1.
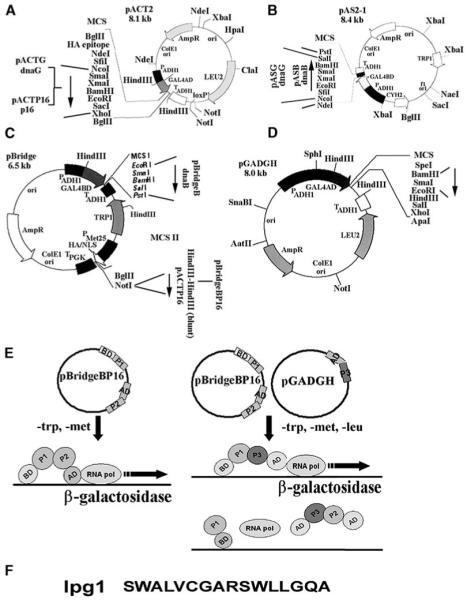
Construction of Plasmids for Y2H and Y3H Experiments
(A) Construction of pACT-based plasmids. The insertion sites of dnaG and p16 genes are indicated.
(B) Construction of pAS-based plasmids. The insertion sites for the dnaG and dnaG genes are indicated.
(C) Construction of pBridge-based plasmids. The insertion sites for the dnaB and p16 genes are indicated.
(D) The pGADGH plasmid carrying the random peptide library at the BamHI-EcoRI, as indicated. In all cases the direction of the inserted genes is indicated by arrows.
(E) Schematic representation of the Y2H system with both proteins (P1, P2) on the same pBridge plasmid (left panel). The plasmid is selected in trp− media. P1 is expressed constitutively, whereas P2 is expressed only in met− media. A schematic representation of the reverse Y3H system is presented in the right panel. The pGADGH plasmid (carrying the peptides, P3) is selected in leu− media and P3 is expressed constitutively. A P2-P3 interaction will give a negative signal, whereas a P1-P3 interaction will give a positive signal. Therefore, if P3 interacts with P2 and disrupts the P1-P2 interaction, it can be detected as a negative signal in the reverse Y3H system.
(F) The sequence of the lpg1 peptide.
We screened approximately 6000 clones from a commercial MATCHMAKER random peptide library from Clontech, cloned into the pGADGH plasmid (Figure 1D). Selection for antagonist peptides was carried out in trp−/met−/leu− media. Potential clones were isolated for antagonist functions from the primary Y3H screen and verified for binding to the primase DnaG (or its P16 helicase-interacting domain) by a secondary Y2H screen (data not shown). One clone was isolated from these screens and sequenced. It was found to code for a unique 16-mer peptide that was named lpg1. The sequence of lpg1 did not resemble any amino acid sequence motifs (or other amino acid sequence parts) of the DnaB sequence and is presented in Figure 1F. We proceeded to test its antagonist function, not only against the DnaB-P16 interaction, but also against the full-length DnaB-DnaG interaction applying again our reverse Y3H approach, and confirmed that, indeed, it inhibited this interaction (Figures 2A–2C). Our data show that lpg1 clearly inhibits the DnaB-DnaG and DnaB-P16 interactions in our reverse Y3H system.
Figure 2.

Lpg1 Antagonist Activity in Yeast Hybrid Systems
Verification of the antagonist activity of lpg1 against the DnaB-DnaG (A) and DnaB-P16 (B) interactions in the Y3H system. In both panels, positive control experiments for the DnaB-DnaG and DnaB-P16 interactions are indicated by the blue coloration, as indicated. Additional controls for the traditional positive interaction between the SV40 large T antigen and p53, and the negative control between SV40 large T antigen and DnaB, are shown in (C). Verification of the lpg1-DnaG interaction by the filter-lift method in a traditional Y2H system is shown in (D) (yeast patches) and (E) (filter lifts). Lpg1 exhibits clear DnaG binding activity. The positive control indicates the DnaB-DnaG interaction and the negative control indicates no interaction between DnaB and SV40 large T antigen.
The configuration of our reverse Y3H system ensured that the GAL4AD-peptide would interact exclusively with the GAL4AD-P16 for antagonist activity to be apparent in our screens (Figure 1E). An interaction between GAL4AD-peptide and GAL4BD-DnaB will simply result in β-galactosidase expression. Our system also offers the flexibility for selecting antagonist peptides that will interact exclusively with the other partner in the complex. For example, a GAL4BD-peptide fusion would result in negative signal only if it interacts with GAL4BD-DnaB, whereas, in this case, an interaction with GAL4AD-P16 will result in positive signal. Therefore, the antagonist activity of the isolated lpg1 peptide must be due to specific interactions with P16 (or DnaG) that exclude DnaB from the complex. To verify this we confirmed the lpg1-DnaG interaction with the conventional Y2H system, using the pASG and pGADGH-lpg1 plasmids in tpr−/leu− media. As expected, lpg1 was found to interact with DnaG (Figures 2D and 2E). In contrast, lpg1 did not interact with DnaB when tested in a similar manner using the pASB instead of the pASG plasmid (data not shown).
Antagonist Activity In Vitro
Confirmation of the antagonist activity of lpg1 against the DnaB-DnaG complex formation was verified by examining its effect on the DnaG-mediated stimulation of the ATPase activity of DnaB in vitro. Purified B. stearothermophilus DnaB and DnaG proteins form a stable complex that can be isolated by gel filtration [20, 21]. The ATPase activity of DnaB is stimulated significantly when in complex with DnaG [20, 21]. However, the DnaB-DnaG system is particularly complex, because (1) the stoichiometry of the complex is not absolute and (2) the ATPase kinetic behavior of DnaB is still unresolved. Although the majority of the complexes exist in a DnaB6-DnaG3 stoichiometry, a significant percentage of complexes with DnaB6-DnaG2 and DnaB6-DnaG1 stoichiometry also exist [20, 21]. The complexity of this system is amplified further by the ATPase kinetic behavior of DnaB. It does not obey first order Michaelis Menten kinetics [20, 21] and the relative kinetic behavior of the heterogeneous complexes is far from certain.
Despite the complexity of this system, we argued that if lpg1 reversibly competes with DnaB for binding to DnaG, the stimulatory effect on the DnaB ATPase activity by DnaG should be inhibited in the presence of lpg1, because lpg1 will bind to DnaG and prevent the DnaB-DnaG complex formation. Obviously, we cannot be certain whether all three DnaG molecules need to be disengaged from the complex or if only one or two DnaG molecules need to be disengaged before inhibition of the stimulatory effect can be observed. Indeed DnaB6-DnaG2 and DnaB6-DnaG1 complexes may still exhibit stimulated ATPase activity. Despite these complications, we would anticipate that removing all three DnaG molecules from the complex will lead to inhibition of the DnaG-mediated stimulatory effect on DnaB ATPase activity.
We tested this hypothesis employing two different ATPase assays: a continuous NADH-linked assay that indirectly measures the rate of ATP hydrolysis by monitoring the oxidation of NADH to NAD+ and an end-point TLC-based assay that directly measures the conversion of ATP to ADP and inorganic phosphate (Pi). Both assays indicated that lpg1 does, indeed, inhibit the DnaG-mediated stimulation of the DnaB ATPase activity. In fact, in the TLC-based assay, lpg1 exhibited good inhibitory activity from 100- to 3.12-fold molar excess over DnaG and appeared to lose its inhibitory effect if diluted further at 1.5-fold molar excess or at half the concentration of DnaG (Figure 3A). We should emphasize that the TLC-based assay provides a qualitative rather than an absolute quantitative assay. In fact in this assay the competitive inhibitory effect of lpg1 is likely to be magnified. With this assay we are measuring the percentage of ATP that has been hydrolysed in 10 min. As ATP is hydrolysed its concentration is reduced with time thus affecting the complex kinetic behavior of DnaB. Moreover, since we are not measuring a continuous ATPase rate the lpg1 dose-response in the former experiment will appear better than the robust end-point assay. Lpg1 actually reduced the stimulatory effect of DnaG on the DnaB ATPase activity by 30%–50% as determined by the TLC-based end-point ATPase assay. Interestingly, a truncated version of lpg1 missing two amino acid residues ([des-Ser1-Trp2] lpg1) from its N terminus exhibited significantly reduced antagonist activity in the same TLC-based assay compared to the full length lpg1 (Figure 3B). It inhibited marginally the stimulatory effect of DnaG by ~10% (compare Figures 3A and 3B).
Figure 3.
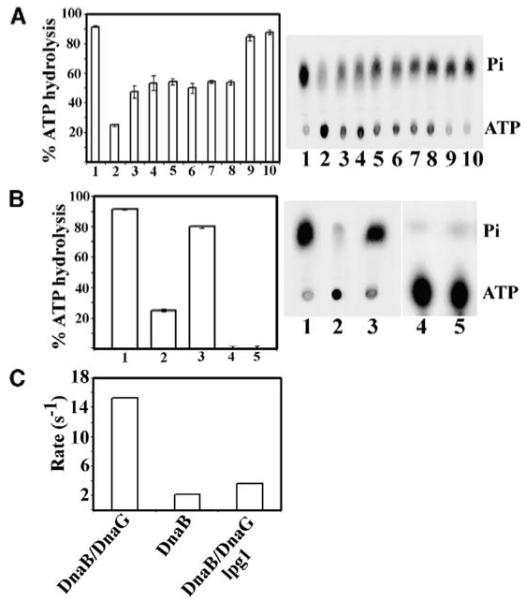
Thin-Layer Chromatography-Based ATPase Assays
(A) A TLC-based end-point ATPase assay showing the inhibitory effect of lpg1 at different concentrations: 100-, 50-, 25-, 12.5-, 6.12-, 3.12-, 1.5-, and 0.5-fold molar excess of lpg1 over DnaG as indicated in bars 3–10, respectively. Bars 1 and 2 indicate the ATPase activities of the DnaB-DnaG mixture and the DnaB protein alone, respectively. Triplicate experiments were carried out, but only one representative TLC plate is shown. The standard deviation is indicated by error bars.
(B) The results from a similar assay, but using 100-fold molar excess of the N-terminally truncated version (des-Ser1Trp2)lpg1 of lpg1 missing the first two N-terminal residues (bar 3). Bar 1 and 2 indicate the ATPase activities of the DnaB-DnaG mixture and the DnaB protein alone, respectively. Bars 4 and 5 indicate controls of DnaG and ATP with no protein, respectively. Experiments were carried out in triplicate and error bars indicate the standard deviation.
(C) A synopsis of the steady-state ATPase data collected using the continuous NADH-linked ATPase assay. Bar 1 indicates the ATPase rate of the DnaB (9.2 nM hexamer)-DnaG (55.2 nM monomer) mixture, bar 2 the rate of the DnaB (9.2 nM hexamer) alone, and bar 3 the rate of the DnaB (9.2 nM hexamer)-DnaG (55.2 nM monomer) in the presence of 5.52 μM of lpg1. All reactions were carried out at 2.0 mM (ATP).
The antagonist effect of lpg1 was also evident in the continuous NADH-linked assay that measures the steady-state rate of ATP hydrolysis. This assay measures real-time continuous rate because the ATP is continuously regenerated. The ATPase profile of DnaB with this assay in the presence and absence of DnaG is complicated, with an initial phase that resembles first order kinetics from 0 to 2.0 mM ATP, followed by an increase between 2.0 and 3.5 mM, and a sharp inhibition at higher concentrations of ATP [20, 21, 24]. This complex kinetic behavior is still unresolved, but has been attributed to negative cooperativity of the monomers around the hexameric ring [20]. However, this suggestion is still purely speculative and the absolute kinetic mechanism behind this complex behavior is still a mystery. Initially, we examined the effect of lpg1 on the ATPase activity at a fixed 2.0 mM ATP and found that the ATPase rate of the DnaB-DnaG mixture was reduced from approximately 15 · s−1 to less than 4 · s−1, comparable to the rate for DnaB alone, approximately 2 · s−1 (Figure 3C).
In order to obtain more reliable quantitative information for the “dose response” of the lpg1 peptide, we investigated the ATPase profile of the DnaB and DnaB-DnaG mixtures in the presence and absence of different concentrations of lpg1 at varying concentrations of ATP up to 2 mM, corresponding to the initial phase of the complicated ATPase profile of DnaB (Figure 4). The presence of DnaG in the reaction mixture in the absence of lpg1 results in a marked stimulation of the DnaB ATPase activity (Figure 4A). Significantly, inclusion of a large excess of the truncated ([des-Ser1-Trp2] lpg1) or mutant ([Ala2]lpg1) peptides resulted only in marginal differences in the ATPase profile without an obvious suppression of the DnaG-mediated stimulatory effect (Figure 4B). In contrast, the inclusion of lpg1 in the reaction mixture resulted in a marked suppression of the DnaG-mediated stimulatory effect, suggesting that it inhibits the DnaB-DnaG complex formation (Figure 4C).
Figure 4.

NADH-Linked ATPase Assays
(A) A comparison of the ATPase profile of the DnaB hexamer (9.2 nM) in the presence (closed triangles) and absence (open squares) of DnaG (55.2 nM) from 0 to 2.0 mM (ATP).
(B) The ATPase profile of the DnaB hexamer (9.2 nM) in the presence of DnaG (55.2 nM) and 27.6 μM of either the truncated lpg1 (open squares) or the mutant lpg1 (closed triangles).
(C) The ATPase profile of the DnaB hexamer (9.2 nM) in the presence of DnaG (55.2 nM) and varying concentrations of lpg1, 2.76 μM (closed circles), 5.52 μM (open triangles), 11 μM (closed squares), and 27.6 μM (open squares) corresponding to 50, 100, 200, and 500 molar excess of lpg1 over DnaG, respectively. All the graphs have the same scale axes for direct comparison, and error bars indicate the standard deviation from duplicate experiments.
Because of the complexity of the actual stoichiometry of the DnaB-DnaG mixtures and the presence of heterogeneous complexes with either three, two, or one DnaG molecule per DnaB hexamer in all our ATPase assays, we used an excess of DnaG (55.2 nM) over the DnaB hexamer (9.2 nM), but still cannot ensure absolutely that all the complexes will be of the DnaB6-DnaG3 stoichiometry. Also, any excess of free DnaG is likely to sequester lpg1 from the solution. The complexity of this system makes it impossible to engage in the simple protein-ligand type of analysis for the determination of a meaningful IC50 dose response for lpg1.
Despite these problems, we carried out a semiquantitative analysis of the lpg1-mediated inhibition, and established that, under our experimental conditions, there was no effect observed at 50 molar excess of lpg1 (2.76 μM) over the DnaG (55.2 nM) in the reaction mixture, whereas the inhibitory effect gradually increased as we raised the concentration of lpg1 to 5.52, 11, and 27.6 μM, corresponding to 100, 200, and 500 molar excess of lpg1 over the DnaG in the reaction mixture, respectively (Figure 4C).
We also verified direct binding of lpg1 to DnaG by isothermal titration calorimetry (ITC), but the interaction was very strong and an accurate KB value could not be calculated (in the Supplemental Data available with this article online). With tight binding (KB > 109 M−1), ITC titrations lose their curvature and KB cannot be measured accurately [25].
The N Terminus of lpg1 Is Critical for Antagonist Activity
In our search for a minimal sequence within lpg1 that might be critical for its antagonist activity, we discovered that a truncated lpg1 missing the first two N-terminal serine and tryptophan residues has lost most of its antagonist activity. In fact, it suppresses the DnaG-stimulatory effect on the DnaB ATPase activity only marginally in both TLC-based (Figure 3B) and NADH-linked ATPase assays (Figure 4B). Interestingly, when we substituted the tryptophan residue close to the N terminus for an alanine we found that the mutant ([Ala2]lpg1) has also lost most of its antagonist activity, with only very weak activity exhibited by this mutant lpg1 in the NADH-linked assay (Figure 4B). In fact, (Ala2)lpg1 was marginally worse than the truncated (des-Ser1-Trp2) lpg1 (compare the two curves in Figure 4B). The fact that both (Ala2)lpg1 and (des-Ser1-Trp2)lpg1 peptides exhibit loss of antagonist activity indicates that the antagonist activity of lpg1 is specific and that the N-terminal segment of lpg1 is critical. A subsequent search through the protein database (PDB) revealed that lpg1 has sequence homology to helices found in the mammalian mevalonate kinase (pdb: 1KVK) and in a viral polymerase (pdb: 1MUK) (Figures 5A and 5B) [26, 27]. Interestingly, in both cases the homology was confined to the N-terminal half of the lpg1 peptide (Figure 5C), indicating that this region of the lpg1 peptide has the propensity to adopt a helical secondary structure. The homologous helices in these two proteins do not seem to have any functional significance and only in the case of the mammalian mevalonate kinase is the homologous helix α8 close to a classic four helix bundle, composed of helices α9 and α10, which constitutes the dimerization interface of this protein [26]. Detailed structural analysis will be required to establish whether lpg1 does indeed adopt a stable secondary structure and whether the integrity of this structure is essential for antagonist activity.
Figure 5.
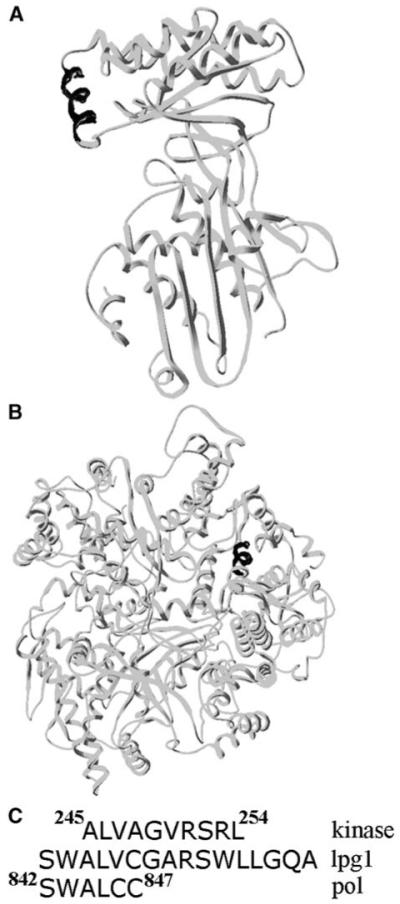
Sequence Similarities from the Structural Database
The N-terminal half of the lpg1 peptide is homologous to helices found in the mammalian mevalonate kinase (A) and in the reovirus RNA polymerase lambda3 (B). In both cases the relevant helices are indicated in black. The actual sequences and their homology to the lpg1 sequence are shown in (C).
Lpg1 Inhibits the Stimulatory Effect of DnaG on the DnaB Helicase Activity
Having established that lpg1 inhibits the stimulatory effect of DnaG on the ATPase activity of DnaB, we then examined the effect on the helicase activity of DnaB. Using a well-established in vitro helicase assay, we showed that lpg1 does indeed inhibit the stimulatory effect of DnaG on the helicase activity of DnaB (Figure 6). DnaB has a relatively low helicase activity level that is stimulated significantly by DnaG (Figure 6A). In the presence of an excess of lpg1, the stimulatory effect of DnaG is inhibited significantly, whereas no significant effect on the activity of DnaB alone is observed (Figure 6B). These data are consistent with our ATPase data and, together, provide strong evidence that lpg1 does indeed act as an antagonist inhibitor of the DnaG-meidated stimulation of the DnaB activity.
Figure 6.
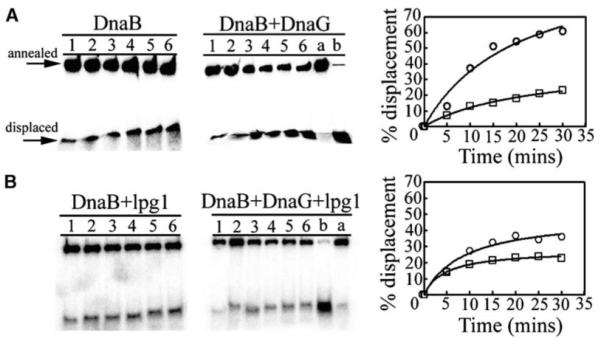
Effects on the Helicase Activity
DnaG stimulates the helicase activity of DnaB (A), whereas lpg1 inhibits the stimulatory effect of DnaG (B). Lanes 1–6 represent helicase reaction time points 5, 10, 15, 20, 25, and 30 min, whereas lanes a and b represent annealed and boiled controls, respectively. The actual gels labeled appropriately are shown on the left and the quantitative data are shown as graphs on the right. In (A), data represent helicase reactions of DnaB in the presence (circles) and absence (squares) of DnaG. In (B), data represent helicase reactions of DnaB + lpg1 in the presence (circles) and absence (squares) of DnaG. Annealed and displaced oligonucleotide is indicated with labeled arrows for clarity.
Lpg1 Disrupts the DnaB-DnaG Complex Formation In Vitro
The B. stearothermophilus DnaB-DnaG complex, in contrast to that of Escherichia coli, is stable enough to be detected by simply mixing purified DnaB and DnaG proteins together and then resolving the mixture through a gel filtration column (Figure 7A) [20, 21]. Three DnaG molecules are believed to interact with a single DnaB hexamer, although minor species of DnaB6-DnaG2 and DnaB6-DnaG1 complexes may also exist [20, 21]. We investigated the direct effect of lpg1 on the complex by gel filtration. The complex elutes from the column much earlier (10.21 ml elution volume) than DnaG (13.8 ml elution volume). The resolving power of the Superdex S-200 column is insufficient to resolve clearly the complex (MW, 504 kDa) from the DnaB hexamer (303 kDa). We discovered that adding an excess of lpg1 (175 μM) in the DnaB (0.5 μM hexamer)-DnaG (0.67 μM monomer) mixture resulted in the release of a substantial amount of DnaG from the complex, confirming that lpg1 competes with DnaB for binding to DnaG. The antagonist activity of lpg1 is apparent in Figure 7B, with an increase of DnaG eluted free from the complex and also a comparative decrease in the amount of DnaG remaining in the complex (compare relative amounts of DnaB and DnaG in the complex in Figure 7A, lanes 1–3 and Figure 7B, lanes 1–4). Lpg1 has to compete with the DnaB helicase and displace it from the preformed DnaB-DnaG complex. Although the affinity of lpg1 for DnaG is in the nanomolar region (Supplemental Data), apparently DnaB also binds to DnaG with very high affinity, and not all of the DnaB could be displaced from DnaG by lpg1. Also, as the proteins in the mixture are loaded onto the top of the column, they begin to separate almost immediately. Because of its small size, excess lpg1 starts separating from the rest of the proteins almost immediately. The remaining DnaB-DnaG complex moves down the column quicker, and some of the DnaB will remain bound to DnaG. Thus, only partial, but nevertheless significant, displacement of DnaB from the complex by lpg1 is observed.
Figure 7.
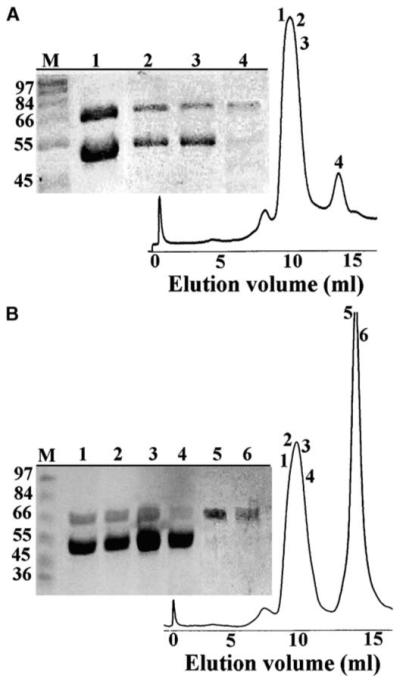
Lpg1 Competes with the Helicase for Binding to the Primase
Analytical gel filtration experiments of the DnaB (0.5 μM hexamers)-DnaG (0.67 μM monomers) mixtures in the absence (A) and presence (B) of lpg1 (175 μM). Samples from the peak fractions were analyzed by SDS-PAGE. The numbers of the lanes in the polyacrylamide gels correspond to the numbers of the peak fractions, as indicated. Lane M shows molecular weight markers, as indicated. lpg1 causes the dissociation of DnaG (67 kDa) from DnaB (50.5 kDa) in a competitive manner.
Our Y3H, Y2H, and in vitro biochemical data for the lpg1 peptide indicate that it competitively binds to DnaG and disengages it from the DnaB-DnaG complex. Because of the complexity of the DnaB-DnaG interaction and the unresolved ATPase kinetics of DnaB, it was not possible to obtain reliable quantitative information for the dose response of lpg1 in this system in a manner analogous to a traditional simple peptide-ligand interaction. However, our combined data provide proof of principle that the novel use of reverse Y3H technology, in screening random peptide libraries for antagonist peptides against protein-protein interactions detectable by traditional Y2H, is a viable method.
Significance
Overall, our data provide proof of principle and show that our reverse Y3H approach can directly select antagonist peptides against a protein-protein interaction that is detectable by the traditional Y2H system. We targeted the bacterial helicase-primase interaction and isolated an antagonist peptide that binds to the primase and disrupts the helicase-primase complex. Despite the complexity of the DnaB-DnaG system, we verified its antagonist activity using a range of in vitro experiments. We have also shown that both a truncated and a point mutant version of this peptide were inactive. Our approach can be adapted to a rapid, high-throughput direct screening assay for the isolation of antagonist peptides against either partner of a binary protein complex, in a similar manner to a traditional Y2H high-throughput screen [28, 29], and can be applied to any protein-protein interaction system that can be detected by the traditional Y2H method. It is a significant advance over existing methods because: (1) it selects antagonist peptides that bind directly at the protein-protein interaction interface (direct antagonists) or peptides that act allosterically from a distant binding site (allosteric antagonists); (2) it allows the specificity of targeting exclusively either partner of a binary protein complex; (3) it can be adapted as a high-throughput method; and (4) once the primary antagonist peptide has been isolated, it can also be used to rapidly investigate a large number of mutant antagonist peptides for improved activity. It will provide a powerful tool for the development of peptide-based drugs that target essential protein-protein interactions in a variety of systems. Isolated antagonist peptides can be used as “chemical seeds” for further development of chemical compounds that mimic their antagonist action and also to probe directly the structural/functional molecular aspects of protein-protein interactions.
Experimental Procedures
Plasmid Constructions for Y2H and Y3H
Our Y2H and 32H methodologies were based upon the MATCH-MAKER Two-Hybrid system 2 from Clontech. The dnaB, dnaG genes, as well as the p16 C-terminal fragment of dnaG coding for a 16 kDa polypeptide (P16) that interacts with DnaB, were all subcloned from their pET expression vectors [20] into the pACT2 and pAS2-1 vectors, as shown in Figures 1A and 1B.
The dnaB gene was cloned into the NcoI-PstI sites of pAS2-1 to give pASB (Figure 1B) that codes for the GAL4 DNA binding domain (BD)-DnaB fusion protein, under the control of the PADH1 constitutive promoter whereas the dnaG and p16 genes were subcloned into the NcoI-XhoI sites of pACT2 to produce the pACTG and pACTP16 plasmids, respectively (Figure 1A). Both pACTG and pACTP16 code for the GAL4 activation domain (AD)-DnaG and AD-P16 fusion proteins, respectively, both under the control of the PADH1 promoter. The dnaG gene was also subcloned as an NcoI-XhoI fragment from the pET21d plasmid [20] into the NcoI-SalI sites of pAS2-1 (XhoI and SalI sites are compatible), to produce the pASG plasmid coding for the GAL4BD-DnaG fusion protein under the control of the PADH1 promoter (Figure 1B).
The dnaB gene was subcloned as NdeI(blunt)-PstI fragment from pASB into the EcoRI(blunt)-PstI sites of pBridge to produce the pBridgeB plasmid, coding for a GALBD-DnaB fusion protein and placing it under the control of the constitutive PADH1 promoter (Figure 1C). The p16 gene was subcloned as HindIII(blunt)-HindIII(blunt) fragment into the NotI(blunt) site of pBridgeB placing the GAL4AD-P16 fusion protein under the control of the inducible PMET25 promoter, to give the pBridgeBP16 plasmid (Figure 1C). The correct orientation was verified by appropriate restriction endonuclease mapping. The dnaG gene was subcloned as a BglII-BglII fragment from pACTG into the same sites of pBridgeBP16, thus replacing the p16 gene to form the pBridgeBG plasmid, placing the GAL4AD-DnaG fusion protein under the control of the PMET25 promoter. Correct orientation was verified by appropriate restriction endonuclease mapping.
The random peptide library was purchased from Clontech and was already cloned into the BamHI-EcoRI sites of pGADGH (Figure 1D), under the control of the constitutive PADH1 promoter, with stop codons in all three frames immediately after the EcoRI site. The library contains 106 independent random peptide clones coding for random peptides of 16 amino acids maximum length fused to GAL4-AD.
Growth Media for Yeast
The AH109 Saccharomyces cerevisiae yeast strain was used in this study. The components of the yeast complete medium (YPD) and the different synthetic dropout (SD) media, as well as the various supplements, were purchased from Becton Dickinson and Co. or Sigma and prepared according to the manufacturer’s instructions. Nutritionally selective media were prepared by omitting leucine, tryptophan, and methionine (or combinations of these) from the growth media. E. coli XL1-Blue bacteria were cultured in Luria-Bertani (LB) media in the presence of tetracycline.
Yeast Transformations
Yeast cells were made either electrocompetent or chemically-competent for transformations with DNA. For electrocompetent yeast preparations, cells were cultured first on a YPD plate at 30°C for 3 days. Single colonies were used to inoculate liquid YPD cultures that were incubated at 30°C overnight with vigorous shaking. Yeast cells were collected by centrifugation and washed repeatedly in filtered-sterilized, ice-clod 1 M sorbitol. Finally, yeast cells were resuspended in 200 μl of 1 M sorbitol and used for transformation by electroporation with a MicroPulser (Bio-Rad), according to the manufacturer’s instructions.
For chemically-competent yeast, single colonies were isolated, as described above, and used to inoculate liquid YPD cultures that were incubated at 30°C overnight with vigorous shaking. The OD600 of the cultures were measured and a volume containing approximately 2.5 × 108 cells was collected and brought up to 50 ml with prewarmed YPD and incubated for a further 4 hr at 30°C under shaking. Yeast cells were then collected by centrifugation, washed three times in double distilled (dd) H2O, resuspended in 1 ml 100 mM LiOAc, and incubated for 10 min at 30°C. Aliquots of 100 μl were used, and from each aliquot, cells were collected by centrifugation. The supernatant was removed and 240 μl 50 w/v PEG, 36 μl of 1 M LiOAc, 50 μl of salmon sperm DNA, 2 μl of plamid DNA, and 32 μl dd H2O were added, in that order. The cells were vortexed vigorously and incubated for 30 min at 30°C. They were then heat shocked at 42°C for 30 min before being collected by centrifugation. The supernatant was removed and the cells were resuspended in 1 ml dd H2O and then loaded onto plates with appropriate SD media.
Y2H and Y3H Screening
Y2H and Y3H screening were carried out by either the agarose overlay [22] or filter-lift methods according to the yeast protocols handbook from Clontech. The pBridgeBP16 plasmid was cotransformed with a random peptide library cloned into pGADGH plasmid. Following transformation, double selective plates were incubated at 30°C for 36 hr. A typical transformation yielded approximately 2000 colonies. An agarose overlay assay was performed, as described previously [22]. This allowed the rapid identification of potential positive white colonies in which the protein-protein interaction appeared to be blocked, thus preventing β-galactosidase activity. Such colonies typically would fail to develop blue coloration overnight. These colonies were then patched out on to double selective plates, cultured for 36 hr at 30°C, and selected again for absent β-galactosidase activity. Colonies that exhibited no β-galactosidase activity were then cultured and plasmid DNA coding for the interfering peptide was prepared. As a further control, a yeast strain containing the pASG plasmid was transformed with the plasmid pGADGH coding for the potentially interfering peptide in order to confirm the interaction of the peptide with its target protein, DnaG.
Peptide Synthesis
Peptide synthesis was carried out using the standard fluoren-9-ylmethoxycarbonyl (Fmoc) solid-phase approach [23] on a 4-[2′, 4′-dimethoxyphenyl(amino)methyl]phenoxyacetamido methyl Novagel (Rink Amide Novagel, 100–200 mesh, 1% crosslinking, 0.61 mmol · g−1) resin. The peptide assembly was achieved in a stepwise manner, in which amino acid residues were incorporated by activation using N-[(dimethylamino)-1H-1,2,3-triazolo[4,5-b]pyridine-1 ylmethylene]-N-methylmethan-aminium hexafluorophosphate N-oxide:7-aza-1-hydroxybenzotriazole:N,N-diisopropylethylamine (HATU:HOAt:DIEA, 1.0:0.5:2.0) in N,N-dimethylformamide (DMF). Exceptionally, the Cys residue was installed by activation using N,N-diisopropylcarbodiimide:HOAt (1:1) in DMF. A 4-fold molar excess of the activated Fmoc-amino acids was used throughout the synthesis and double-couplings (6 hr followed by 18 hr reaction times) were typically required. A solution of 20% (v/v) piperidine in DMF was applied for sequential Fmoc-deprotection.
Following solid-phase peptide assembly, the resin-supported product was collected and dried. This material was then treated with the mixture trifluoroacetic acid (TFA): H2O:triisopropylsilane: ethyl methyl sulfide (90:5:3:2 v/v, 10 ml) for 4 hr at ambient temperature. The reaction mixture was filtered and the filtrate was evaporated to dryness in vacuo to yield, following trituration with diethyl ether, the peptide amide as beige solid, which was dissolved in water (5 ml) and lyophilized. The desired peptide amide was then purified by reversed-phase HPLC on a Kromasil C8 column (150 × 10 mm) at 4.5 ml · min−1. The eluents were: A, 0.06% TFA in water, and B, 0.06% TFA in 90% aqueous acetonitrile. The elution profile was 20%–40% B over a 12 min period, in which the required peptide amide eluted at 9.7 min.
Electrospray-high resolution mass spectrometry (ES-HRMS) analysis (Micromass LCT) of the purified peptide showed the presence of the expected molecular ion (calculated for C78 H122N23O19S [MH+], 1718.0103; found, 1718.7845). Lpg1 was dissolved as a 10 mg/ml stock solution in neat DMSO and then brought down to 1 mg/ml in 10% v/v DMSO by the dropwise addition of dH2O. Aliquots were frozen in liquid N2, stored at −80°C, and used as required.
Both the truncated peptide, (des-Ser1,Trp2)lpg1, and the mutant peptide, (Ala2)lpg1, were also synthesized and purified using the approach outlined above. When analyzed by ES-HRMS, the purified 14-mer peptide showed the presence of the required molecular ion (calculated MH+, 1443.7158; found, 1443.9214 for [des-Ser1-Trp2]lpg1; and calculated MH+, 1602.8784; found 1602.8606 for [Ala2]lpg1). Both peptides were readily soluble as 1 mg/ml stock solutions in dH2O. Aliquots were frozen in liquid N2, stored at −80°C, and used as required.
Protein Purifications
DnaB and DnaG proteins were purified and quantitated as described previously [21].
ATPase Assays and Analytical Gel Filtration
The DNA-independent ATPase activity of DnaB was assayed using either TLC-based or NADH-linked assays.
The end-point TLC-based ATP hydrolysis assay was carried out in 20 mM Tris (pH 7.5), 50 mM NaCl, 10 mM MgCl2, 2.5 mM EDTA, 5 mM DTT, 2 mM ATP, 5 pM of 32P-labeled ATP, 0.6 μM DnaB (referring to hexamers), and in the presence or absence of 1.6 μM DnaG, at 37°C for 10 min. Reactions were also carried out in the presence or absence of lpg1 at various concentrations as indicated in the figure legends. All reactions were terminated by the addition of 50 mM EDTA and run through TLC plates in 1 M formamide and 0.5 M LiCl for 1 hr. TLC plates were visualized and analyzed using a phosphorimager. All reactions were carried out in triplicate with average values plotted.
The continuous NADH-linked ATPase assay was carried out as described previously for the DnaB-DnaG complex [21] in the presence or absence of varying concentrations of lpg1 over DnaG. All reactions were carried out in triplicate and the average values were plotted. All ATPase rates were defined as molecules of ATP hydrolyzed per second per hexamer of DnaB. We should emphasize here that it is unlikely that all monomers in the hexameric complex will be hydrolyzing ATP at the same rate. Also, because the stock lpg1 peptide was dissolved in 10% v/v DMSO, we investigated the effect of increasing concentrations of DMSO on the ATPase activity in both assays and found that, up to 50% v/v, there was no detectable effect on the ATPase activity (data not shown). DnaG does not exhibit an ATPase activity ([20] and Figure 3B) and synthesizes primers only in the presence of an appropriate ssDNA substrate (for a review, see article by Frick and Richardson [30]).
Analytical gel filtration experiments were carried out using a Superdex S-200 gel filtration column (Amersham Pharmacia Biotech) as described previously [20, 21], using 0.5 μM DnaB hexamer, 0.67 μM DnaG, and 175 μM lpg1. In brief, DnaB and DnaG were mixed and left for 10 min on ice to enable complex formation, at which point lpg1 was added, and the mixture of proteins was left for a further 5 min before being loaded onto the sizing column.
ITC
ITC experiments were carried out with a VP-ITC Microcalorimeter and data were analyzed with the associated Origin software. Typically, the appropriate cell was filled with 1.4 ml degassed solution of DnaG (16.4 μM) in 50 mM Tris (pH 7.4), 100 mM NaCl, 10% v/v DMSO. Samples (5 μl) from a 300 μl degassed solution of lpg1 (0.45 mM in 10% v/v DMSO) peptide in the same buffer were injected into the DnaG solution at 240 s intervals and data were collected after each injection. A comparative reference control experiment with 10% v/v DMSO was also carried out, and the reference data were subtracted from the experimental data.
Helicase Assays
Helicase assays were carried out using a well-established in vitro assay system [20, 21, 24, 31]. A radiolabeled oligonucleotide (5′-GTT ATTGCATGAAAGCCCGGCTGACTCTAGAGGATCCCCGGGTACGTTATTGCATGAAAGCCCGGCTG-3′) was annealed onto ssM13mp18 DNA to produce a 3′-5′-tailed DNA substrate. One molecule of DNA substrate is defined as one molecule of ssM13mp18 DNA with one molecule of oligonucleotide annealed to it. DnaB (25 nM) was incubated in the helicase reaction mixture (20 mM Tris [pH 7.5], 20 mM MgCl2, 10% v/v glycerol, 5 mM DTT, and 0.5 nM DNA substrate) at 37°C for 5 min in the presence or absence of 150 nM DnaG and/or 12.5 μM lpg1, as appropriate. Reactions were initiated with the addition of 3 mM ATP and terminated at 5 min intervals by the addition of helicase stop-buffer (0.4% w/v SDS, 40 mM EDTA, 8% v/v glycerol, 0.1% w/v bromophenol blue). Displaced oligonucleotide was separated from annealed oligonucleotide through a 10% nondenaturing polyacrylamide gel at constant voltage (100 V). Gels were dried and quantitative analysis was performed using a phosporimager and associated software (Molecular Imager FX, Bio-Rad). Helicase activity was defined as a percentage of radiolabeled oligonucleotide displaced from M13mp18 DNA.
Supplementary Material
Acknowledgments
We thank Ian Bryenton and Abbigail Coke for their valuable contributions at various stages of this project and Nikos Kouvatsos for helping us with the ITC experiments. This work was supported by a Biotechnology and Biological Sciences Research Council grant (42/B15519) to P.S. and a Medical Research Council grant (G9219778) to W.C.C.
Footnotes
Isothermal titration calorimetry data are available at http://www.chembiol.com/cgi/content/full/12/5/595/DC1/.
References
- 1.Sidhu SS, Badre GD, Boone C. Functional genomics of intracellular peptide recognition domains with combinatorial biology methods. Curr. Opin. Struct. Biol. 2003;7:97–102. doi: 10.1016/s1367-5931(02)00011-x. [DOI] [PubMed] [Google Scholar]
- 2.Vidal M, Legrain P. Yeast forward and reverse ‘n’-hybrid systems. Nucleic Acids Res. 1999;27:919–929. doi: 10.1093/nar/27.4.919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kelleher NL. From primary structure to function: biological insights from large-molecule mass spectra. Chem. Biol. 2000;7:R37–R45. doi: 10.1016/s1074-5521(00)00081-8. [DOI] [PubMed] [Google Scholar]
- 4.Fang Y, Macool DJ, Xue Z, Heppard EP, Hainey CF, Tingey SV, Miao GH. Development of a high-throughput yeast two-hybrid screening system to study protein-protein interactions in plants. Mol. Genet. Genomics. 2002;267:142–153. doi: 10.1007/s00438-002-0656-7. [DOI] [PubMed] [Google Scholar]
- 5.Cochran A. Protein-protein interfaces: mimics and inhibitors. Curr. Opin. Chem. Biol. 2001;5:654–659. doi: 10.1016/s1367-5931(01)00262-9. [DOI] [PubMed] [Google Scholar]
- 6.Turk B, Cantley LC. Peptide libraries: at the crossroads of proteomics and bioinformatics. Curr. Opin. Chem. Biol. 2003;7:84–90. doi: 10.1016/s1367-5931(02)00004-2. [DOI] [PubMed] [Google Scholar]
- 7.Yanofsky SD, Baldwin DN, Butler JH, Holden FR, Jacobs JW, Balasubramanian P, Chinn JP, Cwirla SE, Petrs-Bhatt E, Whitehorn EA, et al. High affinity type I interleukin 1 receptor antagonists discovered by screening recombinant peptide libraries. Proc. Natl. Acad. Sci. USA. 1996;93:7381–7386. doi: 10.1073/pnas.93.14.7381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lowman HB, Chen YM, Skelton NJ, Mortensen DL, Tomlinson EE, Sadick MD, Robinson ICAF, Clark RG. Molecular mimics of insulin-like growth factor 1 (IGF-1) for inhibiting IGF-1:IGF-binding protein interactions. Biochemistry. 1998;37:8870–8878. doi: 10.1021/bi980426e. [DOI] [PubMed] [Google Scholar]
- 9.Fairbrother WJ, Hans WC, Cochran AG, Fuh G, Keenan CJ, Quan C, Shriver SK, Tom JYK, Wells JA, Cunningham BC. Novel peptides selected to bind vascular endothelial growth factor target the receptor-binding site. Biochemistry. 1998;37:17754–17764. doi: 10.1021/bi981931e. [DOI] [PubMed] [Google Scholar]
- 10.Wiesmann C, Christinger HW, Cochran AG, Cunningham BC, Fairbrother WJ, Keenan CJ, Meng G, de Vos AM. Crystal structure of the complex between VEGF and a receptor-blocking peptide interface. Biochemistry. 1998;37:17765–17772. doi: 10.1021/bi9819327. [DOI] [PubMed] [Google Scholar]
- 11.De Lano WL, Ultsch MH, de Vos AM, Wells JA. Convergent solutions to binding at a protein-protein interface. Science. 2000;287:1279–1283. doi: 10.1126/science.287.5456.1279. [DOI] [PubMed] [Google Scholar]
- 12.Ferrer M, Harrison SC. Peptide ligands to human immunodeficiency virus type 1 gp120 identified from phage display libraries. J. Virol. 1999;73:5795–5802. doi: 10.1128/jvi.73.7.5795-5802.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kornberg A, Baker TA. DNA Replication. Second Edition W.H. Freeman & Co.; New York: 1992. [Google Scholar]
- 14.Kunkel TA, Bebenek KK. DNA replication fidelity. Annu. Rev. Biochem. 2000;69:497–529. doi: 10.1146/annurev.biochem.69.1.497. [DOI] [PubMed] [Google Scholar]
- 15.Marians KJ. Prokaryotic DNA replication. Annu. Rev. Biochem. 1992;61:673–719. doi: 10.1146/annurev.bi.61.070192.003325. [DOI] [PubMed] [Google Scholar]
- 16.Yoshida K, Inoue I. Peptide binding to geminin and inhibitory for DNA replication. Biochem. Biophys. Res. Commun. 2004;317:218–222. doi: 10.1016/j.bbrc.2004.03.035. [DOI] [PubMed] [Google Scholar]
- 17.Liu J, Dehbi M, Moeck G, Arhin F, Bauda P, Bergeron D, Callejo M, Ferretti V, Ha N, Kwan T, et al. Antimicrobial drug discovery through bacteriophage genomics. Nat. Biotechnol. 2004;22:185–191. doi: 10.1038/nbt932. [DOI] [PubMed] [Google Scholar]
- 18.Pilger BD, Cui C, Coen DM. Identification of a small molecule that inhibits herpes simplex virus DNA polymerase subunit interactions and viral replication. Chem. Biol. 2004;11:647–654. doi: 10.1016/j.chembiol.2004.01.018. [DOI] [PubMed] [Google Scholar]
- 19.Kley N. Chemical dimerizers and three-hybrid systems: scanning the proteome for targets of organic small molecules. Chem. Biol. 2004;11:599–608. doi: 10.1016/j.chembiol.2003.09.017. [DOI] [PubMed] [Google Scholar]
- 20.Bird LE, Pan H, Soultanas P, Wigley DB. Mapping protein-protein interactions within a stable complex of DNA primase and DnaB helicase from Bacillus stearothermophilus. Biochemistry. 2000;39:171–182. doi: 10.1021/bi9918801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thirlway J, Turner CJ, Gibson CT, Gardiner L, Brady K, Allen S, Roberts CJ, Soultanas P. DnaG interacts with a linker region that joins the N- and C-domains of DnaB and induces the formation of 3-fold symmetric rings. Nucleic Acids Res. 2004;32:2977–2986. doi: 10.1093/nar/gkh628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dumay H, Rubbi L, Sentenac A, Marck C. Interaction between yeast RNA polymerase III and transcription factor TFIIIC via ABC10a and τ131 subunits. J. Biol. Chem. 1999;274:33462–33468. doi: 10.1074/jbc.274.47.33462. [DOI] [PubMed] [Google Scholar]
- 23.Chan WC, White PD. Fmoc Solid Phase Peptide Synthesis: A Practical Approach. Oxford University Press; Oxford: 2000. [Google Scholar]
- 24.Soultanas P. A functional interaction between the putative primosomal protein DnaI and the main replicative DNA helicase DnaB in Bacillus. Nucleic Acids Res. 2002;30:966–974. doi: 10.1093/nar/30.4.966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wiseman T, Williston S, Brandts JF, Lin LN. Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Anal. Biochem. 1989;179:131–137. doi: 10.1016/0003-2697(89)90213-3. [DOI] [PubMed] [Google Scholar]
- 26.Fu Z, Wang M, Potter D, Mizioko HM, Kim JJ. The structure of a binary complex between a mammalian mevalonate kinase and ATP: insights into the reaction mechanism and human inherited disease. J. Biol. Chem. 2002;277:18134–18142. doi: 10.1074/jbc.M200912200. [DOI] [PubMed] [Google Scholar]
- 27.Tao Y, Farsetta DL, Nibert ML, Harrison SC. RNA synthesis in a cage: structural studies of reovirus polymerase lambda3. Cell. 2002;111:733–745. doi: 10.1016/s0092-8674(02)01110-8. [DOI] [PubMed] [Google Scholar]
- 28.Vidalain PO, Boxem M, Ge H, Li S, Vidal M. Increasing specificity in high-throughput yeast two-hybrid experiments. Methods. 2004;32:363–370. doi: 10.1016/j.ymeth.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 29.Li S, Armstrong CM, Bertin N, Ge H, Milstein S, Boxem M, Vidalain PO, Han JD, Chesneau A, Hao T, et al. A map of the interactome network of the metazoan C. elegans. Science. 2004;303:540–543. doi: 10.1126/science.1091403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frick DN, Richardson CC. DNA primases. Annu. Rev. Biochem. 2001;70:39–80. doi: 10.1146/annurev.biochem.70.1.39. [DOI] [PubMed] [Google Scholar]
- 31.Soultanas P, Wigley DB. Site-directed mutagenesis reveals roles for conserved amino acid residues in the hexameric DNA helicase DnaB from Bacillus stearothermophilus. Nucleic Acids Res. 2002;30:4051–4060. doi: 10.1093/nar/gkf527. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.