

Abstract
Targeting the epidermal growth factor receptor kinase (EGFR) with ATP-competitive kinase inhibitors results in dramatic but short-lived responses in patients with EGFR mutant non small cell lung cancer. A series of novel covalent EGFR kinase inhibitors with selectivity for the clinically relevant T790M “gatekeeper” resistance mutation relative to wild-type EGFR were discovered by library screening. A representative compound 3i was obtained through a systematic SAR study guided by mutant EGFR-dependent cellular proliferation assays.
Protein kinases, particularly receptor tyrosine kinases, are estimated as the second largest class of therapeutic targets. There are approximately 518 protein kinase genes encoded in the human genome. Many of these have been observed to become constitutively activated by amplification or mutation, thus becoming oncogenic. A phenomenon that has been termed “oncogene-addiction” renders some cancer cells to be exquisitely sensitive to the inhibitors targeting that particular oncogene 1. For example, patients with non small cell lung cancer (NSCLC) that harbor activating mutations in the EGFR kinase domain such as L858R and exon 19 deletions exhibit excellent objective response rates when treated with EGFR kinase inhibitors such as Gefitinib or Erlotinib.
The epidermal growth factor receptor (EGFR) family of receptor tyrosine kinases regulates cell proliferation, survival, adhesion, migration and differentiation. It consists of EGFR/ErbB-1, HER2/ErbB-2, Her3/Erb-3 and Her4/Erb-4. Among them, the aberrant activity of EGFR and ErbB-2 have been shown to play an important role in the development and growth of tumor cells. The blockade of EGFR and ErbB-2 has been clinically validated as a leading approach to selectively target cancer cells. Currently two small molecule kinase inhibitors, ZD1839 (Iressa, gefitinib) and OSI774 (Tarceva, erlotinib), are in clinical use for the treatment of lung cancer. There are more than ten EGFR inhibitors currently being tested in clinical trials. Among them are several covalent inhibitors such as HKI-3572, HKI-2722, EKB5692, BIBW29923, and PF2998044 which target a unique cysteine 797 residue located at the lip of the EGFR ATP binding site 5.
Point mutations in the kinase domain of EGFR as well as up-regulation of by-pass signaling pathways are frequently observed resistance mechanisms in patients treated with gefitinib and erlotinib6,7. A single point mutation at the gatekeeper position, T790M in EGFR kinase domain accounts for approximately 50 percent of acquired resistance and results in relapse of disease with a median progression free survival of 10–14 months. Inspection of inhibitor-kinase complex structures suggests that replacement of threonine with bulkier methionine would be expected to sterically interfere with inhibitor binding and would result in loss of a key hydrogen-bond. However binding assays have demonstrated that the T790M mutation appears to exert its effects primarily by increasing the affinity for ATP which leads to higher concentrations of the competitive inhibitors being required to effectively suppress kinase activity 8. We focused our efforts on potential covalent compounds because effective targeting of the T790M EGFR mutant with a reversible inhibitor is estimated to require a Kd of 0.05 nM8. Although several C797 directed covalent inhibitors are reported to be active against T790M EGFR2, the concentrations required to inhibit this mutant are considerably higher and unlikely to be achieved in patients being treated with these agents. To date, there have been no clinical reports of responses of T790M patients to first-generation covalent inhibitors. As the gatekeeper position is known to be one of the most important selectivity determinants for kinase inhibitors, we reasoned that a fundamentally different scaffold relative to the common quinazoline or quinoline nitrile-type EGFR scaffold might be better suited for inhibition of T790M. In order to rapidly survey a variety of scaffolds we prepared a small library consisting of frequently used heterocyclic cores that all contained an acrylamide functionality. Molecular modeling of the presumed binding mode was used to guide selection of where to install the acrylamide to allow covalent bond formation with C797.
The first class of irreversible compounds was based on purines and was synthesized as shown in Scheme 1.
Scheme 1.

Synthesis of purine compound 1a (a) Pyridine, CH2Cl2 (b) NaH, MeI, DMF (c) Pd/C, H2, MeOH (d) 3,4-2H-dihydropyran; 1-Boc-3-aminopiperidine, DIEA, BuOH, 60 oC (e) Pd2(dba)3, 1,3-Bis(2,6-diisopropyphenyl)imidazonium chloride, NaOtBu, 1,4-dioxane, 95oC (f) (i) HCl, EtOH; (ii) acryloyl chloride, DIEA, CH2Cl2.
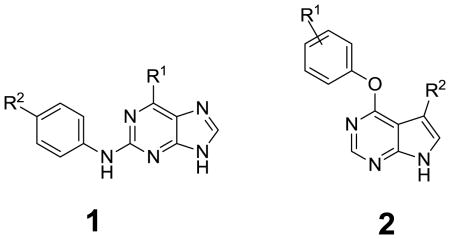
The aniline A1 was obtained in three steps starting with acylation using propionyl chloride, methylation with methyl iodide and hydrogenation of the 4-nitroaniline. Tetrahydropyran (THP) protected 2,6-dichloropurine was regioselectively aminated at the 6-position to afford intermediate A2. Buchwald coupling with A1 in the presence of a carbene ligand provided A3, which was converted into 1a through acidic de-protection of THP, followed by acylation with acryloyl chloride. The purine analogs of 1a were synthesized in an analogous manner using the appropriate amines and anilines.
The compounds listed in Table 1 were evaluated for their antiproliferative effects in cellular assays using cancer cell lines that are known to be dependent on specific EGFR genotypes. Specifically, PC9 cells (EGFR E746_A750) were used to model an activated allele of EGFR while PC9 growth-resistant GR cells (EGFR E746_A750/T790M) were used to model the T790M resistance allele 9. To control for non-specific cytotoxicity, compounds were evaluated against A549 lung adenocarcinoma cells (K-Ras dependent). Evaluation of purine and pyrrolopyrimidine acrylamide sub-libraries revealed that several compounds such as 1e and 2a possessed submicromolar cellular potency which compared favorably with reference inhibitors such as Iressa and PF299804 (Table 1), while they didn’t show inhibition on proliferation of A549 cells. The SAR also revealed that superior potency could be obtained when the C6-position of the purine was substituted with an aryl versus a cycloalkyl group. Variation of water solubility-enhancing groups at the 2-position of purine did not exert a dramatic influence on the antiproliferative activities.
Table 1.
Cellular antiproliferative data
 | ||||
|---|---|---|---|---|
| Compd | R1 | R2 | Cellular antiproliferative Activity (EC50, uM)a |
|
| EGFR del | del/T790M | |||
| Iressa | 0.017 | 7 | ||
| PF299804 | 0.006 | 5 | ||
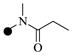
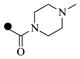
| 1a | 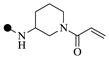 |
 |
7 | 10 |
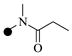
| 1b | 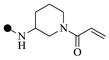 |
1 | 5 | |
| 1c | 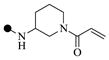 |
 |
10 | 10 |
| 1d | 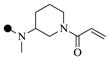 |
 |
0.90 | 6 |
| 1e | 0.30 | 0.60 | ||
| 2a | 3-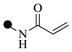
|
0.50 | 0.50 | |
| 2b | 3-
|
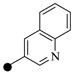 |
6 | 8 |
| 2c | 3-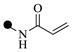
|
6 | 6 | |
| 2d | 4-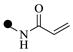
|
6 | 6 | |
| 2e | 4-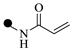
|
 |
10 | 10 |
n≥ 2
A second sub-library of pyrrolopyrimdine-derived acrylamides was synthesized as depicted in Scheme 2. Regioselective iodination of 6-chlorodeazapurine was achieved with N-iodosuccinimide. The resulting intermediate was protected with SEM. Selective Suzuki coupling at the C5 position of B1 gave rise to B2. Intermediate B3 was obtained by nucleophilic substitution at the C4 position. Compound 2a was obtained after acidic deprotection of B3, followed by acylation with acryloyl choride and sequential removal of hydroxymethyl group.
Scheme 2.

Synthesis of pyrrolopyrimidine compound 2a (a) (i) NIS, CH2Cl2; (ii) NaH, SEM-Cl, DMF (b) Pyridine-3-boronic acid, Pd(dppf)Cl2, dppf, Na2CO3(aq)/Dioxane (1:2) (c) 3-Boc-aminophenol, K2CO3, DMSO, 100oC (d) (i) TFA, CH2Cl2; (ii) acryloyl chloride, DIEA, CH2Cl2; (iii) NaHCO3(aq), THF.
In the pyrrolopyridine and quinoline series, the compounds with an electrophile at the meta- position exhibited superior potency relative to para-substituted compounds (Compare 2a with 2d). Larger substituents such as 4-methoxyphenyl substituent at the 3-position appear to be too bulky to fit in the ATP binding site.
A third sub-library was prepared using 6-membered heterocycles including 4,6-pyrimidines, triazine (data not shown) and 5-chloro-1,3-pyrimidines The synthetic scheme of one typical compound 3h is shown in Scheme 3. 3-Nitrophenol was regioselectively introduced to the C4 position of 2,4,5-trichloropyrimidine to afford intermediate C1. Amination of C1 with 4-(4-methylpiperazin-1-yl) aniline was accomplished in the presence of trifluoroacetic acid at 100°C. The nitro group in C2 was reduced by PtO2/H2 without concominant reduction of the C5 chloride. Acylation of C3 by acryloyl chloride gave rise to 3h. The pyrimidine analogs of 3h were obtained in a similar manner using the appropriate phenols and anilines.
Scheme 3.

Synthesis of pyrimidine compound 3h (a) K2CO3, DMF (b) 4-(4-methylpiperazin-1-yl) aniline, TFA, 2-BuOH, 100oC (c) PtO2, H2, MeOH (d) acryloyl chloride, DIEA, CH2Cl2.
The compounds were evaluated for their antiproliferative activity against PC9 and PC9 GR cells (Table 2). Only compounds with meta-substituted acrylamides are shown because the corresponding para-substituted acrylamides were less active. We initially investigated SAR for variation of substituents at the C2-aniline of the pyrimidine, which we would expect to be directed towards solvent based upon the co-structures of related 2,4-disubstituted pyrimidine with kinases (e.g. PDB code 2JKK). The potency of simple C2-fluoroaniline compound 3a may have been limited by its poor water solubility (Table 2). The replacement of the fluoro group with functionality that would be expected to improve water solubility such as morpholine 3c, 4-hydroxypiperidine 3d, N-methylpiperidine 3e, 3f, 3g, piperazine 3i, piperazine derivative 3j resulted in 5–40 fold increases in potency. The analogs which possess amide groups at the para position of the aniline (3k, 3l, 3m, 3n, 3o) exhibited no significant improvement in activity relative to fluoro or pyridinyl substituted compounds. We next explored SAR at the ortho position of C2-aniline moiety. Replacement of methoxy with CF3 (3u vs. 3i) resulted in 6-fold loss of potency. Removal of the methoxy group (3h) improved potency to the single digital nanomolar EC50 range. However, 3i is more selective for EGFR than 3h 10 when evaluated in biochemical and cellular assays of other kinases. A one carbon extension of the piperazine analog 3q is slightly less active than 3h. Introduction of CF3 to the meta position of C2-aniline (3r) resulted in a significant decrease in cellular potency. Two carbon extension of the piperazine analog 3t had approximately the same potency as 3h.
Table 2.
Cellular antiproliferative data
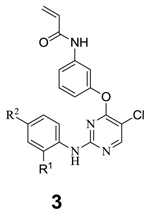 | ||||
|---|---|---|---|---|
| Compd | R1 | R2 | Cellular antiproliferative activity (EC50, uM)a |
|
| EGFR del | del/T790M | |||
| 3a | MeO | F | NDb | 0.80 |
| 3b | MeO | ND | 0.90 | |
| 3c | MeO | ND | 0.15 | |
| 3d | MeO | 0.20 | 0.80 | |
| 3e | MeO | 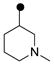 |
0.10 | 0.20 |
| 3f | MeO | 0.10 | 0.04 | |
| 3g | MeO | ND | 0.20 | |
| 3h | H | 0.015 | 0.003 | |
| 3i | MeO | 0.036 | 0.014 | |
| 3j | MeO | 0.08 | 0.10 | |
| 3k | H | 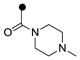 |
0.80 | 0.80 |
| 3l | MeO | 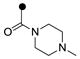 |
>1 | >1 |
| 3m | MeO | 0.50 | ||
| 3n | H | 0.80 | 1 | |
| 3o | MeO | 0.50 | 0.80 | |
| 3p | MeO | 5- |
ND | >10 |
| 3q | H | 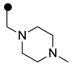 |
ND | 0.03 |
| 3r | m-CF3 | 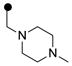 |
ND | 0.42 |
| 3s | H | 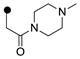 |
ND | 0.050 |
| 3t | H | ND | 0.008 | |
| 3u | CF3 | ND | 0.090 | |
n≥ 2
Not Detected
Next we optimized the heteroatom linkage at the pyrimidine 4-position. The ether linkage was replaced with sulfur (4a, 4b) and NH (4c) (Table 3). Both replacements were tolerated as evidenced by 3i, 4a, and 4c. The same trend was observed for analogs without substituents at the ortho position of C2-aniline (3i vs. 4b). Introduction of a chloro group at the ortho position of oxygen linkage (4d) resulted in a loss of activity. In contrast, introduction of a smaller fluoro-substitutent at the ortho or meta positions (4e, 4f) maintained similar potency as 3i.
Table 3.
Cellular antiproliferative data
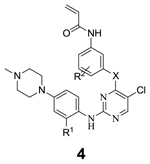 | |||||
|---|---|---|---|---|---|
| Compd | X | R1 | R2 | Cellular antiproliferative Activity (EC50, uM) |
|
| EGFR del | del/T790M | ||||
| 3i | O | OMe | H | 0.036 | 0.014 |
| 4a | S | OMe | H | 0.020 | 0.020 |
| 4b | S | H | H | 0.006 | 0.008 |
| 4c | NH | OMe | H | 0.040 | 0.020 |
| 4d | O | OMe | 2-Cl | >1 | >1 |
| 4e | O | OMe | 2-F | ND | 0.031 |
| 4f | O | OMe | 3-F | ND | 0.020 |
We next investigated how various substituents at the pyrimidine 5-position change their potency and selectivity against EGFR T790M. The analogs of 3h with Cl replaced by H, Br, CF3 were synthesized using the same route as depicted in scheme 3. Among them 3h is the most potent (Table 4) because it is likely that the chloro group engages in a dipole interaction with the thioether of the methionine at position 790 of EGFR10. 3h and 3i were profiled against a panel of 400 kinases using the Ambit Kinome screening platform. In addition to EGFR, several kinases demonstrated strong binding particularly those kinases possessing cysteines at the same position as EGFR. Potent inhibition of 3i against EGFR T790M mutant was also observed using enzymatic assays10,11. To confirm whether the observed binding activity translated into cellular activity, 3h and 3i were assayed against Ba/F3 cells transformed with TEL fusions of Bmx, Blk, Jak2 and Jak3. 3h and 3i both showed no inhibition of Jak2 while 3i, which possesses an ortho-methoxy group at C2-aniline substituent, is much weaker at inhibiting Jak3, Blk, Bmx, and thus more selective for EGFR than 3h10.
Table 4.
Cellular antiproliferative data
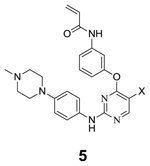 | |||
|---|---|---|---|
| Compd | X | Cellular antiproliferative Activity (EC50, uM) |
|
| EGFR del | Del/T790M | ||
| 3h | Cl | 0.015 | 0.003 |
| 5a | H | 0.08 | >1 |
| 5b | CF3 | 0.70 | 0.80 |
| 5c | Br | 0.80 | 0.90 |
To investigate if our EGFR inhibitors bind irreversibly, we constructed EGFR transformed Ba/F3 cell lines where the reactive cysteine residue is replaced with a serine (C797S). The C797S mutation resulted in a dramatic 400 and 1650-fold reduced potency for 3i in the L858R and E746_A750 backgrounds respectively. For example, 3i possesses an EC50 of 8 nM in L858R/T790M relative to an EC50 of >3300 nM in L858R/T790M/C797S and an EC50 of 2 nM in E746_A750_del/T790M relative to an EC50 of >3300 nM in E746_A750_del/T790M/C797S EGFR-transformed Ba/F3 cells. In addition the reversible analog of 3i, which has a saturated C-C bond instead of C=C bond and therefore is unable to undergo a Michael addition reaction with C797 was synthesized. Its EC50 value in cellular proliferation assays against E746_A750/T790M EGFR Ba/F3 cells was 1290 nM, which is 645-fold less potent than 3i (EC50=2 nM). Furthermore, analysis of recombinant EGFR T790M kinase incubated with 3h by electrospray mass spectrometry revealed stoichiometric addition of one inhibitor molecule to the protein. Analysis of a pepsin digest of the modified protein by tandem mass spectrometry identified Cys797 as the site of modification, thus verifying covalent bond formation between 3h and EGFR10.
In summary we successfully developed a series of covalent inhibitors for EGFR-T790M by a structure based drug design. Those inhibitors exhibited great potency and mutant selectivity against drug resistant EGFR-T790M. A representative compound 3i blocked proliferation of EGFR-T790M PC-9 cells with an EC50 in the two digit nanomolar range. The approach of combining cellular screens against clinically relevant mutant kinases with structure-based drug design, should be useful for numerous important cancer targets. Further work will be reported on the pharmacokinetic properties and in vivo activity for EGFR inhibitors in due course.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Weinstein IW. Science. 2002;297:63. doi: 10.1126/science.1073096. [DOI] [PubMed] [Google Scholar]
- 2.Kwak E, Sordella R, Bell D, Godin-Heymann N, Okimoto R, Brannigan B, Harris P, Driscoll D, Fidias P, Lynch T, Rabindran S, McGinnis J, Wissner A, Sharma S, Isselbacher K, Settleman J, haber D. Proc Natl Acad Sci. 2005;102(21):7665. doi: 10.1073/pnas.0502860102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Agus DB, Terlizzi E, Stopfer P, Amelsberg A, Gordon MS. J Clin Oncol. 2006;24(18S):2074. [Google Scholar]
- 4.Engelman J, Zejnullahu K, Gale C, Lifshits E, Gonzales A, Shimamura T, Zhao F, Vincent P, Naumov G, Bradner J, Althaus I, Gandhi L, Shapiro G, Nelson J, Heymach J, Meyerson M, Wong K, Janne P. Cancer Res. 2007;67(24):11924. doi: 10.1158/0008-5472.CAN-07-1885. [DOI] [PubMed] [Google Scholar]
- 5.Zhang J, Yang P, Gray N. Nat Rev Cancer. 2009;9:28. doi: 10.1038/nrc2559. [DOI] [PubMed] [Google Scholar]
- 6.Engelman J, Janne P. Clin Cancer Res. 2008;14(10):2895–2903. doi: 10.1158/1078-0432.CCR-07-2248. [DOI] [PubMed] [Google Scholar]
- 7.Li D, Shimamura T, Ji H, Chen L, Haringsma H, McNamara K, Liang M, Perera S, Zaghlul S, Borgman C, Kubo S, Takahashi M, Sun Y, Chirieac L, Padera R, Lindeman N, Janne P, Thomas R, Meyerson M, Eck M, Engelman J, Shapiro G, Wong K. Cancer Cell. 2007;12:81. doi: 10.1016/j.ccr.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 8.Yun C, Mengwasser K, Toms A, Woo M, Greulich H, Wong K, Meyerson M, Eck M. Proc Natl Acad Sci. 2008;105:2070. doi: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ogino A, Kitao H, Hirano S, Uchida A, Ishiai M, Kozuki T, Takigawa N, Takata M, Kiura K, Tanimoto M. Cancer Res. 2007;67(17):2807. doi: 10.1158/0008-5472.CAN-07-0681. [DOI] [PubMed] [Google Scholar]
- 10.Zhou W, Ercan D, Chen L, Yun C, Li D, Capelletti M, Cortot A, Chirieac L, Lacob R, Padera R, Engen J, Wong K, Eck M, Gray N, Janne P. Nature. 2009;462:1070. doi: 10.1038/nature08622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kleman-leyer K. Drug Disc Dev. 2003;6:81. [Google Scholar]
- 12.The synthetic procedure of lead compound 3i was reported in reference 10. 1d 1H NMR 600 MHz (DMSO-d6) δ 9.18(s, 1H), 8.00(s, 1H), 7.72(d, J=6.4 Hz, 2H), 7.12(d, J=6.4 Hz, 2H), 6.83(m, 1H), 6.08(q, 1H), 5.60(dd, J=10.8 Hz, 1H), 4.20(m, 1H), 4.05(m, 1H), 3.0(s, 3H), 2.96(m, 5H), 1.98(m, 6H), 0.97(t, J=8.4 Hz, 3H) 2a 1H NMR 600 MHz (DMSO-d6) δ 12.60(s, 1H), 10.35(s, 1H), 9.00 (s, 1H), 8.52(d, J=10.8 Hz, 1H), 8.20(d, J=8.4 Hz, 1H), 7.90(s, 1H), 7.70(s, 1H), 7.44(m, 2H), 7.00(d, 1H), 6.50(q, 1H), 6.30(d, 1H), 5.80(d, 1H) 3i 1H NMR 600 MHz (DMSO-d6) δ 10.3 (s, 1H), 8.33 (s, 1H)), 8.09 (s, 1H)), 7.62 (d, J=7.8 Hz, 1H), 7.54(m, 1H), 7.39 (t, 1H), 7.22 (d, J=10.5 Hz, 1H), 6.95(d, J=7.8 Hz, 1H), 6.50(d, J=2.4 Hz, 1H), 6.41(q, 1H), 6,26(dd, J=1.8, 16.8 Hz, 1H), 5.76(dd, J=1.8, 10.2 Hz, 1H), 3.72 (s, 3H), 3.01(m, 4H), 2.40(m, 4H), 2.20(s, 3H) 4c 1H NMR 600 MHz (DMSO-d6) δ 10.1 (s, 1H), 9.88 (s, 1H), 9.08(s, 1H), 8.06(s, 1H), 7.80(s, 1H), 7.58(d, J=8.8 Hz, 1H), 7.40(d, J=7.2Hz, 1H), 7.20 (m, 2H), 6.60(s, 1H), 6.40(q, 1H), 6.28(m, 2H), 5.70(d, J=8.4 Hz, 1H), 3.70(s, 3H), 3.40(m, 4H), 3.02(m, 4H), 2.80(s, 3H) 4e 1H NMR 600 MHz (DMSO-d6) δ 10.20(s, 1H), 9.80(s, 1H), 8.33(s, 1H), 7.78(s, 1H), 7.60(s, 1H), 7.40(m, 1H), 7.20 (d, J=8.8 Hz, 1H), 6.6 (s, 1H), 6.40 (m, 1H), 6.28(m, 1H), 5.80(dd, J=10 Hz, 2Hz, 1H), 3.78(s, 3H), 3.43(m, 4H), 3.10(m, 4H), 2.92(s, 3H)