

Abstract
Thermanaerovibrio acidaminovorans (Guangsheng et al. 1997) Baena et al. 1999 is the type species of the genus Thermanaerovibrio and is of phylogenetic interest because of the very isolated location of the novel phylum Synergistetes. T. acidaminovorans Su883T is a Gram-negative, motile, non-spore-forming bacterium isolated from an anaerobic reactor of a sugar refinery in The Netherlands. Here we describe the features of this organism, together with the complete genome sequence, and annotation. This is the first completed genome sequence from a member of the phylum Synergistetes. The 1,848,474 bp long single replicon genome with its 1765 protein-coding and 60 RNA genes is part of the Genomic Encyclopedia of Bacteria and Archaea project.
Keywords: strictly anaerobic, amino acid fermentation, thermophile, oxidative decarboxylation, lithotrophic, co-culture with Methanobacterium thermoautotrophicum, Synergistales, Synergistetes
Introduction
Strain Su883T (= DSM 6589 = ATCC 49978) is the type strain of the species Thermanaerovibrio acidaminovorans, which represents the type species of the two species containing genus Thermanaerovibrio [1]. Strain SU883T is of particular interest because it is able to ferment quite a number of amino acids [2,3], and because its metabolism is greatly enhanced in the presence of the hydrogen scavenger Methanobacterium thermoautotrophicum, from which several single substrates solely hydrogen is formed as reduced fermentation product [3]. The physiological properties of the organism have been studied in detail [2,3].
Here we present a summary classification and a set of features for T. acidaminovorans strain SU883T, together with the description of the complete genome sequencing and annotation.
Classification and features
Until now, strain SU883T was the only strain known from this species. Uncultured clones with a rather high degree of 16S rRNA similarity to the sequence of strain SU883T (AF071414) have been obtained from mesophilic and thermophilic bioreactors treating pharmaceutical wastewater [4] (AF280844, 97.5%; AF280820, 97.7%). The sequence similarities to environmental metagenomic libraries [5,6] were below 81%, indicating a rather poor representation of closely related strains in the analyses habitats (status July 2009).
Figure 1 shows the phylogenetic neighborhood of T. acidaminovorans strain Su883T in a 16S rRNA based tree. The three 16S rRNA gene sequences in the genome of strain Su883T differed from each other by up to three nucleotides, and by up to 29 nucleotides (2%) from the previously published 16S rRNA sequence, generated from DSM 6589 (AF071414). The significant difference between the genome data and the reported 16S rRNA gene sequence, which contains ten ambiguous base calls, is most likely due to sequencing errors in the previously reported sequence data.
Figure 1.

Phylogenetic tree highlighting the position of T. acidaminovorans strain Su883T relative to the other type strains within the phylum Synergistetes. The tree was inferred from 1,333 aligned characters [7,8] of the 16S rRNA gene sequence under the maximum likelihood criterion [9], and was rooted with the type strains of the genera within the phylum ‘Thermotogae’. The branches are scaled in terms of the expected number of substitutions per site. Numbers above branches are support values from 1,000 bootstrap replicates if larger than 60%. Strains with a genome sequencing project registered in GOLD [10] are printed in blue; published genomes in bold.
T. acidaminovorans cells are curved rods of 0.5-0.6 × 2.5-3.0 µm in size (Table 1 and Figure 2), with round ends, occur singly, in pairs, or in long chains when grown in a complex medium [3]. The organism is Gram-negative, non-spore-forming, moderately thermophilic, motile by means of a tuft of lateral flagella at the concave side, and strictly anaerobic for growth [1]. Interestingly, it tolerates flushing with air for at least one hour, and it produces catalase [3]. While being exposed to air, strain Su883T loses its motility [3]. Strain Su883T is able to grow by oxidative decarboxylation of succinate to propionate. A mechanism for reductive propionate formation could be excluded [3]. Glutamate, α-ketoglutarate, histidine, arginine, ornithine, lysine, and threonine are fermented to acetate and propionate. Serine, pyruvate, alanine, glucose, fructose, xylose, glycerol and citrate are fermented to acetate. Branched-chain amino acids are converted to branched-chain fatty acids. Hydrogen is the only reduced end product [3]. The growth and the substrate conversion are strongly enhanced by co-cultivation with methanogens, e.g., M. thermoautotrophicum [3]. Strain Su883T contains b-type cytochromes [3]. Originally, it was reported that in strain Su883T thiosulfate, nitrite, sulfur and fumarate are not reduced [3]. However, a more recent study shows that, although elemental sulfur (1%) inhibits the growth of strain Su883T on glucose, strain Su883T could grow lithoheterotrophically with H2 as electron donor, S0 as electron acceptor, and yeast extract as carbon source [16]. The catablolism of arginine has been studied in detail. Apparently, degradation of arginine occurs by the arginine deiminase (ADI) pathway [2]. No activity of arginase, a key enzyme of the arginase pathway, could be detected [2]. No growth was observed on glycine, aspartate, gelatin, xylose, ribose, galactose, lactose, sucrose, mannose, lactate, ethanol, methanol, acetoin, betaine, malonate, and oxalate [3]. With either succinate, α-ketoglutarate or glutamate, the following enzyme activities were measured in cell free extracts: propionyl CoA:succinate IISCoA transferase, propionate kinase, acetate kinase, glutamate dehydrogenase, pyruvate dehydrogenase, α-ketoglutarate dehydrogenase, malate dehydrogenase, citrate lyase and hydrogenase [3]. The following enzymes were not detected: succinate thiokinase, fumarate reductase, succinate dehydrogenase, β-methylaspartase, hydroxyglutarate dehydrogenase, isocitrate dehydrogenase and formate dehydrogenase [3]. Unfortunately, no chemotaxonomic data are currently available for T. acidaminovorans strain Su883T.
Table 1. Classification and general features of T. acidaminovorans strain Su883T according to the MIGS recommendations [11].
| MIGS ID | Property | Term | Evidence code |
|---|---|---|---|
| Current classification | Domain Bacteria | TAS [12] | |
| Phylum Synergistetes | TAS [13] | ||
| Class Synergistia | TAS [13] | ||
| Order Synergistales | TAS [13] | ||
| Family Synergistaceae | TAS [13] | ||
| Genus Thermanaerovibrio | TAS [1] | ||
| Species Thermanaerovibrio acidamonovorans | TAS [1] | ||
| Type strain Su883 | TAS [1] | ||
| Gram stain | negative | TAS [3] | |
| Cell shape | curved rods, 0.5-0.6 × 2.5-3.0 µm | TAS [3] | |
| Motility | motile, lateral flagella | TAS [3] | |
| Sporulation | non-sporulating | TAS [3] | |
| Temperature range | 40-58°C | TAS [3] | |
| Optimum temperature | 55°C | TAS [3] | |
| Salinity | no NaCl required for growth, upper tolerance border unknown | TAS [1] | |
| MIGS-22 | Oxygen requirement | strictly anaerobic | TAS [3] |
| Carbon source | succinate, glucose, fructose, amongst others (see text) | TAS [3] | |
| Energy source | carbohydrates, amino acids | TAS [3] | |
| MIGS-6 | Habitat | granular methanogenic sludge | TAS [3] |
| MIGS-15 | Biotic relationship | free living | NAS |
| MIGS-14 | Pathogenicity | unknown | |
| Biosafety level | 1 | TAS [14] | |
| Isolation | sludge sample taken from an upflow anaerobic sludge bed (UASB) reactor of a sugar refinery | TAS [3] | |
| MIGS-4 | Geographic location | Breda, The Netherlands | TAS [3] |
| MIGS-5 | Sample collection time | 1992 or before | TAS [3] |
| MIGS-4.1 MIGS-4.2 | Latitude, Longitude | 51.589, 4.774 | NAS |
| MIGS-4.3 | Depth | not reported | |
| MIGS-4.4 | Altitude | not reported |
Evidence codes - IDA: Inferred from Direct Assay (first time in publication); TAS: Traceable Author Statement (i.e., a direct report exists in the literature); NAS: Non-traceable Author Statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). These evidence codes are from the Gene Ontology project [15]. If the evidence code is IDA, then the property should have been directly observed for a living isolate by one of the authors, or an expert mentioned in the acknowledgements.
Figure 2.

Scanning electron micrograph of T. acidaminovorans strain Su883T
Genome sequencing and annotation
Genome project history
This organism was selected for sequencing on the basis of its phylogenetic position, and is part of the Genomic Encyclopedia of Bacteria and Archaea project. The genome project is deposited in the Genomes OnLine Database [10] and the complete genome sequence in GenBank NOT YET. Sequencing, finishing and annotation were performed by the DOE Joint Genome Institute (JGI). A summary of the project information is shown in Table 2.
Table 2. Genome sequencing project information.
| MIGS ID | Property | Term |
|---|---|---|
| MIGS-31 | Finishing quality | Finished |
| MIGS-28 | Libraries used | Three genomic libraries: two Sanger libraries (8 kb pMCL200 and fosmid pcc1Fos) and one 454 pyrosequence standard library |
| MIGS-29 | Sequencing platforms | ABI3730, 454 GS FLX |
| MIGS-31.2 | Sequencing coverage | 9.7x Sanger; 9.9× pyrosequence |
| MIGS-30 | Assemblers | Newbler version 1.1.02.15, phrap |
| MIGS-32 | Gene calling method | Prodigal, GenePRIMP |
| INSDC ID | CP001818 | |
| Genbank Date of Release | November 19, 2009 | |
| GOLD ID | Gc01091 | |
| INSDC project ID | 29531 | |
| Database: IMG-GEBA | 2501651200 | |
| MIGS-13 | Source material identifier | DSM 6589 |
| Project relevance | Tree of Life, GEBA |
Growth conditions and DNA isolation
T. acidaminovorans strain Su883T, DSM 6589, was grown anaerobically in DSMZ medium 104 (modified PYG medium) [17] at 55°C. DNA was isolated from 1-1.5 g of cell paste using Qiagen Genomic 500 DNA Kit (Qiagen, Hilden, Germany) following the manufacturer’s protocol without modification according to Wu et al. [18].
Genome sequencing and assembly
The genome was sequenced using a combination of Sanger and 454 sequencing platforms. All general aspects of library construction and sequencing performed at the JGI can be found at the JGI website (http://www.jgi.doe.gov/). 454 Pyrosequencing reads were assembled using the Newbler assembler version 1.1.02.15 (Roche). Large Newbler contigs were broken into 2,046 overlapping fragments of 1,000 bp and 1,838 of them entered into the final assembly as pseudo-reads. The sequences were assigned quality scores based on Newbler consensus q-scores with modifications to account for overlap redundancy and to adjust inflated q-scores. A hybrid 454/Sanger assembly was made using the parallel phrap assembler (High Performance Software, LLC). Possible mis-assemblies were corrected with Dupfinisher or transposon bombing of bridging clones [19]. Gaps between contigs were closed by editing in Consed, custom primer walk or PCR amplification. A total of 401 Sanger finishing reads were produced to close gaps, to resolve repetitive regions, and to raise the quality of the finished sequence. The error rate of the completed genome sequence is less than 1 in 100,000. Together all sequence types provided 19.6 ×coverage of the genome. The final assembly contains 19,461 Sanger and 358,573 pyrosequencing reads.
Genome annotation
Genes were identified using Prodigal [20] as part of the Oak Ridge National Laboratory genome annotation pipeline, followed by a round of manual curation using the JGI GenePRIMP pipeline (http://geneprimp.jgi-psf.org/) [21]. The predicted CDSs were translated and used to search the National Center for Biotechnology Information (NCBI) nonredundant database, UniProt, TIGRFam, Pfam, PRIAM, KEGG, COG, and InterPro databases. Additional gene prediction analysis and functional annotation was performed within the Integrated Microbial Genomes - Expert Review (http://img.jgi.doe.gov/er) platform [22].
Genome properties
The genome is 1,848,474 bp long and comprises one main circular chromosome with a 63.8% GC content. (Table 3, Figure 3). Of the 1,825 genes predicted, 1,765 were protein coding genes, and 60 RNAs. In addition, 27 pseudogenes were identified. The majority of genes (79.3%) were assigned a putative function while the remaining ones were annotated as hypothetical proteins. The distribution of genes into COGs functional categories is presented in Table 4.
Table 3. Genome Statistics.
| Attribute | Value | % of Total |
|---|---|---|
| Genome size (bp) | 1,848,474 | 100.00% |
| DNA Coding region (bp) | 1,745,505 | 94.43% |
| DNA G+C content (bp) | 1,179,189 | 63.79% |
| Number of replicons | 1 | |
| Extrachromosomal elements | 0 | |
| Total genes | 1,825 | 100.00% |
| RNA genes | 60 | 3.29% |
| rRNA operons | 3 | |
| Protein-coding genes | 1,765 | 96.71% |
| Pseudo genes | 27 | 1.48% |
| Genes with function prediction | 1,447 | 79.29% |
| Genes in paralog clusters | 142 | 7.78% |
| Genes assigned to COGs | 1,483 | 81.26% |
| Genes assigned Pfam domains | 1,484 | 81.32% |
| Genes with signal peptides | 275 | 15.07% |
| Genes with transmembrane helices | 404 | 22.14% |
| CRISPR repeats | 0 |
Figure 3.
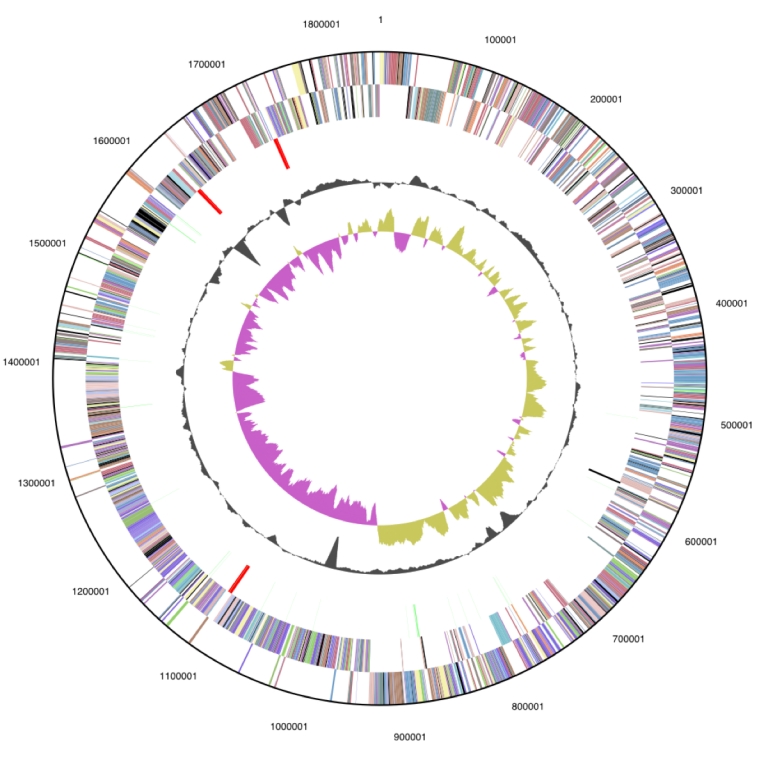
Graphical circular map of the genome. From outside to the center: Genes on forward strand (color by COG categories), Genes on reverse strand (color by COG categories), RNA genes (tRNAs green, rRNAs red, other RNAs black), GC content, GC skew.
Table 4. Number of genes associated with the general COG functional categories.
| Code | Value | %age | Description |
|---|---|---|---|
| J | 150 | 8.5 | Translation, ribosomal structure and biogenesis |
| A | 0 | 0.0 | RNA processing and modification |
| K | 84 | 4.8 | Transcription |
| L | 71 | 4.0 | Replication, recombination and repair |
| B | 0 | 0.0 | Chromatin structure and dynamics |
| D | 26 | 1.5 | Cell cycle control, mitosis and meiosis |
| Y | 0 | 0.0 | Nuclear structure |
| V | 11 | 0.6 | Defense mechanisms |
| T | 101 | 5.7 | Signal transduction mechanisms |
| M | 97 | 5.5 | Cell wall/membrane biogenesis |
| N | 71 | 4.0 | Cell motility |
| Z | 0 | 0.0 | Cytoskeleton |
| W | 0 | 0.0 | Extracellular structures |
| U | 38 | 2.2 | Intracellular trafficking and secretion |
| O | 53 | 3.0 | Posttranslational modification, protein turnover, chaperones |
| C | 126 | 7.1 | Energy production and conversion |
| G | 86 | 4.9 | Carbohydrate transport and metabolism |
| E | 185 | 10.5 | Amino acid transport and metabolism |
| F | 66 | 3.7 | Nucleotide transport and metabolism |
| H | 97 | 5.5 | Coenzyme transport and metabolism |
| I | 32 | 1.8 | Lipid transport and metabolism |
| P | 63 | 3.6 | Inorganic ion transport and metabolism |
| Q | 18 | 1.0 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 152 | 8.6 | General function prediction only |
| S | 104 | 5.9 | Function unknown |
| - | 282 | 16.0 | Not in COGs |
Acknowledgements
We would like to gratefully acknowledge the help of Susanne Schneider (DSMZ) for DNA extraction and quality analysis. This work was performed under the auspices of the US Department of Energy Office of Science, Biological and Environmental Research Program, and by the University of California, Lawrence Berkeley National Laboratory under contract No. DE-AC02-05CH11231, Lawrence Livermore National Laboratory under Contract No. DE-AC52-07NA27344, and Los Alamos National Laboratory under contract No. DE-AC02-06NA25396, as well as German Research Foundation (DFG) INST 599/1-1.
References
- 1.Baena S, Fardeau ML, Woo TH, Ollivier B, Labat M, Patel BK. Phylogenetic relationships of three amino-acid-utilizing anaerobes, Selenomonas acidaminovorans, 'Selenomonas acidaminophila' and Eubacterium acidaminophilum, as inferred from partial 16S rDNA nucleotide sequences and proposal of Thermanaerovibrio acidaminovorans gen. nov., comb. nov. and Anaeromusa acidaminophila gen. nov., comb. nov. Int J Syst Bacteriol 1999; 49:969-974 [DOI] [PubMed] [Google Scholar]
- 2.Plugge CM, Stams AJM. Arginine catabolism by Thermanaerovibrio acidaminovorans. FEMS Microbiol Lett 2001; 195:259-262 10.1111/j.1574-6968.2001.tb10530.x [DOI] [PubMed] [Google Scholar]
- 3.Guangsheng C, Plugge CM, Roelofsen W, Houwen FP, Stams AJM. Selenomonas acidaminovorans sp. nov., a versatile thermophilic proton-reducing anaerobe able to grow by decarboxylation of succinate to propionate. Arch Microbiol 1992; 157:169-175 [Google Scholar]
- 4.LaPara TM, Nakatsu CH, Pantea L, Alleman JE. Phylogenetic analysis of bacterial communities in mesophilic and thermophilic bioreactors treating pharmaceutical wastewater. Appl Environ Microbiol 2000; 66:3951-3959 10.1128/AEM.66.9.3951-3959.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kurokawa K, Itoh T, Kuwahara T, Oshima K, Toh H, Toyoda A, Takami H, Morita H, Sharma VK, Srivastava TP, et al. Comparative metagenomics revealed commonly enriched gene sets in human gut microbiomes. DNA Res 2007; 14:169-181 10.1093/dnares/dsm018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Venter JC, Remington K, Heidelberg JF, Halpern AL, Rusch D, Eisen JA, Wu D, Paulsen J, Nelson KE, Nelson W, et al. Environmental genome shotgun sequencing of the Sargasso Sea. Science 2004; 304:66-74 10.1126/science.1093857 [DOI] [PubMed] [Google Scholar]
- 7.Lee C, Grasso C, Sharlow MF. Multiple sequence alignment using partial order graphs. Bioinformatics 2002; 18:452-464 10.1093/bioinformatics/18.3.452 [DOI] [PubMed] [Google Scholar]
- 8.Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 2000; 17:540-552 [DOI] [PubMed] [Google Scholar]
- 9.Stamatakis A, Hoover P, Rougemont J. A Rapid Bootstrap Algorithm for the RAxML Web Servers. Syst Biol 2008; 57:758-771 10.1080/10635150802429642 [DOI] [PubMed] [Google Scholar]
- 10.Liolios K, Mavromatis K, Tavernarakis N, Kyrpides NC. The Genomes On Line Database (GOLD) in 2007: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res 2008; 36:D475-D479 10.1093/nar/gkm884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol 2008; 26:541-547 10.1038/nbt1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci USA 1990; 87:4576-4579 10.1073/pnas.87.12.4576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jumas-Bilak E, Roudiere L, Marchandin H. Description of 'Synergistetes' phyl. nov. and emended description of the phylum 'Deferribacteres' and of the family Syntrophomonadaceae, phylum 'Firmicutes'. Int J Syst Evol Microbiol 2009; 59:1028-1035 10.1099/ijs.0.006718-0 [DOI] [PubMed] [Google Scholar]
- 14.Anonymous. Biological Agents: Technical rules for biological agents www.baua.de TRBA 466.
- 15.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000; 25:25-29 10.1038/75556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zavarzina DG, Zhilina T, Tourova T, Kuznetsov B, Kostrikina N, Bonch-Osmolovskaya EA. Thermanaerovibrio velox sp. nov., a new anaerobic, thermophilic, organotrophic bacterium that reduces elemental sulfur, and emended description of the genus Thermanaerovibrio. Int J Syst Evol Microbiol 2000; 50:1287-1295 [DOI] [PubMed] [Google Scholar]
- 17.List of growth media used at DSMZ: http://www.dsmz.de/microorganisms/ media_list.php
- 18.Wu D, Hugenholtz P, Mavromatis K, Pukall R, Dalin E, Ivanova N, Kunin V, Goodwin L, Wu M, Tindall BJ, et al. A phylogeny-driven genomic encyclopedia of Bacteria and Archaea. Nature 2009; 462:1056-1060 10.1038/nature08656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sims D, Brettin T, Detter JC, Han C, Lapidus A, Copeland A, Glavina Del Rio T, Nolan M, Chen F, Lucas S, et al. Complete genome of Kytococcus sedentarius type strain (541T). Stand Genomic Sci 2009; 1:12-20 10.4056/sigs.761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anonymous. Prodigal Prokaryotic Dynamic Programming Genefinding Algorithm. Oak Ridge National Laboratory and University of Tennessee 2009 http://compbio.ornl.gov/prodigal/
- 21.Pati A, Ivanova N, Mikhailova, N, Ovchinikova G, Hooper SD, Lykidis A, Kyrpides NC. GenePRIMP: A Gene Prediction Improvement Pipeline for microbial genomes. (Submitted). [DOI] [PubMed]
- 22.Markowitz VM, Mavromatis K, Ivanova NN, Chen IMA, Chu K, Kyrpides NC. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics 2009; 25:2271-2278 10.1093/bioinformatics/btp393 [DOI] [PubMed] [Google Scholar]