

Abstract
Mitochondrial DNA (mtDNA) mutations have been associated with disease and aging. Since each cell has thousands of mtDNA copies, clustered into nucleoids of five to ten mtDNA molecules each, determining the effects of a given mtDNA mutation and their connection with disease phenotype is not straightforward. It has been postulated that heteroplasmy (coexistence of mutated and wild-type DNA) follows simple probability rules dictated by the random distribution of mtDNA molecules at the nucleoid level. This model has been used to explain how mutation levels correlate with the onset of disease phenotype and loss of cellular function. Nonetheless, experimental evidence of heteroplasmy at the nucleoid level is scarce. Here, we report a new method to determine heteroplasmy of individual mitochondrial particles containing one or more nucleoids. The method uses capillary cytometry with laser-induced fluorescence detection to detect individual mitochondrial particles stained with PicoGreen, which makes it possible to quantify the mtDNA copy number of each particle. After detection, one or more particles are collected into polymerase chain reaction (PCR) wells and then subjected to real-time multiplexed PCR amplification. This PCR strategy is suitable to obtain the relative abundance of mutated and wild-type mtDNA. The results obtained here indicate that individual mitochondrial particles and nucleoids contained within these particles are not heteroplasmic. The results presented here suggest that current models of mtDNA segregation and distribution (i.e., heteroplasmic nucleoids) need further consideration.
Keywords: Mitochondria, Heteroplasmy, Mitochondrial DNA, Fluorescence, PCR, Deletion, Nucleoid
Introduction
Mitochondrial DNA (mtDNA) mutations have been linked to many neurological and muscular degenerative diseases [1, 2] and are believed to have a causal role in aging [3–5]. Investigating the role of mtDNA mutations in disease and aging is complicated by heteroplasmy (i.e., the coexistence of wild-type and mutated mtDNA in the same cell) and by the fact that mtDNA molecules are clustered into nucleoids. These structures are made of tightly bound mtDNA and protein complexes, contain approximately five to ten mtDNA molecules, and appear to replicate once and only once per cell cycle [6].
Since mammalian cells contain hundreds to thousands of mtDNA molecules [7], the effect of a given mutation on mitochondrial bioenergetic status and phenotypic expression may not be detected unless the fraction of mutated mtDNA is higher than a certain threshold [8, 9]. A high threshold requires that nucleoids be heteroplasmic [10]. This postulate is known as the “faithful nucleoid model,” which assumes that nucleoid heteroplasmy reflects that of bulk mtDNA [10].
An approach to further investigate the “faithful nucleoid model” requires determining mtDNA copy number and heteroplasmy in individual nucleoids. A surrogate of this experiment is to determine these measurements in isolated individual mitochondria particles that may have one or more nucleoids per mitochondrial particle. So far, there is only one analysis of mtDNA of individual mitochondrial particles isolated from heteroplasmic cells [11]. This report indicates that at least 75% of the mitochondria analyzed were homoplasmic containing exclusively either wild-type or mutated (tRNA lys8344) mtDNA, which would imply that their nucleoids have at least the same degree of heteroplasmy. Unfortunately, this report could not investigate mtDNA copy numbers and heteroplasmy simultaneously.
We previously reported a technique for quantitation of mtDNA in individual mitochondria [12]. Others used fluorescence microscopy [13] and flow cytometry [11] for similar measurements. In agreement with each other, these reports indicate that individual mitochondria contain 0–21 mtDNA molecules with an average ranging from 2 to 4.6 [11–13]. However, these techniques do not allow quantitation of heteroplasmy.
In this report, we used capillary cytometry with laser-induced fluorescence (LIF) detection to detect and analyze the mtDNA contents of individual mitochondrial particles. Similar to flow cytometry, mitochondrial particles were mobilized by hydrodynamic pressure, and LIF detection was performed on-column to allow direct collection of mitochondria postdetection as they exit the capillary. Transmitochondrial cytoplasmic hybrid (cybrid) cells (ΔH2-1) containing a heteroplasmic, large-scale (7.9 kb) deletion were used as a cell model. Mitochondrial DNA was fluorescently labeled with PicoGreen, and individual, isolated mitochondrial particles were detected by LIF and deposited into polymerase chain reaction (PCR) wells. Following collection, a multiplex real-time PCR assay was used to amplify wild-type and deleted mtDNA and quantitate heteroplasmy [14]. When >24 mtDNA molecules were collected into a single PCR well, heteroplasmy was detected, and for large amounts of mtDNA the percent of deleted mtDNA (89±5%) was similar to that detected in bulk cell lysates (80±13% deleted mtDNA). In contradiction to the “faithful nucleoid model,” individual mitochondrial particles were found to be mainly homoplasmic.
Materials and methods
Reagents
Sucrose was purchased from Fluka (Buchs, Switzerland). Mannitol was purchased from Riedel de-Haën (Seelze, Germany). Trypsin solution (10×, 5.0 g/l trypsin, 2.0 g/l EDTA·4Na, 8.5 g/l NaCl), N-[2-hydroxyethyl]- [piperazine]-N-[ethanesulfonic acid] (HEPES), phosphate-buffered saline (10× PBS, containing 100 mM KH2PO4/Na2HPO4 solution, pH 7.4, 27 mM KCl, 1,370 mM NaCl), high-glucose Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum, uridine, gentamicin, and ethylenediaminetetraacetic acid (EDTA) were purchased from Sigma (St. Louis, MO, USA). Fluorescein was purchased from Invitrogen (Carlsbad, CA, USA).
Buffer preparation
Buffer M contained 70 mM sucrose, 5 mM HEPES, 5 mM EDTA, and 210 mM mannitol, pH 7.4. Buffer M was prepared using Milli-Q deionized water, filtered with a 0.22-μm membrane, and adjusted to pH 7.4 with KOH.
Cell culture
The human transmitochondrial cytoplasmic hybrid (cybrid) cell line (ΔH2-1) was a generous gift from Dr. Carlos Moraes (University of Miami). The cell line harbors a 7,522-bp deletion spanning positions 7,982–15,504. The construction of the ΔH2-1 cell line has been described previously [15].
ΔH2-1 cybrid cells were cultured in 0.22-μm-filtered DMEM medium containing 10% (v/v) fetal bovine serum, 50 μg/ml uridine, and 10 μg/ml gentamicin stored at 4 °C. The ΔH2-1 cybrid cells require a high-glucose DMEM medium to compensate for their dysfunctional oxidative phosphorylation system. All cells were cultured in 75-cm2 vented culture flasks at 37 °C and 5% CO2 and split every 3–4 days. Cells were split by rinsing with PBS, lifted with 0.25 g/l trypsin for 5 min, and diluted in fresh growth medium.
Labeling mitochondrial DNA with PicoGreen
The PicoGreen reagent, as received from the manufacturer, was diluted 3:1,000 in warm growth medium. Adhered ΔH2-1 cybrid cells were labeled with the PicoGreen solution for 60 min at 37 °C and 5% CO2. Excess PicoGreen was removed after labeling by washing the cells twice with growth medium.
Isolation of mitochondria from cell culture
Confluent cells were harvested by adding 6 ml of growth medium to 6 ml of cells (approximately 5×105 cells per milliliter). Cells were washed three times by centrifuging at 1,000×g for 10 min at 4 °C followed by resuspension in 1-ml ice-cold buffer M. Cells were lysed in a nitrogen cavitator (Parr Instruments, Moline, IL, USA) at 500 psi for 15 min at 0 °C. Whole cells and nuclei were sedimented at 1,400×g for 10 min at 4 °C. The postnuclear supernatant was removed, and mitochondria were sedimented by centrifugation at 16,000×g for 10 min at 4 °C. The mitochondrial pellet was washed and resuspended in 1 ml buffer M. The mitochondrial preparation contains other contaminating organelles, but they do not interfere with the detection because: (1) PicoGreen labels double-stranded DNA with little interference from RNA [16] and (2) mitochondria are the only organelle in the fraction that contains DNA. The relatively high g-force used to sediment whole cells and nuclei ensures that the mitochondrial fraction is not contaminated with nuclei or whole cells.
Capillary cytometry
The custom-built capillary cytometer uses a detector similar to that of a previously described capillary electrophoresis with laser-induced fluorescence (CE-LIF) instrument [17]. The differences between the instruments are that LIF detection was performed on-column and that mitochondria were mobilized by hydrodynamic pressure for capillary cytometry. The instrument consisted of a 15-mW Ar-ion laser (Melles Griot, Carlsbad, CA, USA) that was focused to approximately 50 μm onto the capillary using a 10× long-working-distance objective (Mitutoyo, Aurora, IL, USA). The capillary (50 μm i.d. fused silica capillary, Polymicro Technologies, Phoenix, AZ, USA) was placed inside a sheath flow cuvette (Hellma, Plainview, NY, USA), and the outlet of the capillary protruded from the cuvette to allow collection of mitochondria. To minimize adsorption of mitochondria to the capillary inner walls, poly(acryloylaminopropanol) was covalently bound to this surface [18]. To allow excitation and collection of the fluorescence from individual mitochondria, the outer polyimide coating of the capillary was removed with hot sulfuric acid. The fluorescence was collected at 90° with respect to the excitation source with a ×40, 0.55 NA objective (New Focus, San Jose, CA, USA). Scatter from the excitation source was optically removed with a 488-nm-long pass filter (Semrock, Rochester, NY, USA) and spatially removed with a pinhole. The fluorescence was further filtered with a band pass filter (520/35 nm, Semrock) before being detected with a PMT (R1477, Hamamatsu, Bridgewater, NJ, USA). The output from the PMT was digitized at 500 Hz with a NiDaq I/O board (PCI-MIO-16XE-50, National Instruments, Austin, TX, USA) and the data displayed and saved using an in-house written data acquisition program (LabView, National Instruments). The capillary cytometry instrument was aligned by continuously introducing 1×10−9 M fluorescein through the capillary, at approximately 1.5 psi, while optimizing the position of the cuvette to maximize the fluorescence signal.
Although the capillary was housed in a sheath flow cuvette, on-column LIF detection was performed, as opposed to off-column detection, to enable collection of mitochondria from the capillary outlet postdetection. The limit of detection (LOD), based on single injections of 1×10−10 M fluorescein, was 4±2 zmol (4×10−21 mol).
Postdetection collection
For collection of mitochondria, the capillary was inserted through the sheath flow cuvette so that approximately 1.5 cm protruded from the end of the cuvette. This allowed deposition of mitochondria into the bottom of a PCR well after on-column detection. The detection and collection schemes are illustrated in Fig. 1.
Fig. 1.

On-column detection and collection of mitochondria. Mitochondria are detected individually by on-column LIF. A section of a PCR plate is placed on a collection tray that allows positioning of the capillary inside PCR wells to collect mitochondria. Mitochondrial particles observed in ∼10-s time windows were collected in the same vial as they reached the end of the capillary
Following LIF detection of a mitochondrial particle, there is a delay of approximately 25 s before the particle reaches the end of the capillary. The delay time was calculated based on the velocity of mitochondria (approximately 0.16 cm/s), which was controlled by the hydrodynamic pressure using a regulator, and the distance between the detection point and the capillary outlet.
A 3 well × 4 well section of a PCR plate was placed onto a custom-built collection tray. The size of the PCR plate was limited by the physical dimensions of the LIF detector. The position of the collection tray was controlled in three dimensions using three Turbo Servo motors (BT-0172EFB-C8PN-NNN, API Motion, Amherst, NY, USA) that were controlled by in-house written software. The software calibrated the position of the collection tray to the PCR plate prior to collection of mitochondria to allow addressable positioning of the capillary outlet into specific PCR wells. The motion and position of the collection tray was visually monitored with a video camera. To collect mitochondria, the outlet of the capillary was immersed in 6.5 μl of water inside a PCR well for a given period of time (e.g., 10 s). After collection for the specified amount of time, the PCR plate was lowered and repositioned to collect a new fraction in another PCR well. Repositioning of the collection tray was automatically performed by the software after the new collection well was specified by the user. After collecting 10-s fractions of mitochondria, the number of mitochondria that were collected in each PCR well was determined from the cytogram by taking into account the delay time between detection and exit from the capillary.
Multiplex real-time PCR analysis
Wild-type and deleted mtDNA were simultaneously amplified using a three-primer amplification strategy [14]. Briefly, the PCR product is formed between the forward primer and reverse primer 1 for wild-type mtDNA. Conversely, PCR product is formed between the forward primer and reverse primer 2 for deleted mtDNA (Table 1, Fig. 2). No PCR product forms between the forward primer and reverse primer 2 in the presence of wild-type mtDNA since the duration of the extension step is relatively short (i. e., 30 s). Real-time multiplex detection of wild-type and deleted mtDNA PCR products was performed using molecular beacons. MB1 hybridizes to the wild-type PCR product, and MB2 hybridizes to the deleted mtDNA PCR product. The primers were synthesized at the Microchemical Facility (University of Minnesota), and the molecular beacons were synthesized by Integrated DNA Technologies (Coralville, IA, USA). The sequences and hybridization positions are shown in Table 1.
Table 1. Primers and molecular beacons.
| Position | Sequence | |
|---|---|---|
| Primers | ||
| Forward (F1) | 7,798–7,820 | 5′-CATCCTAGTCCTCATCGCCCTCC |
| Reverse 1 (R1) | 8,186–8,209 | 5′-GGGCATGAAACTGTGGTTTGCTCC |
| Reverse 2 (R2) | 15,727–15,749 | 5′-GAATGAGGAGGTCTGCGGCTAGG |
| Molecular beacons | ||
| MB1a | 8,072–8,092 | FAM-5′-CCAGCG GCCTAATGTGGGGA CAGCTCACGCTGG-BHQ1 |
| MB2a | 15,534–15,553 | HEX-5′-CCAGCG CTTGATGTGGGGAG GGGTGTCGCTGG-BHQ1 |
See Fig. 2 for primer binding details.
The stem sequence is shown in bold.
Fig. 2.
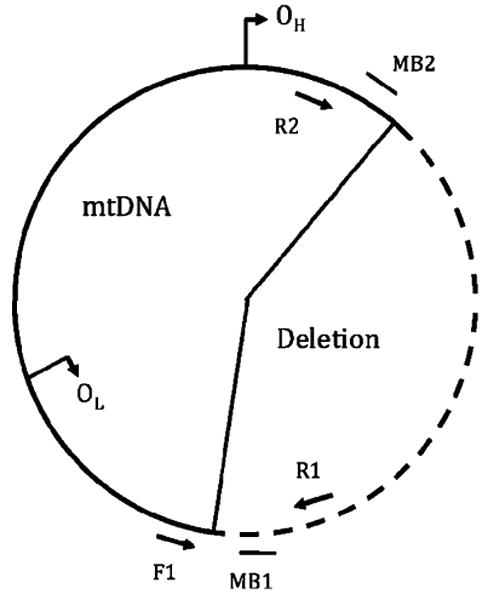
Multiplex three-primer PCR strategy for simultaneous detection of wild-type and deleted mtDNA. Wild-type mtDNA is amplified using the forward and reverse primers F1 and R1, respectively; amplicon detection accomplished with the molecular beacon MB1. Deleted mtDNA (indicated by the dashed line) is amplified using the forward and reverse primers F1 and R2, respectively; amplicon detection is accomplished with the molecular beacon MB2. OH and OL indicate the origin of replication of the heavy and light DNA strands, respectively. Sequences for F1, F2, R1, MB1, and MB2 are shown in Table 1. Other details are published elsewhere [14]
The 6-carboxyfluorescein (FAM) and hexachlorofluorescein (HEX) fluorescence was quenched by Black Hole Quencher-1 (BHQ1) when the molecular beacons were not hybridized to DNA. PCR was performed using FastStart TaqMan Probe Master Mix with ROX (Roche Diagnostics, Indianapolis, IN, USA) and contained 300 nM of each molecular beacon and primer. The PCR reagents were added to the PCR wells following collection of mitochondria to a final volume of 25 μl. The volume of buffer that was collected from the capillary for each mitochondrial fraction was negligible (≪1 μl). Contamination of the PCR wells was minimized by performing all additions of reagents in a laminar flow hood, and PCR wells were capped when not in use. The capillary cytometry instrument was housed in a room with HEPA-filtered forced air, and the instrument was cleaned prior to collection. Thermal cycling was performed by a Mx3000P™ QPCR system (Stratagene) as follows: one cycle of 95 °C for 10 min and 50 cycles of 95 °C for 30 s, 60 °C for 1 min, 72 °C for 30 s. The fluorescence was measured in real time after the annealing stage in each cycle.
In previous experiments, preincubation with Triton X-100 and proteinase K resulted in 100% PCR amplification efficiency [14]. Here, the preincubation was omitted as it resulted in the detection of false positives for both the wild-type and deleted mtDNA products in no-template controls. By omitting the incubation step, the PCR efficiency decreased to approximately 80%, but there were no nonspecific PCR products detected in the no-template controls. Consequently, the PCR assay can no longer be used for absolute quantitation of mtDNA copy numbers but still reports the ratio of deleted mtDNA to wild-type mtDNA as shown in Eq. 1;
| (1) |
where Eff is the amplification efficiency (0.8) and WTCt and ΔCt are the threshold cycles for wild-type and deleted mtDNA, respectively.
Data analysis
Cytograms were analyzed using Igor Pro version 5.03 (Wavemetrics, Lake Oswego, OR, USA). Background shifts and broad peaks were removed from the cytograms using median filtering. The appropriate median filter size was determined empirically to minimize the number of negative peaks in the cytogram while eliminating background shifts. The fluorescence intensity and time for each fluorescent event with intensity greater than five times the standard deviation in the background (S/N ratio >5) was tabulated with an in-house written PickPeak procedure. The average peak width was 22±11 ms (full width at half maximum (FWHM), n=945).
PCR amplification plots were analyzed using the Mx3000P version 2.0 software (Stratagene). The fluorescence thresholds were adjusted manually for both fluorescence channels but remained constant for all samples within an experiment after they were optimized. The cycle number at which the fluorescence reaches the user-defined threshold is the threshold cycle (Ct). It was recorded for both fluorescence channels in each well.
The total mtDNA content in a given PCR well was calculated using Eq. 2 as follows:
| (2) |
where FT is the total fluorescence intensity of all detected events in a given collection window, and fWT and fΔ are fluorescence factors that relate the fluorescence intensity measured by capillary cytometry with the number of wild-type and deleted mtDNA molecules, respectively. The factor fWT was approximated from a previous report [12], and fΔ was calculated by correcting fWT for the size of the deleted mtDNA molecule. NWT and NΔ are the number of wild-type and deleted mtDNA molecules in the PCR well, respectively. To determine the total number of mtDNA molecules in each PCR well, Eq. 2 was solved using the ratio of deleted mtDNA to wild-type mtDNA calculated from Eq. 1.
Results
Quantitation of heteroplasmy in samples containing mitochondria required: (1) labeling mtDNA with PicoGreen, (2) detection of individual mitochondria by LIF, (3) collection of mitochondria post-LIF detection, and (4) quantitation of heteroplasmy with a multiplex PCR assay.
Capillary cytometry of individual mitochondria
Mitochondria were fluorescently labeled with PicoGreen (λex=502 nm, λem=522 nm when bound to mtDNA). PicoGreen undergoes an approximately 1,980-fold fluorescent enhancement when bound to double-stranded DNA [19] and interference with RNA is minimal [16]. Furthermore, contamination with nuclear DNA is not anticipated due to the relatively high g-force used to sediment whole cells and nuclei in the mitochondria isolation procedure. Therefore, fluorescence events that are detected are expected to be mitochondria that contain mtDNA.
The capillary cytometry instrument uses the same LIF detector described previously [12], except that detection is performed on-column as opposed to off-column. The LOD for on-column LIF detection of fluorescein was 4±2 zmol (n=3), which is similar to a previously reported LOD (<5 zmol) for an off-column LIF detector that was used to quantitate mtDNA in individual mitochondria [12]. Therefore, it is expected that mitochondria containing a single copy of wild-type mtDNA labeled with PicoGreen is detectable. Capillary cytometry was performed by continuously introducing a suspension of PicoGreen-labeled mitochondria hydrodynamically into the capillary. For collection of individual mitochondrial events, the suspension was diluted until a few single events were detected every 30 s. The first column of Fig. 3 displays four representative cytograms showing detection of individual mitochondrial events.
Fig. 3.

Detection of isolated mitochondria by capillary cytometry. Mitochondria from ΔH2-1 cybrid cells were hydrodynamically driven through a poly(acryloylaminopropanol)-modified capillary at approximately 0.16 cm/s. Laser-induced fluorescence detection was performed on-column, and mitochondria were collected into PCR wells. PCR reagents were added to each PCR well following collection, and 50 cycles of amplification were performed. Each cytogram (left column) has a box overlaid to indicate the collection window and the number of mitochondria detected in each window. Beside each cytogram is the corresponding real-time PCR amplification plot. In each amplification plot, the wild-type mtDNA trace is offset by 0.03 fluorescence units for clarity, and the dashed line represents the fluorescence threshold used to determine the threshold cycle
Although the cytograms displayed in Fig. 3 are similar to previously presented CE-LIF electropherograms (e.g., Fig. 1a, [12]), there are two notable differences. First, since the mitochondria are mobilized hydrodynamically and not electrokinetically, there is no electrophoretic separation of mitochondria. The frequency of detected events depends solely on the number of mitochondria suspended in the sample and the flow rate through the capillary. Secondly, the velocity of mitochondria through the capillary was approximately two times faster (0.16 cm/s) than when using an electric field (0.08 cm/s). Consequently, the peak widths are smaller (22±11 ms FWHM, n=945) than the peak widths normally observed in CE-LIF (approximately 50 ms).
Amplification of mtDNA
mtDNA was amplified and quantitated from collected mitochondria following the PCR strategy described previously [14]. However, the PCR assay used here omitted the incubation with Triton X-100 and proteinase K because their use caused detection of false positives for both the wild-type and deleted mtDNA PCR products in no-template controls. As previously reported, the use of Triton X-100 and proteinase K is necessary for absolute quantitation of mtDNA copy numbers [14]. Since these reagents were omitted from the PCR mixture, absolute quantitation was not feasible. Instead, we report only relative quantitation of mtDNA (i.e., ratio of deleted to wild-type mtDNA) based on Eq. 1. Based on relative quantitation, four examples of PCR results are (Fig. 3, right column): (1) collection of an individual mitochondrial particle that contains 100% deleted mtDNA, (2) collection of an individual mitochondrial particle that contains 100% wild-type mtDNA, (3) collection of seven mitochondrial particles that contain both wild-type and deleted mtDNA, and (4) detection of mtDNA PCR assay but not in the LIF detector. In these plots, a given type of DNA is present when fluorescence intensity (y-axis) increases with PCR cycle number (x-axis) and crosses the respective threshold (horizontal dashed line). For instance, the first plot is positive only for deleted DNA, while the second plot down is positive only for wild-type DNA.
Table 2 shows the results of LIF detection, which includes number of mtDNA molecules in the sample (column 1), total intensity of PicoGreen signal (column 2), and number of collected mitochondria (column 4). Table 2 also shows the results of PCR amplification of the collected material (i.e., percent of deleted mtDNA, column 3). These results are graphically shown in Fig. 5. For large numbers of collected mitochondria (>20), the percent deleted mtDNA was 89±5%, which is slightly higher than previously published results for single ΔH2-1 cells (75±16%) and bulk lysates (80±13%) [14]. Heteroplasmy was detected in all eight PCR samples that contained >13 mitochondria. In contrast, heteroplasmy was not detected in any samples that contained less than four mitochondria, and no individual mitochondrial particles were heteroplasmic. This agrees with the findings of Cavelier et al., in which they demonstrated that individual mitochondria were predominantly homoplasmic for an mtDNA point mutation [11].
Table 2. Real-time PCR amplification of wild-type and deleted mtDNA from mitochondria collected after laser-induced fluorescence detection.
| No. of mtDNA moleculesa | Total PicoGreen signal | Deleted mtDNA (%)b | No. of collected mitochondria |
|---|---|---|---|
| 0 | 0.00 | 100.0 | 0 |
| 0 | 0.00 | 100.0 | 0 |
| 0 | 0.00 | 0.0 | 0 |
| 0 | 0.00 | 100.0 | 0 |
| 3 | 0.06 | 0.0 | 1 |
| 3 | 0.03 | 100.0 | 1 |
| 3 | 0.06 | 0.0 | 1 |
| 4 | 0.07 | 0.0 | 2 |
| 6 | 0.06 | 100.0 | 2 |
| 7 | 0.12 | 0.0 | 1 |
| 8 | 0.08 | 100.0 | 2 |
| 8 | 0.14 | 0.0 | 3 |
| 10 | 0.18 | 0.0 | 2 |
| 10 | 0.18 | 0.0 | 2 |
| 10 | 0.18 | 0.0 | 3 |
| 11 | 0.11 | 100.0 | 1 |
| 18 | 0.17 | 100.0 | 2 |
| 18 | 0.18 | 100.0 | 3 |
| 20 | 0.35 | 0.0 | 1 |
| 23 | 0.23 | 100.0 | 5 |
| 24 | 0.23 | 100.0 | 6 |
| 25 | 0.31 | 66.7 | 6 |
| 27 | 0.26 | 100.0 | 8 |
| 28 | 0.27 | 100.0 | 2 |
| 28 | 0.46 | 18.2 | 6 |
| 35 | 0.34 | 100.0 | 4 |
| 38 | 0.37 | 100.0 | 7 |
| 39 | 0.69 | 0.0 | 2 |
| 40 | 0.59 | 39.7 | 9 |
| 42 | 0.27 | 100.0 | 5 |
| 46 | 0.45 | 100.0 | 3 |
| 50 | 0.49 | 100.0 | 5 |
| 57 | 0.64 | 84.1 | 7 |
| 58 | 1.05 | 0.0 | 7 |
| 59 | 0.58 | 100.0 | 7 |
| 73 | 0.72 | 100.0 | 6 |
| 79 | 1.04 | 59.3 | 7 |
| 83 | 0.97 | 76.4 | 6 |
| 88 | 0.87 | 100.0 | 4 |
| 90 | 0.91 | 97.1 | 8 |
| 100 | 1.80 | 0.0 | 5 |
| 101 | 1.00 | 100.0 | 4 |
| 107 | 1.29 | 73.3 | 8 |
| 122 | 1.20 | 100.0 | 13 |
| 126 | 1.25 | 98.9 | 12 |
| 146 | 2.62 | 0.0 | 9 |
| 148 | 2.66 | 0.0 | 11 |
| 154 | 1.52 | 100.0 | 3 |
| 157 | 1.70 | 87.8 | 7 |
| 170 | 1.92 | 82.1 | 9 |
| 175 | 1.73 | 100.0 | 12 |
| 181 | 3.26 | 0.0 | 1 |
| 192 | 2.24 | 78.0 | 11 |
| 208 | 2.18 | 92.1 | 12 |
| 209 | 3.76 | 0.0 | 2 |
| 210 | 2.07 | 100.0 | 9 |
| 239 | 2.57 | 88.6 | 16 |
| 295 | 3.45 | 77.4 | 8 |
| 415 | 4.46 | 88.8 | 21 |
| 420 | 4.37 | 93.0 | 5 |
| 430 | 4.27 | 98.9 | 11 |
| 537 | 6.44 | 73.6 | 12 |
| 615 | 6.06 | 100.0 | 10 |
| 617 | 6.61 | 89.4 | 7 |
| 664 | 7.47 | 82.8 | 7 |
| 808 | 8.99 | 84.3 | 15 |
| 811 | 8.25 | 95.8 | 29 |
| 822 | 8.56 | 92.9 | 32 |
| 988 | 17.78 | 0.0 | 8 |
| 1,516 | 16.62 | 86.3 | 46 |
| 1,818 | 20.35 | 83.4 | 64 |
| 2,001 | 22.16 | 84.9 | 64 |
Mitochondria were isolated from ΔH2-1 cybrid cells, harboring a heteroplasmic mtDNA deletion, and labeled with PicoGreen. Mitochondria were hydrodynamically mobilized through a capillary and detected on-column using laser-induced fluorescence. Mitochondria were then collected from the outlet of the capillary, and real-time PCR was performed to amplify the wild-type and deleted mtDNA. Of the collected samples, 35% were PCR negative
The number of total mtDNA molecules was determined using Eq. 2
Italics indicate samples displaying heteroplasmy
Fig. 5.

Predicted percentages of deleted mtDNA based on the number of mtDNA molecules in the sample. The expected percent of deleted mtDNA was calculated using the binomial distribution, assuming mtDNA is randomly distributed (triangles) or that individual mitochondria are homoplasmic (squares) and contains on average 4.6 mtDNA molecules per mitochondrion [13]
Discussion
Technological developments
We have built a capillary cytometry instrument that allows detection and collection of isolated mitochondrial particles. It mobilizes mitochondrial particles through a capillary using pressure-driven flow that are then detected by LIF. The components of this detector are identical to those used in LIF, but the fluidics is compatible for collection of mitochondrial particles postdetection. Although it was expected that the fluorescence background would increase due to scatter at the capillary walls and hence increase the LOD, this was not observed. In fact, the LOD for fluorescein was statistically similar to off-column LIF detection (p>0.05). Since the LOD and sensitivity for each detection method, i.e., off-column and on-column, are similar, we applied the previously reported fluorescence factor that correlated the number of wild-type mtDNA copies to the LIF fluorescence intensity [12].
To amplify wild-type and deleted mtDNA, a previously reported multiplex three-primer PCR assay was used [14]. The assay was performed using the previously described primers and molecular beacons. However, an important modification to the procedure was that the incubation with Triton X-100 and proteinase K was omitted because it resulted in nonspecific detection of wild-type and deleted mtDNA in no-template controls (4.3±2.2 wild-type copies; 4.5±2.7 deleted copies, n=4). By performing the PCR directly from the collected mitochondria, without any treatment with Triton X-100 and proteinase K, these false positives in the no-template controls were eliminated.
One complication with collection of individual mitochondrial particles for PCR analysis is false positives (i.e., detection of PCR products when templates are not present). False positives were detected by PCR in seven out of 20 no-template controls. Despite the precautions taken, this appears to be the result of cross-contamination from handling the PCR wells or contamination from the collection environment. The latter would only be expected to result in contamination with wild-type mtDNA from the authors and, indeed, the five out of the seven cases involved wild-type mtDNA. If cross-contamination results in carryover by the capillary, its washing or cleaning between collection intervals might reduce this contamination. The authors did not attempt any such procedure in order to decrease the risk of jarring the on-column detector out of alignment or breaking the capillary tip. In order to address this issue, further developments of the collection design may include automated washes in between collection intervals.
Also of interest, mtDNA was not detected by the PCR assay in approximately 35% of samples that were expected to contain at least a single mitochondrial particle. This may have been caused by adsorption of mitochondria to the capillary wall postdetection or indicate the need for more accurate methods of determining the velocity of particles traveling through the capillary. While further characterization of the detection and collection process will help in explaining the absence of PCR amplification in some instances, this information does not prevent the analysis of the relative abundances of mutated and wild-type mtDNA that is presented below.
mtDNA was also detected in four samples where no mitochondrial particles were detected by capillary cytometry (Table 2). Based on our previous study, the average fluorescence intensity from a single wild-type mtDNA molecule was slightly higher than the detection threshold [12]. Since the detection is based upon the binding of PicoGreen with mtDNA, the fluorescence from deleted mtDNA molecules used in this study is expected to be 45% less than from a wild-type molecule. The reduction in fluorescence makes individual mitochondria carrying a single deleted mtDNA molecule undetectable. This may explain the results in which deleted mtDNA was detected despite no mitochondria being detected by capillary cytometry (first, second, and fourth entries in Table 2). Alternatively, the detection of mtDNA in samples (e.g., wild-type mtDNA) where no mitochondrial particles were detected by capillary cytometry could also be caused by carryover on the capillary tip.
Heteroplasmy models
The “faithful nucleoid” model provides an explanation for the dynamics of mtDNA organization and inheritance [10]. In particular, the “faithful nucleoid” model attempts to resolve the incongruence between the functional complementation observed in heteroplasmic cells, which implies a genetic unit of five to ten mtDNA molecules, and the experimentally determined segregation number (number of independently segregating mtDNA units), which is comparable with the total mtDNA copy number in a cell. The most important requirement for the validity of the “faithful nucleoid” model is that nucleoids are heteroplasmic and that they reflect the total cellular heteroplasmy. For example, if the heteroplasmy level of the cell is 80%, then on average each nucleoid should contain eight mutated and two wild-type mtDNA molecules.
Our results do not support the “faithful nucleoid” model. As shown in Fig. 4 and Table 2, heteroplasmy was not detected until many mtDNA molecules were collected into the same PCR well. In fact, heteroplasmy was not detected unless >24 mtDNA molecules were collected. Only when the number of mtDNA molecules collected in a sample increased did heteroplasmy (89±5%) become similar to the percentage of deleted mtDNA reported for bulk cell lysates (80±13%) [14]. Altogether, these results suggest that nucleoids are homoplasmic (i.e., composed of either 100% wild-type or 100% deleted mtDNA).
Fig. 4.

Detection of heteroplasmy by real-time PCR. The percent of deleted mtDNA as determined by real-time PCR is plotted against the total number of mtDNA molecules detected by capillary cytometry. The number of deleted and wild-type mtDNA molecules was determined using Eq. 2
In order to further investigate heteroplasmy at the nucleoid level, we used a binomial distribution p(x) to predict the level of heteroplasmy as a function of the number mtDNA molecules in a sample. This binomial distribution is:
| (3) |
where p is the probability of detecting deleted mtDNA (i.e., 0.8 detected in bulk analyses), n is the number of samples (either individual mtDNA molecules or individual mitochondrial particles), x is the number of samples containing deleted mtDNA, and
| (4) |
Figure 5 displays the expected percentage of deleted mtDNA versus the number of mtDNA molecules, assuming that mtDNA is distributed randomly (triangles) or that mtDNA in each sampled mitochondrion is homoplasmic (squares). Comparison of these predictions with the experimental results (Fig. 4) clearly indicates that mtDNA is not randomly distributed (i.e., the “faithful nucleoid” model) since the experimentally measured percentage of deleted mtDNA is low until larger numbers of mtDNA molecules are sampled. Instead, the experimental data suggest that individual mitochondrial particles are homoplasmic (cf. squares in Fig. 5), which in turn would imply that their nucleoids are homoplasmic.
Consistent with our findings, there is one report on the quantitation of heteroplasmy in individual mitochondrial particles [11]. Cavelier et al. used fluorescence-activated cell sorting to deposit individual mitochondrial particles into PCR wells. Even when mtDNA copy numbers could not be determined, this study found that 75% of individual mitochondrial particles were homoplasmic. In a second report, Schon and Gilkerson did not observe transcomplementation of DNA among nucleoids, thereby supporting that mtDNA nucleoids do not conform to the “faithful nucleoid” model (i.e., nucleoids do not reflect the overall heteroplasmy of the cell) [20].
Conclusions
We developed a method that is based on detection of individual mitochondria by capillary cytometry and quantitation of heteroplasmy in collected mitochondria with real-time PCR. Using this method, we investigated the distribution of wild-type and mutated mtDNA molecules within isolated mitochondrial particles. The results do not support the “faithful nucleoid” model but suggest that nucleoids are composed of either 100% wild-type or 100% mutated mtDNA. This hypothesis could be further explored by utilizing this method to analyze nucleoid preparations. Such preparations have been reported previously [21].
The method described here could also be adapted to investigate and monitor heteroplasmy in mitochondria from different cell types and tissues in which mutations are well characterized. For instance, heteroplasmy in cells or tissues from mitochondrial-related diseases, cancer, or phylogenetic studies [22, 23] could be investigated at the suborganellar level with the methodology presented here. The method described here could also be further modified to create powerful technologies that are applicable to single-cell studies [24], direct sampling from tissue cross sections [25, 26], or powerful sequencing technologies [27].
Acknowledgments
This work was supported by the National Institutes of Health (R03-AG18099 and R01-AG20866). B.G.P. acknowledges support from the NIH Chemistry/Biology Interface Training Grant (T32-GM08700).
Contributor Information
Bobby G. Poe, III, Department of Chemistry, University of Minnesota, Minneapolis, MN 55455, USA.
Ciarán F. Duffy, Department of Chemistry, University of Minnesota, Minneapolis, MN 55455, USA
Michael A. Greminger, Department of Mechanical Engineering, University of Minnesota, Minneapolis, MN 55455, USA
Bradley J. Nelson, Department of Mechanical Engineering, University of Minnesota, Minneapolis, MN 55455, USA
Edgar A. Arriaga, Email: Arriaga@umn.edu, Department of Chemistry, University of Minnesota, Minneapolis, MN 55455, USA
References
- 1.Naviaux RK. Eur J Pediatr. 2000;159(Suppl 3):S219. doi: 10.1007/pl00014407. [DOI] [PubMed] [Google Scholar]
- 2.Schmiedel J, Jackson S, Schafer J, Reichmann H. J Neurol. 2003;250:267. doi: 10.1007/s00415-003-0978-3. [DOI] [PubMed] [Google Scholar]
- 3.Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, Morrow JD, Van Remmen H, Sedivy JM, Yamasoba T, Tanokura M, Weindruch R, Leeuwenburgh C, Prolla TA. Science. 2005;309:481. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- 4.Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H, Wallace DC, Rabinovitch PS. Science. 2005;308:1909. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- 5.Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly YM, Gidlof S, Oldfors A, Wibom R, Tornell J, Jacobs HT, Larsson NG. Nature. 2004;429:417. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 6.Garrido N, Griparic L, Jokitalo E, Wartiovaara J, van der Bliek AM, Spelbrink JN. Mol Biol Cell. 2003;14:1583. doi: 10.1091/mbc.E02-07-0399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.King MP, Attardi G. Science. 1989;246:500. doi: 10.1126/science.2814477. [DOI] [PubMed] [Google Scholar]
- 8.Bentlage HA, Attardi G. Hum Mol Genet. 1996;5:197. doi: 10.1093/hmg/5.2.197. [DOI] [PubMed] [Google Scholar]
- 9.Porteous WK, James AM, Sheard PW, Porteous CM, Packer MA, Hyslop SJ, Melton JV, Pang CY, Wei YH, Murphy MP. Eur J Biochem. 1998;257:192. doi: 10.1046/j.1432-1327.1998.2570192.x. [DOI] [PubMed] [Google Scholar]
- 10.Jacobs HT, Lehtinen SK, Spelbrink JN. Bioessays. 2000;22:564. doi: 10.1002/(SICI)1521-1878(200006)22:6<564::AID-BIES9>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 11.Cavelier L, Johannisson A, Gyllensten U. Exp Cell Res. 2000;259:79. doi: 10.1006/excr.2000.4949. [DOI] [PubMed] [Google Scholar]
- 12.Navratil M, Poe BG, Arriaga EA. Anal Chem. 2007;79:7691. doi: 10.1021/ac0709192. [DOI] [PubMed] [Google Scholar]
- 13.Satoh M, Kuroiwa T. Exp Cell Res. 1991;196:137. doi: 10.1016/0014-4827(91)90467-9. [DOI] [PubMed] [Google Scholar]
- 14.Poe BG, Navratil M, Arriaga EA. Anal Biochem. 2007;362:193. doi: 10.1016/j.ab.2006.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moraes CT, Kenyon L, Hao H. Mol Biol Cell. 1999;10:3345. doi: 10.1091/mbc.10.10.3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ashley N, Harris D, Poulton J. Exp Cell Res. 2005;303:432. doi: 10.1016/j.yexcr.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 17.Poe BG, Navratil M, Arriaga EA. J Chromatogr A. 2006;1137:249. doi: 10.1016/j.chroma.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 18.Gelfi C, Curcio M, Righetti PG, Sebastiano R, Citterio A, Ahmadzadeh H, Dovichi NJ. Electrophoresis. 1998;19:1677. doi: 10.1002/elps.1150191026. [DOI] [PubMed] [Google Scholar]
- 19.Singer VL, Jones LJ, Yue ST, Haugland RP. Anal Biochem. 1997;249:228. doi: 10.1006/abio.1997.2177. [DOI] [PubMed] [Google Scholar]
- 20.Schon EA, Gilkerson RW. Biochim Biophys Acta Gen Subj. 2010;1800:245. doi: 10.1016/j.bbagen.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 21.Gilkerson RW. Int J Biochem Cell Biol. 2009;41:1899. doi: 10.1016/j.biocel.2009.03.016. [DOI] [PubMed] [Google Scholar]
- 22.Chatterjee A, Mambo E, Sidransky D. Oncogene. 2006;25:4663. doi: 10.1038/sj.onc.1209604. [DOI] [PubMed] [Google Scholar]
- 23.DiMauro S, Hirano M, Schon EA. Muscle Nerve. 2006;34:265. doi: 10.1002/mus.20598. [DOI] [PubMed] [Google Scholar]
- 24.Johnson RD, Navratil M, Poe BG, Xiong G, Olson KJ, Ahmadzadeh H, Andreyev D, Duffy CF, Arriaga EA. Anal Bioanal Chem. 2007;387:107–118. doi: 10.1007/s00216-006-0689-6. [DOI] [PubMed] [Google Scholar]
- 25.Ahmadzadeh H, Johnson RD, Thompson L, Arriaga EA. Anal Chem. 2004;76:315. doi: 10.1021/ac034809g. [DOI] [PubMed] [Google Scholar]
- 26.Ahmadzadeh H, Thompson LV, Arriaga EA. Anal Bioanal Chem. 2006;384:169. doi: 10.1007/s00216-005-0171-x. [DOI] [PubMed] [Google Scholar]
- 27.He YP, Wu J, Dressman DC, Iacobuzio-Donahue C, Markowitz SD, Velculescu VE, Diaz LA, Kinzler KW, Vogelstein B, Papadopoulos N. Nature. 2010;464:610. doi: 10.1038/nature08802. [DOI] [PMC free article] [PubMed] [Google Scholar]