

Abstract
3D QSAR studies comparative molecular field analysis (CoMFA) and comparative molecular similarity indices analysis (CoMSIA) were carried out on 26 structurally diverse subcutaneous pentylenetetrazol (scPTZ) active enaminone analogues, previously synthesized in our laboratory. CoMFA and CoMSIA were employed to generate models to define the specific structural and electrostatic features essential for enhanced binding to the putative GABA receptor. The 3D QSAR models demonstrated a reliable ability to predict the CLog P of the active anticonvulsant enaminones, resulting in a q2 of 0.558 for CoMFA, and a q2 of 0.698 for CoMSIA. The outcomes of the contour maps for both models provide detailed insight for the structural design of novel enaminone derivatives as potential anticonvulsant agents.
Keywords: Enaminones, 3D-QSAR, CoMFA, CoMSIA, scPTZ
Introduction
Epilepsy is one of the most frequent neurological afflictions, characterized by excessive temporary neuronal discharges resulting in uncontrolled convulsions.1 Over the years, the field of epilepsy has received a great deal of attention from research investigators in hopes of discovering new drug agents that will elicit better selectivity and potency, and most importantly have minimal toxic effects. Though several new anticonvulsants are introduced, some types of seizures are still not adequately controlled with the current therapy.1,2 Toxicity, intolerance, and lack of efficacy for certain types of seizures are some of the limitations of existing antiepileptic medications.1,3 In view of the broad etiology of the syndrome, epilepsy involves more than one mechanism that may be responsible for the various seizures, thus its response to different therapeutic agents would be expected.4 For instance, the voltage-gated sodium channels (VGSCs) are responsible for the initial inward current during the depolarization phase of the action potential in excitable cells5, reduction of GABA-ergic transmission6, and excessive glutamatergic neurotransmission7 are various prominent physiological abnormalities for triggering epileptic seizures.
A number of structurally diverse anticonvulsant active enaminone analogues have been synthesized in our laboratory for over 15 years. During that time, we have discovered several novel, potent entities that have shown good to moderate protection against the maximal electroshock seizure (MES) test and the subcutaneous pentylenetetrazol (scPTZ) test.8 The scPTZ seizure model identifies compounds that inhibit the GABA antagonistic effects of pentylenetetrazol or raises the seizure threshold.9 Recent studies from another laboratory have shown that a number of our compounds display inhibition against glutamate-mediated excitatory synaptic transmission by modulation of GABAergic transmission.10–11 Based on these findings, we hypothesize there is an essential pharmacophore within the enaminone structure that possibly interacts with the GABA receptor, which is significant for achieving anticonvulsant activity. Even though the exact site and structural requirements for optimal binding are unknown, we believe because of the molecular similarities between the enaminone analogues, the compounds share a common binding pocket on the GABA receptor which explains the probability of eliciting similar biological properties. Theoretically, if the chemical entities are comparable by their 3D features and found to have similar properties, which means that a high quality QSAR can be generated among those compounds, it is likely that the compounds share a common pharmacophore and and interact with the target in the same binding site as a similar mode. Therefore, 26 scPTZ active enaminones previously synthesized in our laboratory were employed to determine a pharmacophore model using the three-dimensional quantitative structure–activity relationship (3D QSAR) techniques of comparative molecular field analysis (CoMFA) and comparative molecular similarity indices analysis (CoMSIA); the results of these studies are presented in this paper.
Computational Method
The molecular modeling and comparative molecular field evaluations were performed using SYBYL version 7.2 running on a Silicon Graphics Tezro. The ChemDraw - Power Mac G5 1 GB memory running ChemDraw Ultra v. 8 was used to compute the CLog P values for each molecule. Default settings for these programs were used unless specified otherwise.
Data Setting
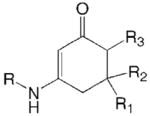
As listed in Table 1, the 26 compounds which form the training set for this study showed good activity against the chemically induced subcutaneous pentylenetetrazol (scPTZ) seizure test.7 The CLog P was calculated for each derivative to determine the ideal lipophilicity which represents the capability of the compounds to cross the blood brain barrier (BBB) to reach the active site.
Table 1.
Training set of diverse scPTZ active enaminone compounds.
| Cyclic enaminones | 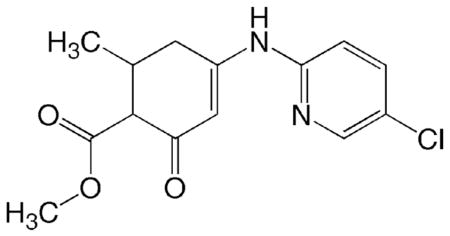 |
||||
|---|---|---|---|---|---|
| Compound ID | R | R1 | R2 | R3 | scPTZ datab |
|
DM 50
Add a 217012 |
C6H4 (p-OCF3) | CH3 | H | CO2CH3 | 2 |
|
E 144
ADD 208075 |
C6H4 (p-CF3) | CH3 | H | CO2CH3 | 2 |
|
E 120
ADD 208040 |
C6H4 (p-F) | CH3 | H | CO2CH3 | 2 |
|
E 139
ADD 208071 |
C6H4 (p-Br) | CH3 | H | CO2CH3 | 2 |
|
LS 16
ADD 226059 |
C6H4 (o-F) | CH3 | H | CO2CH3 | 2 |
|
AT 3
ADD 221002 |
C6H3 (m,p-Cl) | CH3 | H | CO2CH3 | 2 |
|
E 148
ADD 210024 |
C6H4 (p-CF3) | CH3 | CH3 | CO2CH3 | 1 |
|
E 140
ADD 208072 |
C6H4 (p-Cl) | CH3 | CH3 | CO2CH3 | 2 |
|
E 196
ADD 226074 |
C6H4 (p-CN) | CH3 | H | CO2C2H5 | 1 |
|
E 191
ADD 226070 |
C6H4 (m-OCF3) | CH3 | H | CO2C2H5 | 2 |
|
E 183
ADD 226063 |
C6H3 (m, p-Cl) | CH3 | H | CO2C2H5 | 2 |
|
E 190
ADD 226069 |
C6H3 (o, p-Cl) | CH3 | H | CO2C2H5 | 2 |
|
E 167
ADD 224032 |
C6H3 (2′-OCH3, 5′-CH3) | CH3 | H | CO2C2H5 | 2 |
|
DM 27
ADD 211031 |
C6H4 (p-Cl) | CH3 | H | H | 2 |
|
K-4-16
ADD 300078 |
C6H4 (p-OCF3) | CH3 | H | H | 2 |
|
K-4-33
ADD 326078 |
C6H4 (p-F) | CH3 | H | H | 2 |
|
K-4-32
ADD 326077 |
C6H4 (m-Cl) | CH3 | H | H | 2 |
|
K-4-30
ADD 326075 |
C6H4 (m-Br) | CH3 | H | H | 2 |
|
JF-1
ADD 231041 |
- | H | H | H | 1 |
|
| |||||
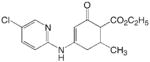
|
E 170
ADD 224035 |
|
2 | |||
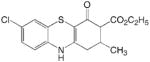
| MLL-5 |
|
2 | |||
|
| |||||
| Acyclic enaminones |
|
||||
|
| |||||
| Compound ID | X | scPTZ data b | |||
|
| |||||
|
DM 28
ADD 211005 |
C2H5 | 2 | |||
|
DM 31A
ADD 211032 |
OCH3 | 2 | |||
|
DM 34
ADD 211026 |
Cl | 2 | |||
|
DM 33
ADD 211025 |
Br | 2 | |||
|
E 157
ADD 216059 |
I | 2 | |||
ADD: Antiepileptic Drug Development Program;
Phase 1 in mice activity – class 1 = activity at 100 mg/kg or <; class 2 = activity between 100 and 300 mg/kg
Generation of enaminone structures
CoMFA and CoMSIA studies require the coordinates of molecules to be aligned according to the reasonable bioactive conformation. So far neither the GABA receptor nor a GABA-agonist receptor complex has been solved at the structural level. In this respect, having the x-ray structure of the active anti-scPTZ compound DM 26 (class 2 activity; Figure 1), gave us a possibility to establish a reasonable rule for the structural alignment in the 3D-QSAR study. The structure of DM 26 is very similar to the group of compounds studied here except, DM 26 has a para-chloro pyridine moiety at the secondary amine whereas the other derivatives have a mono- or di-substituted phenyl ring attached to the secondary amine or amide functional group. In the 3D structure of DM26 there is an H-bond located between the nitrogen of the pyridine ring and the vinyl proton of the enaminone system, which keeps the central part of the molecule (the enaminone system (HN-C=C-CO) and the pyridine ring) flat in both the minimized and crystal structures. However, the other molecules with the exception of MLL-5 have the substituted phenyl ring instead of the pyridine ring and because they lack the H-bond, the molecules tend to twist at an angle. Hence, when the compounds were aligned in their minimized conformation, the highest q2 we were able to obtain was 0.353, indicating the 3D QSAR model was not a good predictor (Figure S1 in supplementary material). Therefore, the crystal structure of DM26 was chosen as a template to establish the conformation of the compounds in the database.
Figure 1.
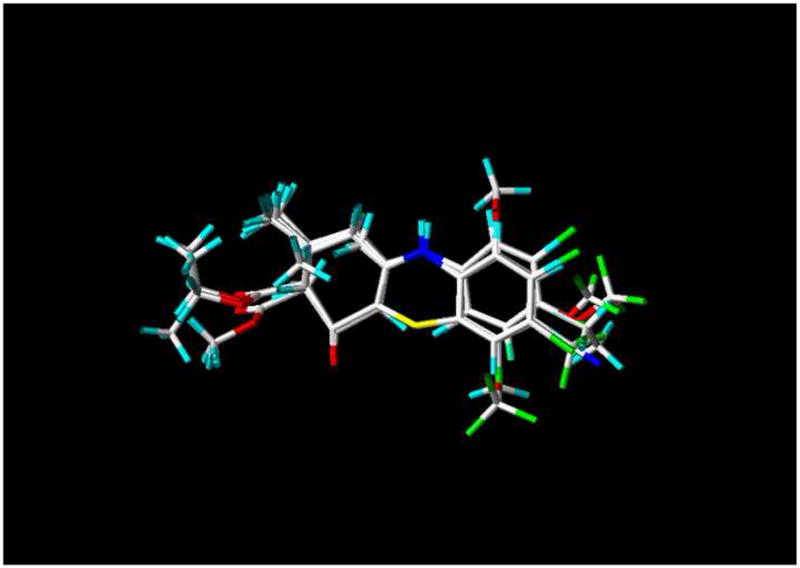
Alignment of the diverse scPTZ active enaminone compounds used in the training set for CoMFA and CoMSIA analysis.
The starting coordinates of the DM 26 (Scheme 1) x-ray crystal structure12 was imported into SYBYL. Energy minimization was carried out in SYBYL using the Tripos force field and the Gasteiger-Huckel charge with distance-dependent dielectric and conjugate gradient method. The convergence criterion was 0.01 kcal/mol Å. The results showed the minimized structure of DM 26 to be similar to its crystal structure. For consistency, the energy minimized structure of DM 26 was used to construct all compounds in the data set and was later saved into the database for alignment. For compounds that shared the common cyclic enaminone backbone with DM 26, the structural modifications were performed by deletion and replacement of different side chains on the cyclic enaminone backbone via the molecular sketch option in SYBYL, followed by a systematic (conformational) search and energy minimization via calculation options in SYBYL. The latter two methods allowed the structures side chains to rotate freely. For compounds with a different backbone, DM 26 was first loaded into the molecular area and modified to the new backbone. Subsequently, the modification of the side chains, the structural systematic search, and the energy minimization of the new structures were performed as previously described.
Scheme 1.
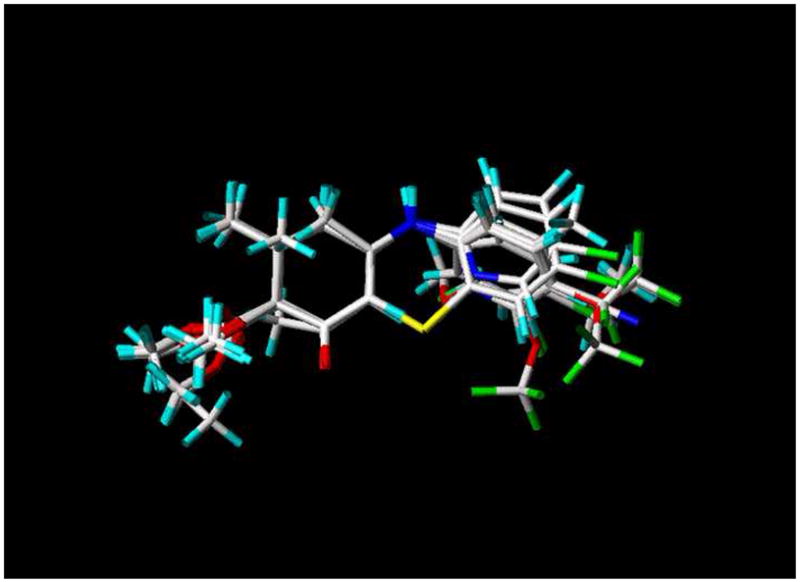
DM 26 (ADD 207092)
Alignment
The minimized structure of each enaminone analogue was imported into SYBYL. All conformers were aligned together by least-squares fitting the five atoms of the common functional moiety of all the structures (Scheme 2). Before performing CoMFA and CoMSIA field calculations, the partial charges for each compound were calculated using the Gasteiger-Huckel method. Figure 1 depicts the alignment of structures used in the training set for CoMFA and CoMSIA analyses.
Scheme 2.

Core structure used for superimposition
CoMFA
As a 3D QSAR technique, CoMFA is based on the relationship between biological activity and the structural properties of the tested compounds. This computational technique is mainly applied when the receptor site is unknown. In this study, the aligned molecules (Figure 1) were kept in a three-dimensional grid of 2 Å in the x, y, and z directions. The steric and electrostatic properties were calculated at various grid points using an sp3 carbon probe atom with a charge of +1.0 and a van der Waals radius of 1.52 Å. The default value of 30 kcal/mol was set as a maximum steric and electrostatic energy cutoff. The regression analysis was carried out using the full cross-validated partial least-square (PLS) leave one out (LOO) method to give the q2 value. The cross-validated q2 indicates the predictive power of the analysis. The final model was developed with the optimal number of components determined in the regression analysis to yield the non-cross-validated PLS correlation coefficient, r2, indicating the quality of data fit in the equation.
CoMSIA
In CoMSIA, five fields are calculated: steric, electrostatic, hydrophobic, hydrogen bond acceptor, and hydrogen bond donor by using the standard settings (probe with charge +1, radius 1 Å and hydrophobicity +1, hydrogen-bond donating +1, hydrogen-bond acceptor +1, attenuation factor α of 0.3 for Gaussian type distance, and grid spacing of 2 Å). This technique has several advantages over CoMFA, such as greater robustness, no application for energy cutoff, and more interpretable contour maps.13,14 The alignment in Figure 1 was used to compute the similarity index fields for CoMSIA and the statistical analysis was carried out as previously stated for CoMFA.
Results and Discussion
Our extensive research and structure-activity relationship (SAR) studies with enaminones8,15–20 have uncovered remarkable anticonvulsant activity for several of our analogues. The cellular mechanism studies conducted by Kombian and co-workers showed that some anticonvulsant aniline and benzylamino enaminones reversibly suppressed glutamate-mediated excitatory postsynaptic currents (EPSCs) recorded in the nucleus accumbens and hippocampus regions which is analogous to GABA activity.10,11 The results from the study encouraged us to reinvestigate the library of scPTZ active compounds which were previously synthesized to determine a practical pharmacophore model that could be utilized to develop novel anticonvulsant enaminones by means of the 3D-QSAR techniques, CoMFA and CoMSIA.
A 3D-QSAR CoMFA study requires the superimposition of a set of active molecules that are kept in a 3D grid space as the steric and electrostatic fields are calculated at various grid points.13 With CoMSIA, the steric and electrostatic fields are computed as in CoMFA, but additionally the hydrophobic, hydrogen-bond donor and hydrogen-bond acceptor fields are included in the analysis. In Table 1, the training set which consists of the 26 active anti-scPTZ derivatives were developed on a structure-based design approach to identify and characterize the minimal molecular requirements for anticonvulsant activity. Previously, we reported on the correlation between CLog P and the anticonvulsant activity of our enaminone derivatives. It is known that the lipophilicity of various compounds is associated with the biological activity.18 Thus, the numerical data used in this study to develop the CoMFA and CoMSIA models were the CLog P values for each molecule.8,21 We decided to use these data values for two reasons 1) the ED50 values were not available for all of the compounds, and 2) the CLog P data provided better values for the computational analysis than the scPTZ data (denoted as class 1 or 2, shown in Table 1). In addition, the wide ranged CLogP values of these compound (−0.5 to 4.98) not only show their different capability to penetrate through the cell membrane into the suitable position of the binging site, but also offer a good sample group for the QSAR study.
By having the X-ray crystal structure of the active compound DM 26 (ADD 207092),7 (shown in Scheme 1) an “alignment rule”22 was established for the superimposition of the diverse set of compounds to perform both the CoMFA and CoMSIA studies (see Scheme 2 and Figure 1). 3D QSAR studies with a q2 greater than 0.3 are considered to be statistically significant, although values greater than 0.4 are preferred.13
For the CoMFA study, a cross-validated partial least square (PLS) analysis was performed using the leave-one-out option and the SAMPLS program determined the optimal number of components to be used (five). The PLS analysis predictive power was clearly statically significant with a q2 (cross-validated r2) of 0.558. Using the optimal number of components found earlier, the non-cross-validated PLS analysis resulted in an r2 (non-cross-validated) of 0.895, a SEE (standard error of estimate) of 0.380, and an F value of 34.224. The relative contributions to this CoMFA model for the steric and electrostatic fields were 57.1% and 42.9%, respectively. This is an indication that the steric properties of the model have more impact on the anticonvulsant activity than the electrostatic properties.
With CoMSIA combined properties steric, electrostatic, hydrophobic, hydrogen-bond donor, and hydrogen-bond acceptor fields, gave a highly significant q2 of 0.698 with an r2 of 0.991. This suggests that the CoMSIA model is also a reliable predictor. Since the hydrophobicity property had greater contribution in the model (42.5%) compared to the other properties calculated, a CoMSIA analysis was performed for this field only. The hydrophobic field also showed a significant q2 value of 0.688 and an r2 of 0.939, indicating that lipophilicity plays a major role in the activity of the anticonvulsant enaminones. The results for both the CoMFA and CoMSIA studies are summarized in Table 2, and the calculated results are shown in Table 3.
Table 2.
Statistical Results for CoMFA and CoMSIA Models Based on 26 Active Enaminone Derivatives CLog P
| Cross-validated data | Conventional data | HDPfh | HBDfi | HBAfj | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| q2a | ONCb | r2c | SEEd | Fe | Sff | Efg | ||||
| CoMFA | 0.558 | 5 | 0.895 | 0.380 | 34.224 | 0.571 | 0.429 | - | - | - |
| CoMSIA | 0.698 | 7 | 0.991 | 0.118 | 281.802 | 0.134 | 0.263 | 0.452 | 0.063 | 0.087 |
| CoMSIA HDP only | 0.688 | 5 | 0.939 | 0.290 | 61.667 | - | - | - | - | - |
q2, cross-validated r2;
ONC, optimum number of components;
r2, non-cross validated;
SEE, standard error estimate;
F, F value;
Sf, steric field;
Ef, electrostatic field;
HDPf, hydrophobic field,
HBDf, hydrogen-bond donor field,
HBAf, hydrogen-bond acceptor field
Table 3.
Predicted CLog P (PCLP) vs. Calculated CLog P and Residuals (Res) by CoMFA and CoMSIA
| Compound | CLog P | CoMFA | CoMSIA | CoMSIAa | |||
|---|---|---|---|---|---|---|---|
| PCLP | Res | PCLP | Res | PCLP | Res | ||
| DM 50 | 3.14 | 3.55 | −0.41 | 2.96 | 0.18 | 3.23 | −0.09 |
| E 144 | 4.46 | 4.47 | −0.01 | 4.52 | −0.06 | 3.95 | 0.51 |
| E 120 | 2.25 | 2.34 | −0.09 | 2.42 | −0.17 | 2.42 | −0.17 |
| E 139 | 2.97 | 2.60 | 0.37 | 3.29 | −0.32 | 3.36 | −0.39 |
| LS 16 | 2.25 | 2.24 | 0.01 | 2.16 | 0.09 | 2.09 | 0.16 |
| AT 3 | 3.41 | 3.57 | −0.16 | 3.50 | −0.09 | 3.34 | 0.07 |
| E 148 | 4.98 | 4.69 | 0.29 | 4.81 | 0.17 | 4.52 | 0.46 |
| E 140 | 3.34 | 3.43 | −0.09 | 3.26 | 0.08 | 3.62 | −0.28 |
| E 196 | 2.07 | 2.39 | −0.32 | 2.24 | −0.17 | 2.24 | −0.17 |
| E 191 | 3.67 | 3.71 | −0.04 | 3.53 | 0.14 | 3.63 | 0.04 |
| E 183 | 3.94 | 2.90 | 1.04 | 3.72 | 0.22 | 3.89 | 0.05 |
| E 190 | 4.06 | 4.26 | −0.20 | 4.25 | −0.19 | 4.23 | −0.17 |
| E 167 | 3.06 | 3.18 | −0.12 | 3.02 | 0.04 | 2.86 | 0.20 |
| DM 27 | 2.96 | 2.99 | −0.03 | 2.98 | −0.02 | 2.92 | 0.04 |
| K-4-16 | 3.27 | 3.04 | 0.23 | 3.49 | −0.22 | 3.17 | 0.10 |
| K-4-33 | 2.39 | 2.93 | −0.54 | 2.40 | −0.01 | 2.30 | 0.09 |
| K-4-32 | 2.96 | 2.88 | 0.08 | 2.78 | 0.18 | 2.87 | 0.09 |
| K-4-30 | 3.11 | 2.91 | 0.20 | 3.07 | 0.04 | 3.26 | −0.15 |
| JF 1 | −0.35 | −0.40 | 0.05 | −0.32 | −0.03 | −0.23 | −0.12 |
| E 170 | 1.94 | 2.32 | −0.38 | 1.90 | 0.04 | 2.32 | −0.38 |
| MLL-5 | 4.19 | 4.17 | 0.02 | 4.15 | 0.04 | 4.35 | −0.16 |
| DM 28 | 2.51 | 2.47 | 0.04 | 2.74 | −0.23 | 3.01 | −0.50 |
| DM 31A | 1.58 | 2.15 | −0.57 | 1.48 | 0.10 | 1.08 | 0.50 |
| DM 34 | 2.50 | 2.54 | −0.04 | 2.31 | 0.19 | 2.29 | 0.21 |
| DM 33 | 2.65 | 2.54 | 0.11 | 2.67 | −0.02 | 2.73 | −0.08 |
| E 157 | 2.91 | 2.33 | 0.58 | 2.88 | 0.03 | 2.76 | 0.15 |
CoMSIA, only hydrophobic field
In the study, we observed when the database included DM 26, the highest q2 for CoMFA only reached 0.378 indicating the model had poor predictive capability while on the other hand the CoMSIA analysis gave a q2 of 0.671, suggesting the model has good predictive power. We believe the difference in the predictability for the two models is mainly due to the fact that CoMSIA considers hydrophobicity as a relative contributor whereas CoMFA does not. Therefore, to maintain consistency in this 3D-QSAR study the DM 26 was not included in the second training set, shown in Figure 1 and Table 1.
To test the predictive ability of the CoMFA and CoMSIA models, the CLog P was computed for E 121–123 and JF 37, 43, 93, and 102, which were not included in the training set. The calculated and predicted CLog P values were in good agreement for both analyses (Table 4, rFigure 2). The CoMFA model revealed a correlation coefficient 2 of 0.969 and the CoMSIA model gave an r2 of 0.905. These results are a clear indication that our models have good predictive ability (Figure 2).
Table 4.
Test set data for the 3D CoMFA and CoMSIA.
| Compounds ID | Structure | scPTZb | CLogP | Predicted |
|||
|---|---|---|---|---|---|---|---|
| CoMSIA | CoMFA | ||||||
| CLogP | Residual | CLogP | Residual | ||||
|
JF 102
ADD a 275006 |
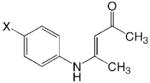
|
1 | 2.42 | 1.82 | 0.6 | 2.09 | 0.33 |
|
E 123
ADD 206043 |
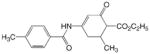
|
2 | 2.78 | 2.55 | 0.23 | 2.53 | 0.25 |
|
E 121
ADD 206041 |
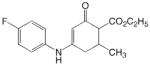
|
1 | 3.35 | 3.02 | 0.33 | 3.15 | 0.20 |
|
JF 37
ADD 261108 |
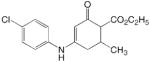
|
1 | 0.97 | 1.23 | −0.26 | 0.949 | 0.021 |
|
JF 93
ADD 273008 |
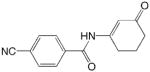
|
1 | 1.97 | 1.84 | 0.13 | 2.06 | −0.09 |
|
JF 43
ADD 261110 |
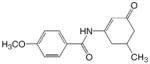
|
1 | 2.01 | 1.97 | 0.04 | 1.97 | 0.04 |
ADD, Antiepileptic Drug Development;
Phase 1 in mice activity – class 1 = activity at 100 mg/kg or <; class 2 = activity between 100 and 300 mg/kg
Figure 2.

Test set observed and predicted CLog P for CoMFA (A) and CoMSIA (B) models. CoMFA (A), correlation coefficient r2 = 0.9697; CoMSIA (B), correlation coefficient r2 = 0.9056.
According to Golbraikh and Tropsha23, performing both internal and external validations for 3D QSAR models are very important for obtaining reliable results. Further analytical CoMFA calculations were carried out for the validation of our model. All 32 compounds, those of the training set and the test set, were combined as a single, internally validated set. The results from the cross-validation gave a q2 of 0.571, the optimal number of components (ONC) as 8, an r2 of 0.977, and a SEE of 0.182. This is a clear indication that the data is highly statistically significant. For the external validation, we ran the CoMFA analyses 130 times. The 32 compounds were randomly divided into a training set (26 compounds) and a test set (6 compounds). Among those 130 calculations, 60 CoMFA calculations generated q2s higher than 0.5, and of the 60 there were four runs that gave q2s higher than 0.7, (Figure S2 in Supplementary material). However, when the model was evaluated with the test set, some of them did poorly to predict the training set. For example, the top four q2 values were 0.727, 0.714, 0.709, and 0.703 respectively, and the correlation coefficients for the observed CLog P and the predicted CLog P were lower than 0.655. The CoMFA model we presented above is by far the best model which can fulfill the stringent requirements for a statically valid mathematical model set forth by Golbraikh and Tropsha.
CoMFA contour maps
The contributions of the steric and electrostatic fields for the CoMFA results were graphically displayed in contour maps with E140 acting as the reference molecule (Figure 3). The abundance of the yellow-green region to the red-blue region indicates a greater contribution of the steric field towards anticonvulsant activity than the electrostatic field. This trend is shown in the statistical data in Table 2 where the relative contribution for the steric field is 57.1% whereas the contribution of the electrostatic field is 42.9%. Based on the contour plots, the following structural optimizations are essential for enhancing the binding of diverse cyclic enaminone derivatives to the proper site on the GABA receptor to elicit anticonvulsant activity. In the steric contour plot (Panel A of Figure 4), the yellow polyhedra areas are primarily on the left end of the structure around the methyl ester moiety and the 6′-methyl, indicating that these areas of the molecule are where the addition of bulky groups might cause a decrease in activity. This is also true in the region of the para-chloro on the aromatic ring; a larger, more bulky substituent at that particular position may result in a loss of activity. The two green regions implies just the opposite of the yellow regions; bulky groups in these areas are favored and may increase anticonvulsant activity. The small green area around the 6′-methyl shows the possibility of extending the methyl group to a propyl group before moving into the yellow region. The electrostatic contour (Panel B of Figure 4) shows majority of its contribution on the opposite side of the molecule. The blue polyhedra favor electropositive and hydrogen bond donor moieties, whereas the red polyhedra are the areas where electronegative and hydrogen bond acceptor moieties are favored.
Figure 3.
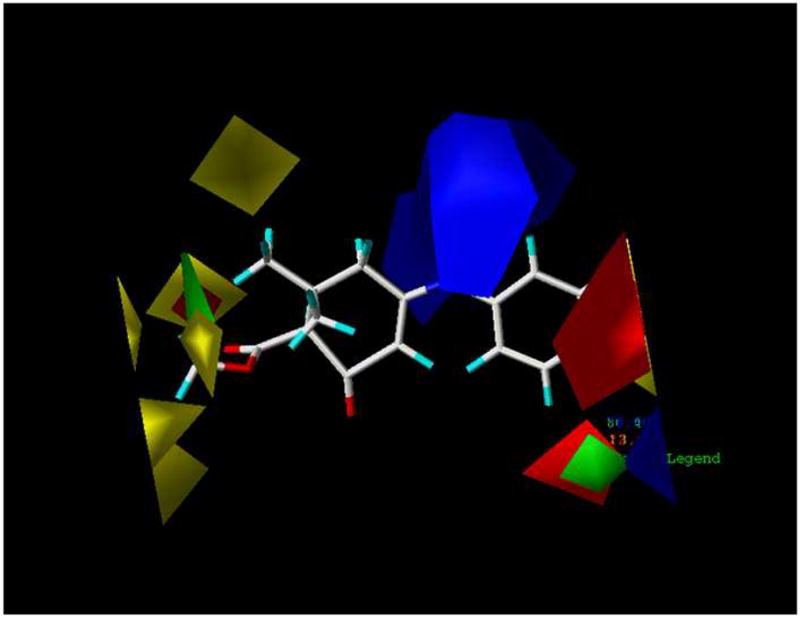
Contour STDEV*COEFF map from CoMFA analysis with 2 Å grid spacing displayed with E140 as the reference molecule.
Figure 4.

Comparison of contour STDEV*COEFF maps of CoMFA and CoMSIA with E140 as reference molecule (A) Steric field distributions of CoMFA. (B) Electrostatic field distribution of CoMFA. (C) Steric field distributions of CoMSIA. (D) Electrostatic field distribution of CoMSIA. The E140 molecule is shown as the stick structure. Green contour indicate regions were bulky groups are favored to improve the activity. Yellow contour indicate regions were bulky groups are disfavored and can decreased activity. Blue contour indicate regions were electropositive substituents are favored for improving the activity. Red contour indicate regions were electronegative substitutents are favored.
CoMSIA contour maps
For the CoMSIA steric and electrostatic plots the colors represent similar meanings as in CoMFA (Figure 4). In the CoMSIA steric map (Panel C of Figure 4), the green contour about the para position of the aromatic ring indicates a region where a bulky substituent would be favorable and possibly improve activity. The yellow regions in the CoMSIA map are focused around similar areas as in the CoMFA steric map. Therefore, less bulky groups at the 6′ position on the cyclic enaminone ring and about the aromatic ring would be favorable for enhancing activity. With the electrostatic contours, the blue regions refer to areas where increased positive charged groups are favored for activity, and the red regions represent areas where electronegative substituents would be favorable for activity, are shown in Panel D of Figure 4. Panel A of Figure 5 shows the contributions of the hydrophobic field. The yellow polyhedron spanning the meta and para position of the benzene ring refer to an area where hydrophobic interactions are favorable for anticonvulsant activity, while the white region, which has a greater size than the yellow region, represent areas where hydrophilic interactions are favorable for activity. The contour maps for the H-bond donor and acceptor fields in Panel B of Figure 5, shows the cyan-color region to be favored for H-bond donor interactions, and the magenta region to be favorable for H-bond acceptor interactions. According to Figure 5, the secondary amine moiety has the possibility to act as a H-bond donor to the binding site of the receptor that may be significant for activity.
Figure 5.

Contour STDEV*COEFF maps of CoMSIA with E140 as reference molecule. (A) Contour map of the hydrophobic field distribution. (B) Contour map of hydrogen-bond donor field and hydrogen-bond acceptor field contributions. In panel A, the yellow contour is a hydrophobic favored area and the hydrophilic favored area is surrounded by the white contour. In panel B, the cyan contour favors hydrogen- bond donor interactions and the magenta contour favors the hydrogen-bond acceptor interactions.
Conclusion
This paper provides the first predictable 3D-QSAR CoMFA and CoMSIA experiments with the enaminone pharmacophore for scPTZ results. The 3D-QSAR CoMFA and CoMSIA models demonstrated a good ability to predict the CLog P of the scPTZ active enaminone analogues. The results suggest within the designed compounds there is a common pharmacophore that shares the same binding site and interacts with the target receptor in a similar mode. The outcomes from the contour maps have provided insight for the design of a novel series of anticonvulsant agents that will have greater activity by identifying significant regions for steric, electrostatic, hydrophobic, H-bond donor and H-bond acceptor interactions.
Supplementary Material
Acknowledgments
This research was supported by National Institutes of Health Grants 1R21 GM63494 to K.R.S. and 2 G12 RR003048 from the RCMI Program, Division of Research Infrastructure, National Center for Research Resources through Howard University, and the American Foundation for Pharmaceutical Education. The authors are indebted to Mr. James P. Stables, Epilepsy Branch, Division of Convulsive, Developmental and Neuromuscular Disorders, National Institute of Neurological Disorders and Stroke for helpful discussions and initial data.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.World Health Organization Report. 2001 http://www.who.int/inf/en/pr-2002-30.html.
- 2.Edafiogho IO, Kombian SB, Ananthalakshmi KV, Salama NN, Eddington ND, Wilson TL, Alexander MS, Jackson PL, Hanson CD, Scott KR. Enaminones: Exploring additional therapeutic activities. J Pharm Sci. 2007;96(10):2509–31. doi: 10.1002/jps.20967. [DOI] [PubMed] [Google Scholar]
- 3.Thenmozhiyal JC, Wong PTH, Chui W-K. Anticonvulsant activity of phenylmethelenehydantoins: A structure-activity relationship study. J Med Chem. 2004;47:1527–35. doi: 10.1021/jm030450c. [DOI] [PubMed] [Google Scholar]
- 4.Anger T, Madge DJ, Mulla M, Riddall D. Medicinal chemistry of neuronal voltage-gated sodium channel blockers. J Med Chem. 2001;44:115–37. doi: 10.1021/jm000155h. [DOI] [PubMed] [Google Scholar]
- 5.Denac H, Mevissen M, Scholtsik GN. Structure, function and pharmacology of voltage-gated sodium channels. Nauyn-Schmiededeberg’s Arch Pharmacol. 2000;326(6):453–79. doi: 10.1007/s002100000319. [DOI] [PubMed] [Google Scholar]
- 6.Ragavendran JV, Sriram D, Kotapati S, Stables J, Yogeeswari P. Newer GABA derivatives for the treatment of epilepsy including febrile seizures: A bioisosteric approach. Eur J Med Chem. 2008;XX:1–6. doi: 10.1016/j.ejmech.2008.01.036. [DOI] [PubMed] [Google Scholar]
- 7.Bough KJ, Paquet M, Pare JF, Hassel B, Smith Y, Hall R, Dingledine R. Evidence against enhanced glutamate transport in the anticonvulsant mechanism of the ketogenic diet. Epilepsy Res. 2007;74:232–236. doi: 10.1016/j.eplepsyres.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 8.(a) Edafiogho IO, Hinko CN, Chang H, Moore JA, Mulzac D, Nicholson JM, Scott KR. Synthesis and anticonvulsant activity of enaminones. J Med Chem. 1992;35:2798–805. doi: 10.1021/jm00093a012. [DOI] [PubMed] [Google Scholar]; (b) Scott KR, Edafiogho IO, Richardson EL, Farrar VA, Moore JA, Tietz EI, Hinko CN, Chang H, El-Assadi A, Nicholson JM. Synthesis and anticonvulsant activity of enaminones. 2. Further structure activity correlations. J Med Chem. 1993;36:1947–55. doi: 10.1021/jm00066a003. [DOI] [PubMed] [Google Scholar]; (c) Mulzac D, Scott KR. The profile of anticonvulsant activity and minimal toxicity of methyl 4-[(p-chlorophenyl) amino]-6methyl-2-oxocyclohexen-3-en-1-oate and some prototype antiepileptic drugs in mice and rats. Epilepsia. 1993:1141–6. doi: 10.1111/j.1528-1157.1993.tb02147.x. [DOI] [PubMed] [Google Scholar]; (d) Scott KR, Rankin GO, Stables JP, Alexander MS, Edafiogho IO, Farrar VA, Kolen KR, Moore JA, Sims LD, Tonnu AD. Synthesis and anticonvulsant activity of enaminones. 3. Investigations on 4′-, 3′-, and 2′-substituted and polysubstituted anilino compounds, sodium channel binding studies, and toxicity evaluations. J Med Chem. 1995;38:4033–43. doi: 10.1021/jm00020a019. [DOI] [PubMed] [Google Scholar]; (e) Eddington ND, Cox DS, Roberts RR, Butcher RJ, Edafiogho IO, Stables JP, Cooke N, Goodwin AM, Smith CA, Scott KR. Synthesis and anticonvulsant activity of enaminones. 4. Investigations on isoxazole derivatives. Eur J Med Chem. 2002;37:635–48. doi: 10.1016/s0223-5234(02)01377-6. [DOI] [PubMed] [Google Scholar]; (f) Laws ML, Roberts RR, Nicholson JM, Butcher R, Stables JP, Goodwin AM, Smith CA, Scott KR. Synthesis, characterization, and anticonvulsant activity of enaminones. Part 5: investigations on 3-carboalkoxy-2-methyl-2,3-dihydro-1H-phenothiazin-4[10H]-one derivatives. Bioorg Med Chem. 1998;6:2289–99. doi: 10.1016/s0968-0896(98)80009-6. [DOI] [PubMed] [Google Scholar]; (g) Foster JE, Nicholson JM, Butcher R, Stables JP, Edafiogho IO, Goodwin AM, Henson MC, Smith CA, Scott KR. Synthesis, characterization and anticonvulsant activity of enaminones. Part 6: Synthesis of substituted vinylic benzamides as potential anticonvulsants. Bioorg Med Chem. 1998;6:2289–99. doi: 10.1016/s0968-0896(99)00185-6. [DOI] [PubMed] [Google Scholar]; (h) Eddington ND, Cox DS, Khurana M, Salama NN, Stables JP, Harrison SJ, Negussie A, Taylor RS, Tran UQ, Moore JA, Barrow JC, Scott KR. Synthesis and anticonvulsant activity of enaminones Part 7. Synthesis and anticonvulsant evaluation of ethyl 4-[(substituted phenyl)amino]-6-methyl-2-oxocyclohex-3-ene-1-carboxylates and their corresponding 5-methylcyclohex-2-enone derivatives. Eur J Med Chem. 2003;38:49–64. doi: 10.1016/s0223-5234(02)00006-5. [DOI] [PubMed] [Google Scholar]; (i) Anderson AJ, Nicholson JM, Bakare O, Butcher RJ. Enaminones 9. Further studies on the anticonvulsant activity and potential type IV phosphodiesterase inhibitory activity of substituted vinylic benzamides. Bioorg Med Chem. 2006;14:997–1006. doi: 10.1016/j.bmc.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 9.Stables JP, Kupferberg HJ. The NIH Anticonvulsant Drug Development (ADD) Program: preclinical anticonvulsant screening project. In: Avanzini G, Tanganelli P, Avoli M, editors. Molecular and Cellular Targets for Anti-epileptic Drugs. John Libbey & Co; 1997. pp. 191–198. [Google Scholar]
- 10.Kombian SB, Edafiogho IO, Ananthalakshmi KVV. Anticonvulsant enaminones depress excitatory synaptic transmission in the rat brain by enhancing extracellular GABA levels. Br J Pharmacol. 2005;145:945–953. doi: 10.1038/sj.bjp.0706250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ananthalakshmi KVV, Edafiogho IO, Kombian SB. Concentration-dependent effects of anticonvulsant enaminone methyl 4-(4′-bromophenyl)aminocyclohex-3-en-6-methyl-2-oxo-1-oate on neuronal excitability in vitro. Neuroscience. 2006;141:345–356. doi: 10.1016/j.neuroscience.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 12.Kubicki M, Bassyouni HAR, Codding PW. Hydrogen bonding in three anticonvulsant enaminones. J Molecular Structure. 2000;525:141–152. [Google Scholar]
- 13.Dixit A, Kashaw SK, Gaur S, Saxena AK. Development of CoMFA, advance CoMFA and CoMSIA models in pyrroloquinazolines as thrombin receptor antagonist. Bioorg Med Chem. 2004;12:3591–3598. doi: 10.1016/j.bmc.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 14.Lee JH, Kang NS, Yoo S. Docking-based 3D-QSAR study for 11β-HSD1 inhibitors. Bioorganic and Medicinal Chem Letts. 2008;18:2479–2490. doi: 10.1016/j.bmcl.2008.02.042. [DOI] [PubMed] [Google Scholar]
- 15.Khurana M, Salama NN, Scott KR, Nemieboka NN, Bauer KS, Jr, Eddington ND. Preclinical evaluation of the pharmacokinetics, brain uptake and metabolism of E 121, and antiepileptic enaminone ester, in rats. Biopharm Drug Dispos. 2003;24:397–407. doi: 10.1002/bdd.376. [DOI] [PubMed] [Google Scholar]
- 16.Anderson AJ, Nicholson JM, Bakare O, Butcher RJ, Scott KR. A base-catalyzed solution-phase parallel synthesis of substituted vinylic benzamides from 3-amino-2-cyclohexanones. J Comb Chem. 2004;6:950–954. doi: 10.1021/cc0498837. [DOI] [PubMed] [Google Scholar]
- 17.Salama NN, Scott KR, Eddington ND. DM 27, and enaminone, modifies the in vitro transport of antiviral therapeutic agents. Biopharm Drug Dispos. 2004;25:227–236. doi: 10.1002/bdd.404. [DOI] [PubMed] [Google Scholar]
- 18.Wilson TL, Jackson PL, Hanson CD, Xue Z, Eddington ND, Scott KR. QSAR of the anticonvulsant enaminones; Molecular modeling aspects and other assessments. Med Chem. 2005;1:371–381. doi: 10.2174/1573406054368710. [DOI] [PubMed] [Google Scholar]
- 19.Scott KR, Butcher RJ, Hanson CD. 5-Methyl-3-(5-methylisoxazol-3-yl)cyclohex-2-enone. Acta Cryst. 2006;E62:o215–o217. (online) [Google Scholar]
- 20.Scott KR, Butcher RJ, Hanson CD. 5,5-Dimethyl-3-(5-methylisoxazol-3-yl)cyclohex-2-enone. Acta Cryst. 2006;E62:o218–o220. (online) [Google Scholar]
- 21.Edafiogho IO, Alexander MS, Moore JA, Farrar VA, Scott KR. Anticonvulsant enaminones: With emphasis on methyl 4-[(p-chlorophenyl)-amino]-6-methyl-2-oxocyclohex-3-en-1-oate (ADD 196022) Curr Med Chem. 1994;1:159–175. [Google Scholar]
- 22.Johnson SL, Jung D, Forino M, Chen Y, Satterthwait A, Rozanov DV, Strongin AY, Pellecchia M. Anthrax lethal factor protease inhibitors: Synthesis, SAR, and structure-based 3D QSAR studies. J Med Chem. 2006;49:27–30. doi: 10.1021/jm050892j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Golbraikh A, Tropsha A. Beware of q2! J Mol Graphics Modell. 2002;20:269–276. doi: 10.1016/s1093-3263(01)00123-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.