

Abstract
Slackia heliotrinireducens (Lanigan 1983) Wade et al. 1999 is of phylogenetic interest because of its location in a genomically yet uncharted section of the family Coriobacteriaceae, within the deep branching Actinobacteria. Strain RHS 1T was originally isolated from the ruminal flora of a sheep. It is a proteolytic anaerobic coccus, able to reductively cleave pyrrolizidine alkaloids. Here we describe the features of this organism, together with the complete genome sequence, and annotation. This is the first complete genome sequence of the genus Slackia, and the 3,165,038 bp long single replicon genome with its 2798 protein-coding and 60 RNA genes is part of the Genomic Encyclopedia of Bacteria and Archaea project.
Keywords: Gram-positive coccus, anaerobic, asaccharolytic, pyrrolizidine alkaloids, Coriobacteriaceae
Introduction
Strain RHS 1T (= DSM 20476 = ATCC 29202 = JCM 14554) is the type strain of the species Slackia heliotrinireducens and was originally described by Lanigan in 1976 as Peptococcus heliotrinreducans (sic) [1] and validly published following an orthographic correction as Peptococcus heliotrinreducens in 1983 [2,3]. The strain was later transferred to the genus Peptostreptococcus on the basis of its G+C content [4]. 16S rRNA gene sequence analysis indicated that it should not be assigned to the genus Peptostreptococcus and therefore the strain was subsequently allocated to the novel genus Slackia as S. heliotrinireducens [5,6]. The three species of the genus Slackia, S. exigua, S. faecicanis, and S. heliotrinireducens form a distinct cluster within the Coriobacteriaceae, located in the neighborhood to the genera Cryptobacterium and Collinsella.
Five additional strains identified as S. heliotrinireducens based on their proteolytic enzyme profiles have been isolated from human polymicrobial abscesses [7], but these strains were dissimilar from the type strain as shown by pyrolysis mass spectrometry [8]. With 94% sequence identity (16S rRNA gene), S. exigua, the type strain of the closest related species represents the only meaningful (>91%) hit in nucleotide sequence database searches, indicating a complete lack of cultivated and even uncultivated relatives of strain RHS 1T in accessible microbiological diversity. Screening of environmental samples and surveys reported at NCBI BLAST server indicated no closely related phylotypes that can be linked to the species (as of July 2009). Here we present a summary classification and a set of features for S. heliotrinireducens RHS 1T Together with the description of the complete genomic sequencing and annotation.
Classification and features
Figure 1 shows the phylogenetic neighborhood of S. heliotrinireducens strain RHS 1T in a 16S rRNA based tree. The sequence of one of the two 16S rRNA genes differs in two nucleotides from the other copy and from the previously published 16S rRNA sequence generated from ATCC 29202 (AF101241).
Figure 1.

Phylogenetic tree highlighting the position of S. heliotrinireducens RHS 1T relative to other type strains within the family Coriobacteriaceae. The tree was inferred from 1,422 aligned 16S rRNA characters [9,10] under the maximum likelihood criterion [11], and rooted with type strains of the genera Collinsella and Coriobacterium. The branches are scaled in terms of the expected number of substitutions per site. Numbers above branches are support values from 1,000 bootstrap replicates, if larger than 60%. Lineages with type strain genome sequencing projects registered in GOLD [12] are shown in blue, published genomes in bold, e.g. the recently published GEBA genomes from Cryptobacterium curtum [13], and Eggerthella lenta [14].
S. heliotrinireducens is Gram-positive, nonmotile, obligatly anaerobic, and does not produce endospores (Table 1). Strain RHS 1 forms cocci or coccobacilli (Figure 2) with a diameter of 0.3 to 0.6 µm and 0.8 x 1.2 µm, respectively [5,6]. The strain grows very slowly on blood agar and forms small translucent, glistening colonies, up to 1 mm in diameter after extensive incubation. It does not utilize carbohydrates, but reduces nitrates and pyrrolizidine alkaloids [5,6]. Reductive cleavage of pyrrolizidines (heliotrine, europine, heleurine, supinine and lasiocarpine) occurs by using hydrogen gas or formate as hydrogen donor [1]. Ammonia is formed from tryptone, yeast extract, adenine, uracil and arginine. Nitrates are completely reduced to ammonia if an appropriate electron donor (H2, formate) is present [19]. The strain is bile-sensitive, indole-negative, hydrolyses arginine but not esculin. Does not produce catalase or urease, but is able to dissimilate arginine. Growth is generally stimulated by addition of 0.5% arginine. Metabolic products from S. heliotrinireducens grown in prereduced PYG broth are acetic acid, isovaleric acid, and butyric acid in trace amounts [4].
Table 1. Classification and general features of S. heliotrinireducens RHS 1T in accordance to the MIGS recommendations [15].
| MIGS ID | Property | Term | Evidence code |
|---|---|---|---|
| Current classification | Domain Bacteria | TAS [16] | |
| Phylum Actinobacteria | TAS [17] | ||
| Class Actinobacteria | TAS [18] | ||
| Order Coriobacteriales | TAS [18] | ||
| Suborder Coriobacteridae | TAS [18] | ||
| Family Coriobacteriaceae | TAS [18] | ||
| Genus Slackia | TAS [5] | ||
| Species Slackia heliotrinireducens | TAS [5] | ||
| Type strain RHS 1 | TAS [5] | ||
| Gram stain | positive | TAS [1] | |
| Cell shape | cocci to coccobacilli | TAS [1] | |
| Motility | nonmotile | TAS [1] | |
| Sporulation | nonsporulating | TAS [1] | |
| Temperature range | mesophile, 30-46°C | TAS [19] | |
| Optimum temperature | 38-42°C | TAS [19] | |
| Salinity | 5g NaCl per l | TAS [5] | |
| MIGS-22 | Oxygen requirement | obligate anaerobic | TAS [1] |
| Carbon source | asaccharolytic | TAS [1] | |
| Energy source | arginine, proteolytic | NAS | |
| MIGS-6 | Habitat | rumen (sheep) | TAS [1] |
| MIGS-15 | Biotic relationship | free living | NAS |
| MIGS-14 | Pathogenicity | assumed | NAS |
| Biosafety level | 1 (+) | TAS [20] | |
| Isolation | rumen of sheep | TAS [1] | |
| MIGS-4 | Geographic location | Australia | NAS |
| MIGS-5 | Sample collection time | about 1974 | TAS [1] |
| MIGS-4.1 MIGS-4.2 | Latitude – Longitude | not reported | |
| MIGS-4.3 | Depth | not reported | |
| MIGS-4.4 | Altitude | not reported |
Evidence codes - IDA: Inferred from Direct Assay (first time in publication); TAS: Traceable Author Statement (i.e., a direct report exists in the literature); NAS: Non-traceable Author Statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). These evidence codes are from the Gene Ontology project [21]. If the evidence code is IDA, then the property should have been directly observed for a living isolate by one of the authors, or an expert mentioned in the acknowledgements.
Figure 2.

Scanning electron micrograph of S. heliotrinireducens RHS 1T
Almost nothing is known about the chemotaxonomy of strain RHS 1T, except that its predominant cellular fatty acid is C18:1 [4].
Genome sequencing information
Genome project history
This organism was selected for sequencing on the basis of its phylogenetic position, and is part of the Genomic Encyclopedia of Bacteria and Archaea project. The genome project is deposited in the Genome OnLine Database [12] and the complete genome sequence is in GenBank. Sequencing, finishing and annotation were performed by the DOE Joint Genome Institute (JGI). A summary of the project information is shown in Table 2.
Table 2. Genome sequencing project information.
| MIGS ID | Property | Term |
|---|---|---|
| MIGS-31 | Finishing quality | Finished |
| MIGS-28 | Libraries used | Three genomic libraries: two Sanger-8kb pMCL200 and fosmid pcc1Fos Sanger libraries and one 454 pyrosequence standard library |
| MIGS-29 | Sequencing platforms | ABI3730, 454 GS FLX |
| MIGS-31.2 | Sequencing coverage | 6x Sanger; 20× pyrosequence |
| MIGS-30 | Assemblers | Newbler version 1.1.02.15, phrap |
| MIGS-32 | Gene calling method | Genemark 4.6b, GenePRIMP, tRNAScan-SE-1.23, infernal 0.81 |
| INSDC ID | CP001684 | |
| Genbank Date of Release | August 28, 2009 | |
| GOLD ID | Gc01094 | |
| NCBI project ID | 20831 | |
| Database: IMG-GEBA | 2500901757 | |
| MIGS-13 | Source material identifier | DSM 20476 |
| Project relevance | Tree of Life, GEBA |
Growth conditions and DNA isolation
S. heliotrinireducens strain RHS 1T, DSM 20476, was grown anaerobically in DSMZ medium 104 (PYG) [22]; at 37°C. DNA was isolated from 1-1.5 g of cell paste using Qiagen Genomic 500 DNA Kit (Qiagen, Hilden, Germany) with a modified protocol for cell lysis (FT), as described in Wu et al. [23].
Genome sequencing and assembly
The genome was sequenced using a combination of Sanger and 454 sequencing platforms. All general aspects of library construction and sequencing performed at the JGI can be found on the JGI website (http://www.jgi.doe.gov/). 454 Pyrosequencing reads were assembled using the Newbler assembler version 1.1.02.15 (Roche). Large Newbler contigs were broken into 3,507 overlapping fragments of 1,000 bp and entered into the assembly as pseudo-reads. The sequences were assigned quality scores based on Newbler consensus q-scores with modifications to account for overlap redundancy and to adjust inflated q-scores. A hybrid 454/Sanger assembly was made using the parallel phrap assembler (High Performance Software, LLC). Possible mis-assemblies were corrected with Dupfinisher or transposon bombing of bridging clones [24]. Gaps between contigs were closed by editing in Consed, custom primer walk or PCR amplification. A total of 1,433 Sanger finishing reads were produced to close gaps, to resolve repetitive regions, and to raise the quality of the finished sequence. The final assembly consists of 21.045 Sanger and 205,234 pyrosequence (454) reads. Together all sequence types provided 26× coverage of the genome. The error rate of the completed genome sequence is less than 1 in 100,000.
Genome annotation
Genes were identified using GeneMark [25] as part of the genome annotation pipeline in the Integrated Microbial Genomes Expert Review system (http://img.jgi.doe.ogv/er) [26], followed by a round of manual curation using the JGI GenePRIMP pipeline (http://geneprimp.jgi-psf.org/) [27]. The predicted CDSs were translated and used to search the National Center for Biotechnology Information (NCBI) nonredundant database, UniProt, TIGRFam, Pfam, PRIAM, KEGG, COG, and InterPro databases. The tRNAScanSE tool [28] was used to find tRNA genes, whereas ribosomal RNAs were found by using the tool RNAmmer [29]. Other non coding RNAs were identified by searching the genome for the Rfam profiles using INFERNAL (v0.81) [30]. Additional gene prediction analysis and manual functional annotation was performed within the Integrated Microbial Genomes (http://img.jgi.doe.gov/) platform [31].
Metabolic network analysis
The metabolic Pathway/Genome Database (PGDB) was computationally generated using Pathway Tools software version 12.5 [32] and MetaCyc version 12.5 [33], based on annotated EC numbers and a customized enzyme name mapping file. It has undergone no subsequent manual curation and may contain errors, similar to a Tier 3 BioCyc PGDB [34].
Genome properties
The genome is 3,165,038 bp long and comprises one main circular chromosome with a 60.2% GC content (Table 3 and Figure 3). Of the 2,858 genes predicted, 2,798 were protein coding genes, and 60 RNAs; 33 pseudogenes were also identified. The majority of the protein-coding genes (70.6%) were assigned with a putative function, while those remaining were annotated as hypothetical proteins. The properties and the statistics of the genome are summarized in Table 3. The distribution of genes into COGs functional categories is presented in Table 4, and a cellular overview diagram is presented in Figure 4, followed by a summary of metabolic network statistics shown in Table 5.
Table 3. Genome Statistics.
| Attribute | Value | % of Total |
|---|---|---|
| Genome size (bp) | 3,165,038 | 100.00% |
| DNA coding region (bp) | 2,756,714 | 87.10% |
| DNA G+C content (bp) | 1,905,720 | 60.21% |
| Number of replicons | 1 | |
| Extrachromosomal elements | 0 | |
| Total genes | 2,858 | 100.00% |
| RNA genes | 60 | 2.03% |
| rRNA operons | 2 | |
| Protein-coding genes | 2,798 | 97.90% |
| Pseudo genes | 33 | 1.15% |
| Genes with function prediction | 2,014 | 70.52% |
| Genes in paralog clusters | 433 | 15.15% |
| Genes assigned to COGs | 1,969 | 68.94% |
| Genes assigned Pfam domains | 1,977 | 69.22% |
| Genes with signal peptides | 562 | 19.66% |
| Genes with transmembrane helices | 123 | 4.30% |
| CRISPR repeats | 0 |
Figure 3.
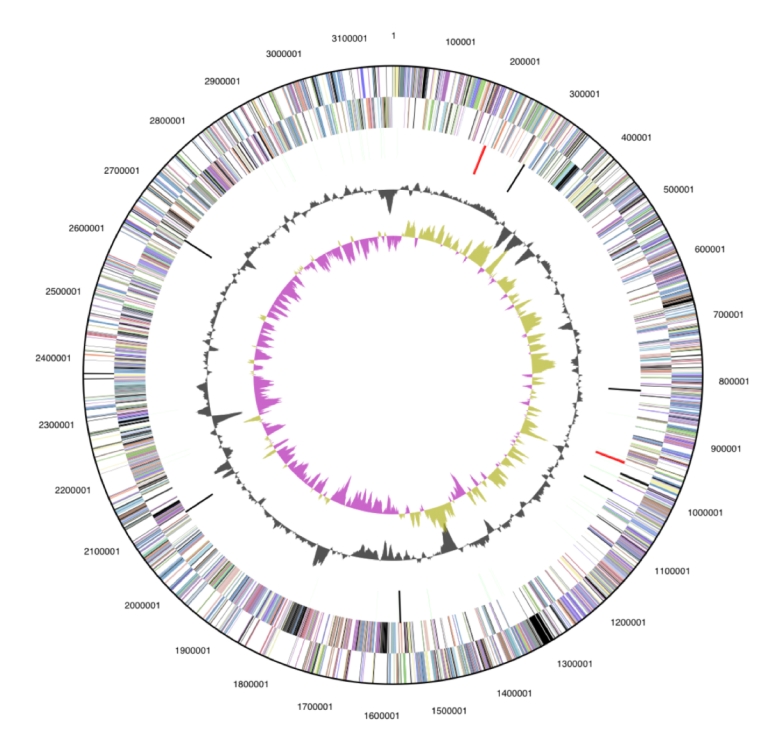
Graphical circular map of the genome. From outside to the center: Genes on forward strand (color by COG categories), Genes on reverse strand (color by COG categories), RNA genes (tRNAs green, rRNAs red, other RNAs black), GC content, GC skew.
Table 4. Number of genes associated with the general COG functional categories.
| Code | Value | % age | Description |
|---|---|---|---|
| J | 139 | 5.0 | Translation, ribosomal structure and biogenesis |
| A | 0 | 0.0 | RNA processing and modification |
| K | 208 | 7.4 | Transcription |
| L | 134 | 4.8 | Replication, recombination and repair |
| B | 1 | 0.0 | Chromatin structure and dynamics |
| D | 25 | 0.9 | Cell cycle control, mitosis and meiosis |
| Y | 0 | 0.0 | Nuclear structure |
| V | 48 | 1.7 | Defense mechanisms |
| T | 107 | 3.8 | Signal transduction mechanisms |
| M | 93 | 3.3 | Cell wall/membrane biogenesis |
| N | 3 | 0.1 | Cell motility |
| Z | 0 | 0.0 | Cytoskeleton |
| W | 0 | 0.0 | Extracellular structures |
| U | 30 | 1.1 | Intracellular trafficking and secretion |
| O | 83 | 3.0 | Posttranslational modification, protein turnover, chaperones |
| C | 229 | 8.2 | Energy production and conversion |
| G | 68 | 2.4 | Carbohydrate transport and metabolism |
| E | 151 | 5.4 | Amino acid transport and metabolism |
| F | 58 | 2.1 | Nucleotide transport and metabolism |
| H | 109 | 3.9 | Coenzyme transport and metabolism |
| I | 66 | 2.4 | Lipid transport and metabolism |
| P | 101 | 3.6 | Inorganic ion transport and metabolism |
| Q | 33 | 1.2 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 319 | 11.4 | General function prediction only |
| S | 155 | 5.5 | Function unknown |
| - | 829 | 29.6 | Not in COGs |
Figure 4.

Schematic cellular overview diagram of all pathways of the S. heliotrinireducens RHS 1T metabolism. Nodes represent metabolites, with shape indicating class of metabolite (see key to right). Lines represent reactions.
Table 5. Metabolic Network Statistics.
| Attribute | Value |
|---|---|
| Total genes | 2,856 |
| Enzymes | 457 |
| Enzymatic reactions | 750 |
| Metabolic pathways | 156 |
| Metabolites | 576 |
Acknowledgements
We would like to gratefully acknowledge the help of Gabriele Gehrich-Schröter (DSMZ) for growing S. heliotrinireducens cultures. This work was performed under the auspices of the US Department of Energy's Office of Science, Biological and Environmental Research Program, and by the University of California, Lawrence Berkeley National Laboratory under contract No. DE-AC02-05CH11231, Lawrence Livermore National Laboratory under Contract No. DE-AC52-07NA27344, and Los Alamos National Laboratory under contract No. DE-AC02-06NA25396, as well as German Research Foundation (DFG) INST 599/1-1.
References
- 1.Lanigan GW. Peptococcus heliotrinreducans, sp. nov., a cytochrome-producing anaerobe which metabolizes pyrrolizidine alkaloids. J Gen Microbiol 1976; 94:1-10 [DOI] [PubMed] [Google Scholar]
- 2.Associate Editor Validation List no. 10. Validation of the publication of new names and new combinations previously effectively published outside the IJSB. Int J Syst Bacteriol 1983; 33:438-440 [Google Scholar]
- 3.Associate Editor Validation List no. 11. Validation of the publication of new names and new combinations previously effectively published outside the IJSB. Int J Syst Bacteriol 1983; 33:672-674 [Google Scholar]
- 4.Ezaki T, Yabuuchi E. Transfer of Peptococcus heliotrinreducens corrig. to the genus Peptostreptococcus: Peptostreptococcus heliotrinireducens Lanigan 1983 comb. nov. Int J Syst Bacteriol 1986; 36:107-108 [Google Scholar]
- 5.Wade WG, Downes J, Dymock D, Hiom SJ, Weightman AJ, Dewhirst FE, Paster BJ, Tzellas N, Coleman B. The family Coriobacteriaceae: reclassification of Eubacterium exiguum (Poco et al. 1996) and Peptostreptococcus heliotrinreducens (Lanigan 1976) as Slackia exigua gen. nov., comb. nov. and Slackia heliotrinireducens gen. nov., comb. nov., and Eubacterium lentum (Prevot 1938) as Eggerthella lenta gen. nov., comb. nov. Int J Syst Bacteriol 1999; 49:595-600 [DOI] [PubMed] [Google Scholar]
- 6.Murdoch DA. Gram-positive anaerobic cocci. Clin Microbiol Rev 1998; 11:81-120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murdoch DA, Mitchelmore IJ. Isolation of Peptostreptococcus heliotinreducens from human polymicrobial abscesses. Lett Appl Microbiol 1989; 9:223-225 10.1111/j.1472-765X.1989.tb00331.x [DOI] [Google Scholar]
- 8.Goodacre R, Hiom SJ, Cheeseman SL, Murdoch DA, Weightman AJ, Wade WG. Identification and discrimination of oral asaccharolytic Eubacterium species by pyrolysis mass spectrometry and artificial neural networks. Curr Microbiol 1996; 32:77-84 10.1007/s002849900014 [DOI] [PubMed] [Google Scholar]
- 9.Lee C, Grasso C, Sharlow MF. Multiple sequence alignment using partial order graphs. Bioinformatics 2002; 18:452-464 10.1093/bioinformatics/18.3.452 [DOI] [PubMed] [Google Scholar]
- 10.Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 2000; 17:540-552 [DOI] [PubMed] [Google Scholar]
- 11.Stamatakis A, Hoover P, Rougemont J. A rapid bootstrap algorithm for the RAxML web-servers. Syst Biol 2008; 57:758-771 10.1080/10635150802429642 [DOI] [PubMed] [Google Scholar]
- 12.Liolios K, Mavrommatis K, Tavernarakis N, Kyrpides NC. The Genomes OnLine Database (GOLD) in 2007: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res 2008; 36:D475-D479 10.1093/nar/gkm884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mavrommatis K, Pukall R, Rohde C, Chen F, Sims D, Brettin T, Kuske C, Detter JC, Han C, Lapidus A, et al. Complete genome of Cryptobacterium curtum type strain (12-3T). Stand Genomic Sci 2009; 1:93-100 10.4056/sigs.12260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saunders E, Pukall R, Abt B, Lapidus A, Galvina Del Rio T, Copeland A, Tice H, Cheng JF, Lucas S, Chen F, et al. Complete genome sequence of Eggerthella lenta type strain (VPI 0255T). Stand Genomic Sci 2009; 1:174-182 10.4056/sigs.33592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol 2008; 26:541-547 10.1038/nbt1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci USA 1990; 87:4576-4579 10.1073/pnas.87.12.4576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garrity GM, Holt J. Taxonomic Outline of the Archaea and Bacteria Bergey's Manual of Systematic Bacteriology, 2nd Ed. In: G.Garrity GM, Boone DR, Castenholz RW Eds. Vol 1 The Archaea, Deeply Branching and Phototrophic Bacteria 2001 pp. 155-166 [Google Scholar]
- 18.Stackebrandt E, Rainey FA, Ward-Rainey NL. Proposal for a new hierarchic classification system, Actinobacteria classis nov. Int J Syst Bacteriol 1997; 47:479-491 [Google Scholar]
- 19.Holdemann Moore LV, Johson JL, Moore WEC. The genus Peptostreptococcus. In: Bergey's Manual of Systematic Bacteriology (ed. PHA Sneath) Vol. 2, 1083-1092. [Google Scholar]
- 20.Anonymous. Biological Agents: Technical rules for biological agents, TRBA 466 <www.baua.de>.
- 21.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000; 25:25-29 10.1038/75556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.List of growth media used at DSMZ: http://www.dsmz.de/microorganisms/ media_list.php
- 23.Wu D, Hugenholtz P, Mavrommatis K, Pukall R, Dalin E, Ivanova N, Kunin V, Goodwin L, Wu M, Tindall BJ, et al. A phylogeny-driven genomic encyclopedia of Bacteria and Archaea. Nature 2009; 462:1056-1060 10.1038/nature08656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sims D, Brettin T, Detter JC, Han C, Lapidus A, Copeland A, Glavina Del Rio T, Nolan M, Chen F, Lucas S, et al. Complete genome of Kytococcus sedentarius type strain (541T). Stand Genomic Sci 2009; 1:12-20 10.4056/sigs.761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Besemer J, Lomsadze A, Borodovsky M. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res 2001; 29:2607-2618 10.1093/nar/29.12.2607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Markowitz VM, Mavrommatis K, Ivanova NN, Chen IMA, Chu K, Kyrpides NC. Expert Review of Functional Annotations for Microbial Genomes. Bioinformatics 2009; 25:2271-2278 10.1093/bioinformatics/btp393 [DOI] [PubMed] [Google Scholar]
- 27.Pati A, Ivanova N, Mikhailova N, Ovchinikova G, Hooper SD, Lykidis A, Kyrpides NC. GenePRIMP: A Gene Prediction Improvement Pipeline for microbial genomes. (Submitted). [DOI] [PubMed] [Google Scholar]
- 28.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 1997; 25:955-964 10.1093/nar/25.5.955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lagesen K, Hallin P, Rødland EA, Staerfeldt HH, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 2007; 35:3100-3108 10.1093/nar/gkm160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Griffiths-Jones S, Moxon S, Marshall M, Khanna A, Eddy SR, Bateman A. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res 2005; 33:D121-D124 10.1093/nar/gki081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Markowitz VM, Szeto E, Palaniappan K, Grechkin Y, Chu K, Chen IMA, Dubchak I, Anderson I, Lykidis A, Mavrommatis K, et al. The Integrated Microbial Genomes (IMG) system in 2007: data content and analysis tool extensions. Nucleic Acids Res 2008; 36:D528-D533 10.1093/nar/gkm846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karp PD, Paley SM, Romero P. The Pathway Tools Software. Bioinformatics 2002; 18:S225-S232 [DOI] [PubMed] [Google Scholar]
- 33.Caspi R, Foerster H, Fulcher CA, Kaipa P, Krummenacker M, Latendresse M, Paley SM, Rhee SY, Shearer A, Tissier C, et al. The MetaCyc Database of metabolic pathways and enzymes and the BioCyc collection of pathway/Genome Databases. Nucleic Acids Res 2008; 36:D623-D631 10.1093/nar/gkm900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karp PD, Ouzounis CA, Moore-Kochlacs C, Goldovsky L, Kaipa P, Ahren D, Tsoka S, Darzentas N, Kunin V, Lopez-Bigas N. Expansion of the BioCyc collection of pathway/genome databases to 160 genomes. Nucleic Acids Res 2005; 33:6083-6089 10.1093/nar/gki892 [DOI] [PMC free article] [PubMed] [Google Scholar]