

Abstract
Kv4.2, as the primary α-subunit of rapidly inactivating, A-type voltage-gated K+ (Kv) channels expressed in hippocampal CA1 pyramidal dendrites, plays a critical role in regulating their excitability. Activity-dependent trafficking of Kv4.2 relies on C-terminal protein kinase A (PKA) phosphorylation. A-kinase-anchoring proteins (AKAPs) target PKA to glutamate receptor and ion channel complexes to allow for discrete, local signaling. As part of a previous study, we showed that AKAP79/150 interacts with Kv4.2 complexes and that the two proteins colocalize in hippocampal neurons. However, the nature and functional consequence of their interaction has not been previously explored. Here, we report that the C-terminal domain of Kv4.2 interacts with an internal region of AKAP79/150 that overlaps with its MAGUK (membrane-associated guanylate kinase)-binding domain. We show that AKAP79/150-anchored PKA activity controls Kv4.2 surface expression in heterologous cells and hippocampal neurons. Consistent with these findings, disrupting PKA anchoring led to a decrease in neuronal excitability, while preventing dephosphorylation by the phosphatase calcineurin resulted in increased excitability. These results demonstrate that AKAP79/150 provides a platform for dynamic PKA regulation of Kv4.2 expression, fundamentally impacting CA1 excitability.
Introduction
CA1 hippocampal pyramidal neurons receive tens of thousands of synaptic inputs each. How information, transmitted by these inputs, is processed in CA1 dendrites to produce meaningful output is not well understood. Dendrites contain an abundance of ion channels which can impact synaptic integration and which are subject to activity-dependent modulation during synaptic plasticity (Shah et al., 2010), the leading model of information storage in neurons.
Excitability in hippocampal CA1 dendrites critically depends on rapidly activating and inactivating, A-type K+ channels. In addition to controlling action potential (AP) backpropagation into dendrites (Hoffman et al., 1997), these channels contribute to synaptic integration (Kim and Hoffman, 2008). Kv4.2 channels, which produce the A-current in CA1 dendrites (Kim et al., 2005), are trafficked out of the membrane upon the induction of long-term potentiation (LTP), a form of synaptic plasticity (Kim et al., 2007; Jung and Hoffman, 2009). In addition to inducing a lasting increase in synaptic strength, LTP also results in an increase in the excitability of CA1 neurons (Bliss and Lomo, 1973). This “intrinsic plasticity” likely involves changes in the function of a number of voltage-gated channels, including Kv4.2 (Frick et al., 2004; Fan et al., 2005; Campanac et al., 2008; Jung and Hoffman, 2009). The specific mechanisms involved with regulating Kv4.2 expression upon LTP induction are unknown but acute activation of protein kinase A (PKA) leads to an increase in dendritic excitability through a downregulation of A-type K+ currents in CA1 dendrites (Hoffman and Johnston, 1998; Narayanan and Johnston, 2007) and a decrease in Kv4.2 surface expression in hippocampal neurons, which requires PKA phosphorylation at Kv4.2 site S552 (Hammond et al., 2008). We therefore hypothesized that Kv4.2 may be associated with A-kinase-anchoring protein (AKAP) complexes which provide restricted, local PKA signaling.
AKAP79/150 has been previously shown to dock PKA near channels and receptors to specifically regulate their phosphorylation state (Colledge et al., 2000; Tavalin et al., 2002; Hoshi et al., 2003, 2005; Oliveria et al., 2007). AKAP79/150 additionally anchors the protein phosphatase 2B [calcineurin (CaN)] and PKC (Coghlan et al., 1995; Klauck et al., 1996; Dell'Acqua et al., 2002). AKAP79/150-anchored PKA and CaN are thought to be important for regulating AMPA receptor currents and membrane trafficking to control synaptic plasticity in hippocampal neurons (Snyder et al., 2005; Smith et al., 2006; Lu et al., 2007; Nie et al., 2007; Tunquist et al., 2008; Bhattacharyya et al., 2009; Weisenhaus et al., 2010).
Here we show that Kv4.2 binding to AKAP79/150 requires a region overlapping with the membrane-associated guanylate kinase (MAGUK)-binding site on AKAP79/150 but is independent of Kv4.2 binding to MAGUK PDZ domains. Signaling through anchored PKA and CaN was found to bidirectionally regulate Kv4.2 surface expression and A-type K+ currents in hippocampal neurons. Consistent with these results, the firing properties of hippocampal neurons were altered according to the AKAP79/150 scaffolding effects on Kv4.2 expression levels. These results show that AKAP79/150, in addition to impacting synaptic function, contributes to the intrinsic excitability of hippocampal neurons through regulation of Kv4.2 surface expression.
Materials and Methods
Constructs.
Kv4.2 deletion mutant (Kv4.2ΔPDZ) lacking the C-terminal PDZ domain, which includes the four amino acids, VSAL, was prepared by PCR using the primers 5′GGAGGAAATATCGTCAGGCCTCCGTCGACGGTACCGC (forward) and 5′-GCGGTACCGTCGACGGAGGCCTGACGATATTTCCTCC (reverse) on construct Kv4.2-GFP (green fluorescent protein). The truncated C-terminal deletions of Kv4.2 were generated by PCR using the forward primer 5′-CGAATTCTGGGGTACACCCTGAAGAGC and reverse primers F (1-560) 5′-GTCGACGGCTGAATTGTACTGAGTTCTTG; F (1-490) 5′-GTCGACGGGTTCGTGGTTTTCTCCAGGCAGTG; F (1-417) 5′-GTCGACGGTTGGTTTTGGTGGTAGATCCG and F (1-406) 5′-GTCGACGGCACGATCACAGGCACGGGTAG and subcloned into the EcoR1-SalI sites of the pEGFP vector. Kv4.2S552A mutation of Kv4.2 C-terminal phosphorylation site at S552 was described by Lin et al. (2010). Briefly, we mutagenized the serine (S) to alanine (A) to remove the phosphorylation of PKA site at site 552 on construct Kv4.2-GFP. Both mutations were performed using the Quick-Change Site-Directed Mutagenesis Kit (Stratagene) and confirmed by sequencing analysis. pcDNA3 or pEGFPN1 vectors encoding AKAP79 Wild-type and mutations AKAP79ΔPKA, AKAP79ΔCaN, AKAP79ΔPIX (deletion of the 7 residues composing the PXIXIT-like motif in CaN-binding region), AKAP79ΔMAGUK, AKAP79 fragments 1-153, 150-427 and AKAP150shRNAi constructs were as previously described and characterized (Dell'Acqua et al., 2002; Gomez et al., 2002; Oliveria et al., 2003; Hoshi et al., 2005; Robertson et al., 2009).
Rat hippocampal neuron culture and expression.
Primary hippocampal neurons cultures were prepared as previously described (Kim et al., 2007). Neurons were infected with Kv4.2Myc Sindbis virus (Hammond et al., 2008) using a modified Sindbis virus expression system (Kim et al., 2004). Briefly, cultured 14 day in vitro (DIV14) hippocampal neurons were infected with Kv4.2Myc virus for 1 h at 37°C. Neurons were transfected by using the Nucleofector System (Amaxa). Cultured hippocampal neurons (6 × 106) were resuspended in 100 μl of rat neuron Nucleofector solution with 10 μg of DNAs, electroporated using the O-03 program, then plated on poly-d-lysine and laminin-coated glass coverslips (BD Biosciences) in a 24-well or 100 mm culture plate in MEM (Invitrogen) supplemented with 10% FBS, penicillin and streptomycin. After 5–6 h, the medium was replaced with Neurobasal medium plus B27 supplements (Invitrogen). Cultures were maintained at 37°C with 10% CO2.
Coimmunoprecipitation and Western blotting.
To confirm an interaction between Kv4.2 and AKAP79/150, we performed coimmunoprecipitation (co-IP) experiments either in native, detergent-solubilized rat brain extracts or COS7 cells cotransfected with various WT and mutant Kv4.2 and AKAP79 constructs for 24–48 h. Rat brain or cells were lysed in lysis buffer: 150 mm NaCl, 20 mm Tris-HCl, 1% NP40, 0.5% SDS and protease inhibitor mixture (Roche). Anti-Kv4.2 (2 μg/500 μg protein, The UC Davis/NIH NeuroMab Facility), nonspecific IgG (Invitrogen) or anti-AKAP79 antibody (2 μg/500 μg protein, Millipore) was then added to the lysate. The mixture was then incubated and rotated at 4°C for overnight. The antibody-antigen complex was immobilized by adsorption onto 50 μl of immobilized protein A (Pierce) and incubated for 2 h at RT. The protein-bead mixtures were washed six times with lysis buffer. The beads were resuspended in reducing SDS sample buffer and analyzed on 10% SDS polyacrylamide gels. The separated proteins were immuonoblotted using Kv4.2 (1:2000) and AKAP79 antibody (1:1000) and visualized by Alexa Fluor 680 secondary antibody (1:10,000, Invitrogen) and Alexa Fluor 800 secondary antibody (1:10,000, Rockland). Immunoreactivity was detected with the Odyssey infrared imaging system (LI-COR Biosciences). Quantification of results was performed using Odyssey software (LI-COR Biosciences).
On-cell Western assays.
Assays were performed as described by Lin et al. (2010). Briefly, 0.1 × 106 of rat or mouse (for Kv4.2−/− experiments) hippocampal neurons were plated into 24-well tissue culture plates. DIV14 cultured hippocampal neurons were treated with either 10 μm Ht31 control or Ht31 peptide for 15 min at 37°C and then were fixed with PBS containing 4% paraformaldehyde. After blocking with Odyssey blocking solution for 1–1.5 h at RT, neurons were incubated with mouse anti-Kv4.2 antibody (1:200, The UC Davis/NIH NeuroMab Facility) or anti-Myc antibody (1:100, Sigma) in Li-COR blocking buffer at 4°C for overnight. Cells were washed and incubated with the secondary antibody IRDye 800 Goat anti-mouse (1:1000, Rockland) at 37°C for 1 h. After a wash in PBS, cells were permeabilized with 0.2% Triton X-100 in PBS for 5 min. We used rabbit anti-β actin antibody (1:1000, Sigma) and goat anti-rabbit secondary antibody IRDye 680 (1:800, Invitrogen) to detect actin. The intensity of the 700 nm and 800 nm infrared signal for each well was quantified using the Odyssey infrared imaging system software (LI-COR Biosciences).
Biotinylation assays.
Biotinylation assays were performed as previously described (Kim et al., 2007). Briefly, either DIV14 culture hippocampal neurons or transfected COS7 cells were rinsed with ice-cold PBS, surface protein were biotinylated with 1.5 mg/ml Sulfo-NHS-SS-Biotin reagent [succinimidyl2-(biotinamido)-ethyl-1,3′-dithiopropionate] (Pierce) in PBS for 30 min on ice. Unbound biotin was quenched with cold 100 mm glycine in PBS. Cells were lysed with ice-cold lysis buffer: 150 mm NaCl, 20 mm Tris-HCl, 1% NP40 and protease inhibitor mixture (Roche), sonicated and centrifuged at 12,000 × g for 15 min. Cell lysates were incubated overnight at 4°C with immobilized-Streptavidin agarose beads (Pierce), after washed 5 times in lysis buffer; the bound proteins were eluted with 2× SDS sample buffer. Surface expressed Kv4.2 was separated on 10% Tris-bis SDS-PAGE (Invitrogen) and transferred to PVDF membranes. Western blots were probed with the following antibodies: mouse anti-Kv4.2 (1:2000, The UC Davis/NIH NeuroMab Facility), rabbit anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (1:1000, Calbiochem) and rabbit anti-β-actin (1:1000, Abcam). Secondary antibodies conjugated to infrared dyes (Rockland Immunochemicals) were detected using Odyssey infrared imaging system (LI-COR Biosciences). Quantification of results was performed using Odyssey software.
Electrophysiology.
For patch-clamp recordings, coverslips containing DIV14 hippocampal primary culture neurons were transferred to a submerged recording chamber with a continuous flow of extracellular solution containing the following (in mm): 145 NaCl, 3 KCl, 10 HEPES, 2 CaCl2, 8 glucose, 2 MgCl2 (pH 7.2 with NaOH). Hippocampal primary culture neurons were visualized by using infrared differential interference contrast videomicroscopy system (Carl Zeiss). The patch pipettes (2∼5 MΩ) were filled with an internal solution containing the following (in mm): 20 KCl, 125 K-gluconate, 10 HEPES, 4 NaCl, 0.5 EGTA, 10 phosphocreatine, 4 ATP, 0.3 TrisGTP (pH 7.2 with NaOH). For some experiments, 50 μm Tris-HCl (no peptide control), 10 μm Ht31 control peptide, 50 nm rapamycin (FK506 control, Sigma), Ht31peptide (Promega) or 4 μm FK506 (Tocris Bioscience) were included in the internal solution to treat neurons for ∼20 min before recording. All patch-clamp recordings were made using a Multiclamp 700B amplifier (Molecular Devices) and Clampex 10.1 software (Molecular Devices). Signals were digitized at 10 kHz with a Digidata 1440A (Molecular Devices) and filtered at 4 kHz.
Nucleated patch recordings of voltage-gated K+ currents were made in voltage-clamp mode at room temperature. TTX (0.5 μm, Tocris Bioscience) was included in the bath solution. Only neurons with holding currents greater than −100 pA were selected for recording or analysis. Membrane potential holding potential was −60 mV while peak current was measured at +60 mV. Current ensemble averages were constructed from 15 to 30 sweeps. Leakage currents were subtracted digitally using a P/6 protocol. Transient current was isolated from sustained current using a 150 ms prepulse step to −20 mV to inactivate transient channels. To determine A-current density in nucleated patches, peak currents were normalized to membrane capacitance.
Neuronal firing properties were recorded in current-clamp mode at 31∼33°C. Series resistance was 5∼12 MΩ. Neurons in which resting membrane potential changed by more than ±5 mV of the initial value were excluded from analysis. Current injection series were repeated 10 times and the results reported as the average of the 10 individual measurements. For current-clamp analyses, the numeric values (AP onset, threshold etc.) were measured using a +200 pA current injection. The data were taken from the first AP initiated upon current injection, which was determined using the first derivative of the voltage trace with respect to time. Peak and steady-state potentials were measured from the maximal hyperpolarization and the voltage at the end of a 1000 ms, −200 pA current step, respectively. “Sag” potential is the difference between peak and steady-state voltages.
All patch-clamp recordings were analyzed using Clampfit 10.1 (Molecular Devices) and Microsoft Excel. Statistical significance was evaluated using Student's t test (unpaired, two tails). p values are reported in the text or in the figure legends with values <0.05 considered significant.
Results
Kv4.2-AKAP79/150 interaction requires a region overlapping with the MAGUK-binding domain of AKAP79/150
To begin investigating the functional role of AKAP-Kv4.2 complexes, we first confirmed endogenous Kv4.2 interaction with AKAP150 (mouse homolog of the human AKAP79) in native rat brain co-IP experiments (Fig. 1B). Next we determined which domains are required for co-IP of AKAP79 and Kv4.2. Confirming our previous work (Lin et al., 2010), an antibody to AKAP79 was able to co-IP wild-type AKAP79 and Kv4.2 from transfected COS7 cells. However, we found that AKAP79 fragments and deletion constructs lacking the MAGUK-binding site between residues 153 and 315 failed to pull-down Kv4.2 (Robertson et al., 2009) (ΔMAGUK, Fig. 1C,F). Members of the postsynaptic density protein 95 (PSD-95) family of MAGUKs [e.g., PSD-95, PSD-93, synapse-associated protein 97 (SAP-97), and SAP-102] target and anchor glutamate receptors to spines, regulating spine size during development and synaptic plasticity (Migaud et al., 1998; Colledge et al., 2000; El-Husseini et al., 2000; Schnell et al., 2002; Rumbaugh et al., 2003; Ehrlich and Malinow, 2004; Elias et al., 2006; Schlüter et al., 2006; Xu et al., 2008; Bhattacharyya et al., 2009; Robertson et al., 2009).
Figure 1.
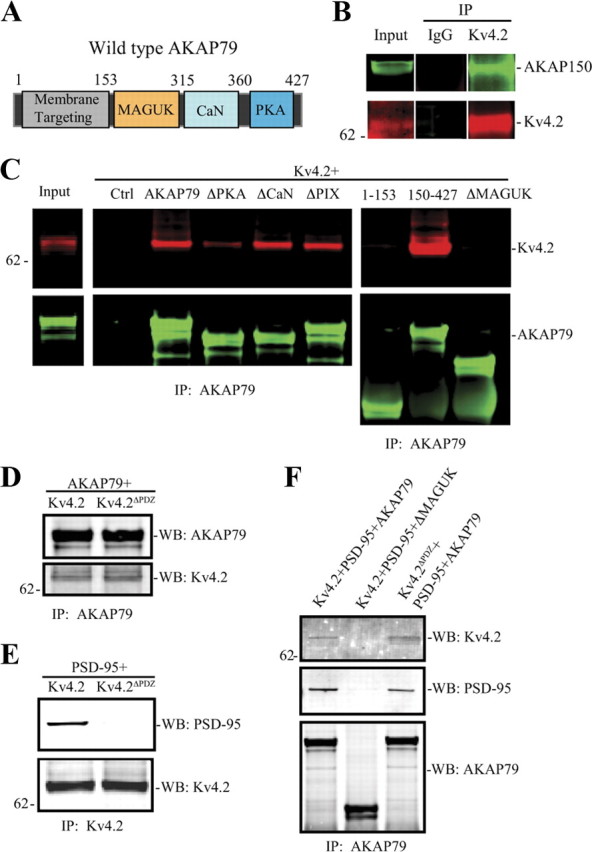
The MAGUK-binding domain of AKAP79/150 is necessary for its interaction with Kv4.2. We used AKAP79 or Kv4.2 deletion mutations to map their interaction. A, Schematic diagram of wild-type human AKAP79 showing the relative positions of various binding domains. B, Native co-IP from rat brain shows endogenous Kv4.2 interacts with AKAP150. C, Co-IP assay in COS7 cells cotransfected Kv4.2 with control, or AKAP79 or AKAP79 deletion mutations. Results show that the MAGUK-binding region (153–315) on AKAP79 is necessary for its interaction with Kv4.2. D, Co-IP of AKAP79 with Kv4.2 or its PDZ domain deletion (Kv4.2ΔPDZ) shows that the binding between the two proteins appears to be direct rather than through a MAGUK intermediate because the PDZ domain of Kv4.2 is not required for interaction. E, PSD-95 with Kv4.2 or its PDZ domain deletion (Kv4.2ΔPDZ) showing that the PDZ domain of Kv4.2 is required for interaction with PSD-95. F, PSD-95 cotransfected with Kv4.2 and AKAP79, Kv4.2, and AKAP79 MAGUK deletion mutation, or Kv4.2ΔPDZ and AKAP79. Cell lysates were pulled down by anti-AKAP79, anti-Kv4.2 antibody, or nonspecific IgG, and probed with anti-Kv4.2 antibody (1:2000), AKAP150 (1:1000, Millipore; for native co-IP), anti-AKAP79 antibody (1:2000), or PSD-95 (1:1000), and visualized by anti-mouse Alexa Fluor 680 and anti-rabbit Alexa Fluor 800 secondary antibody, respectively.
The requirement for MAGUK-binding site of AKAP79 for co-IP with Kv4.2 suggested that their association might occur through MAGUK intermediate binding proteins, analogous to glutamate receptors. Kv4.2 has been shown previously to bind to PSD-95 (Wong et al., 2002; Gardoni et al., 2007; Hammond et al., 2008) and SAP-97 (Gardoni et al., 2007). Subsequent co-IP experiments suggested, however, that Kv4.2-AKAP79 binding does not require MAGUK intermediate proteins as the two proteins were still found in complex after deleting the PDZ domain of Kv4.2 (Fig. 1D,F) which is necessary for PSD-95 interaction with Kv4.2 (Fig. 1E). These experiments suggest a direct interaction with Kv4.2 at the MAGUK-binding site of AKAP79. The possibility of an indirect interaction involving an additional, unknown accessory binding protein, however, cannot be excluded.
Deletion of the PKA domain of AKAP79 resulted in a weaker signal but did not eliminate AKAP79-Kv4.2 complex formation (ΔPKA, Fig. 1C). PKA phosphorylation of Kv4.2 at site S552 was not required for AKAP-Kv4.2 co-IP, however, as mutating this site did not prevent its IP with wild-type AKAP79 using an AKAP79/150 antibody (Fig. 2A). Disruption of PKA anchoring to AKAP79 by applying a stearated, membrane-permeant peptide, Ht31, which resembles the AKAP79-binding site for PKA (Carr et al., 1992), also did not affect AKAP79-Kv4.2 co-IP (Fig. 2B), suggesting that PKA subunits are not required for AKAP79-Kv4.2 interaction. Together, these data indicate that determinants within the MAGUK-binding region of AKAP79 are necessary for AKAP79/150-Kv4.2 complex formation. In addition, it seems that the PKA-binding region of AKAP79 binds transiently or less stably to Kv4.2 than the MAGUK region. PKA anchoring by AKAP79/150 or phosphorylation of Kv4.2 at site PKA S552 are not, however, necessary for AKAP79/150-Kv4.2 interaction.
Figure 2.
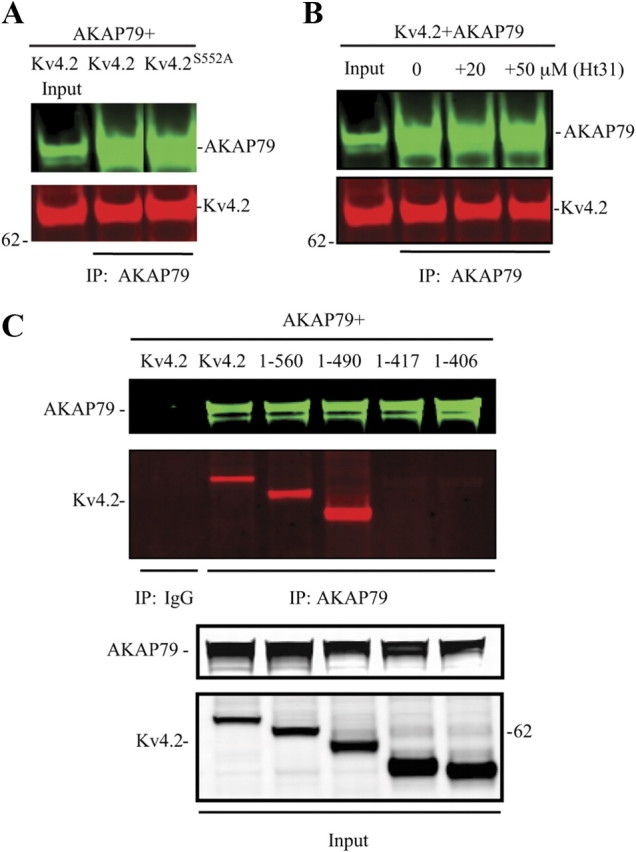
C-terminal Kv4.2 (417–490) is necessary for the interaction of Kv4.2 with AKAP79/150. A, Co-IP assay in COS7 cells cotransfected with AKA79 and Kv4.2 or Kv4.2S552A mutant showing that PKA phosphorylation site ser552 is not necessary for their interaction. B, Co-IP assay of AKAP79 and Kv4.2 with Ht31 treatment (20 or 50 μm for 15 min) showing that PKA anchoring by AKAP79 is not required for AKAP79 interaction with Kv4.2. C, Co-IP assay in COS7 cells cotransfected with AKAP79 and Kv4.2 or Kv4.2 C-terminal truncation mutations. Inputs show the total expression of Kv4.2 and AKAP79/150 in COS7 cells. Cell lysates were pulled down by anti-AKAP79 or nonspecific IgG, and probed with anti-Kv4.2 antibody (1:2000) or anti-AKAP79 antibody (1:2000) and visualized by anti-mouse Alexa Fluor 680 and anti-rabbit Alexa Fluor 800 secondary antibody, respectively.
To determine which region of Kv4.2 was required for co-IP with AKAP79, we made serial C-terminal deletions (Fig. 2C). Association was observed between AKAP79 and Kv4.2 fragments containing aa 1-560 and 1-490 but lost in fragments 1-417 and 1-406 suggesting a required site between aa 417 and 490. These amino acids contain a consensus protein kinase C phosphorylation site (Ser447). However, a phosphorylation mutant of Kv4.2 (S447A) still immunoprecipitated with AKAP79 and PKC activation by phorbol esters did not alter their interaction (data not shown). These data show that phosphorylation at this site was not necessary for AKAP79-Kv4.2 interaction. Note that AKAP79-Kv4.2 co-IP in the absence of the S552 PKA site in these deletion mutants is additional evidence that PKA phosphorylation is not required for their interaction. These results also offer further indication that the PDZ domain of Kv4.2 is not required for association with AKAP79.
Surface expression of Kv4.2 is regulated by AKAP79 in hippocampal neurons
Although PKA activation may not be necessary for AKAP79-Kv4.2 complex formation, their association may provide for local, dynamic regulation of Kv4.2 phosphorylation. As PKA phosphorylation of Kv4.2S552 is necessary for activity-dependent Kv4.2 trafficking we wondered whether PKA anchoring by AKAPs affected Kv4.2 surface expression. We first performed a biotinylation assay using extracts from cultured hippocampal neurons. Application of Ht31 peptide to disrupt AKAP anchoring increased Kv4.2 surface expression (Fig. 3A; neurons+Ht31 = 1.85 ± 0.11 in relation to with control peptide, Ht31C; n = 5; p < 0.05). Surface increase of Kv4.2 by Ht31 was also observed in on-cell Western assays using either an antibody against endogenous Kv4.2 (Fig. 3B; surface expression with Ht31 = 1.34 ± 0.03 in relation to Ht31C control peptide; n = 6; p < 0.05), or in neurons expressing a myc-tagged Kv4.2 (Fig. 3C; Ht31 = 1.44 ± 0.10 normalized to control peptide, Ht31C; n = 6; p < 0.05). No signal was observed in on-cell Western experiments using cultured hippocampal neurons from Kv4.2−/− mice (data not shown) (Chen et al., 2006).
Figure 3.
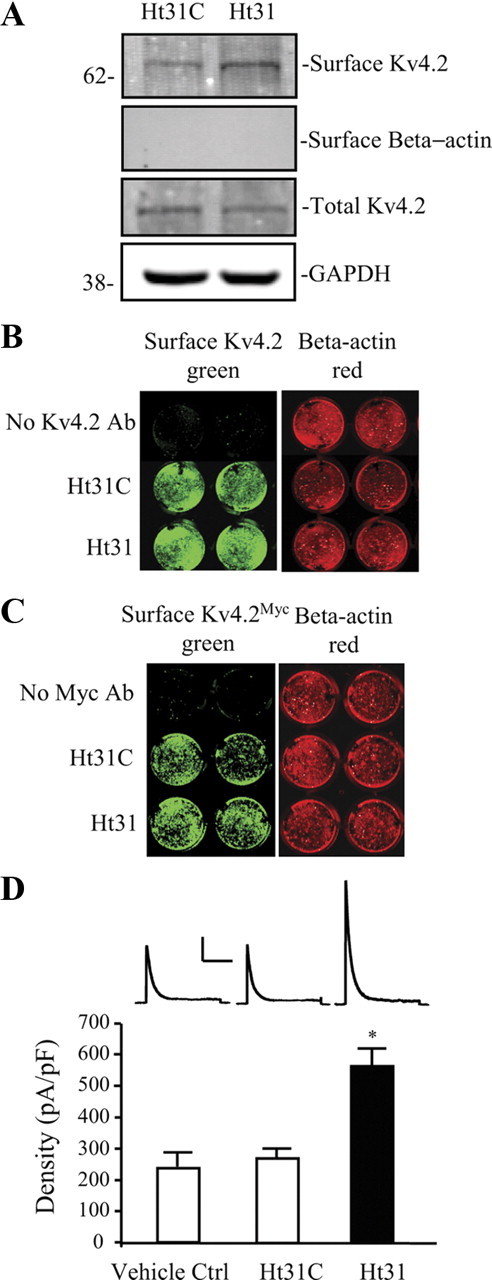
Surface Kv4.2 expression is enhanced after disrupting PKA anchoring in hippocampal neurons. DIV14 cultured embryonic day 18 (E18) rat hippocampal neurons were treated with Ht31 control peptide or Ht31 peptide (10 μm, 15 min). A, Surface proteins were labeled with NHS-SS-Biotin in a biotinylation assay and probed with mouse anti-Kv4.2 (1:2000). Surface level of endogenous Kv4.2 is significantly increased by Ht31 treatment in neurons compared with the Ht31 control. GAPDH served as a loading control. β-Actin is labeled as negative surface control. Ht31 increases surface Kv4.2 by ∼2-fold (n = 5, p < 0.05). B, C, On-cell Western assay of surface endogenous Kv4.2 (B) and overexpressed Kv4.2 by infection with Kv4.2Myc Sindbis virus (C) in hippocampal cultured neurons. The top row is control without Kv4.2 antibody labeled, the second row is treated with Ht31 control peptide (Ht31C), and the last row is treated with Ht31. Intensity levels of surface endogenous or overexpressed Kv4.2 (green) were significantly increased in wells of Ht31-treated compared with Ht31 control. The intensity levels of total β-actin (red) were not different in each well, demonstrating that equal numbers of neurons are found in each well. Data showed a significant increase in surface Kv4.2 with Ht31 treatment (n = 6, p < 0.05). D, In electrophysiological recordings from cultured hippocampal neurons, peak current density of Kv4.2-mediated currents was also significantly increased (∼2-fold) by Ht31 (n = 11, *p < 0.05) peptide application compared with Ht31 control (n = 14). Vehicle control (50 μm Tris-HCl) does not affect A-current density (n = 10). Error bars represent SEM. Calibration: 200 pA, 200 ms.
Our biochemical results were confirmed in electrophysiological recordings from cultured hippocampal neurons (Fig. 3D). In nucleated patch recordings, peak current density of endogenous A-type K+ currents increased ∼2-fold after Ht31 application (n = 11 for Ht31) vs recordings including a control peptide (Ht31C, n = 14; p < 0.05), or vs vehicle control (vehicle Ctrl, n = 10; p > 0.1, compared with Ht31C). The increase in A-current density in the presence of Ht31 was not accompanied by a change in the rate of current inactivation or in the voltage dependence of channel activation (decay τ: Ht31C = 36.9 ± 1.4 ms, n = 14; Ht31 = 37.8 ± 1.3 ms, n = 11; p > 0.1; V1/2: Ht31C= −6.4 ± 2.4 mV, n = 10; Ht31 = −9.3 ± 2.7 mV, n = 16; p > 0.1). Together these data from three different experimental approaches confirm Kv4.2 surface expression enhancement after disrupting PKA anchoring by AKAPs.
If disruption of PKA anchoring results in greater membrane expression, we might find that altering AKAP expression would affect Kv4.2 steady-state membrane localization. We investigated this first by overexpression of AKAP79 in hippocampal neurons. In a biotinylation assay, AKAP79-transfected hippocampal neurons showed a significant decrease in the level of Kv4.2 surface expression compared with control (Fig. 4A; AKAP79 = 0.708 ± 0.10 normalized to control; n = 5; p < 0.05). This result was again replicated in both on-cell Western (Fig. 4B,C; n = 6; p < 0.05) and for endogenous A-type K+ currents in electrophysiology experiments (Fig. 4D; n = 14 for Ctrl; n = 11 for AKAP79; p < 0.05). In cells overexpressing AKAP79, Ht31 peptide was again able to increase surface expression of Kv4.2 (Fig. 4C) and endogenous A-type K+ currents (Fig. 4D).
Figure 4.
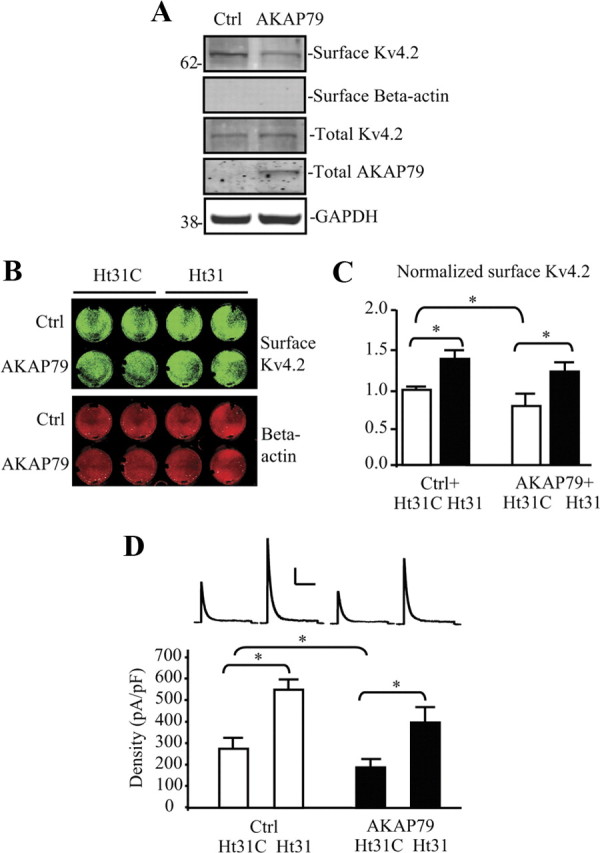
Surface expression of Kv4.2 is regulated by AKAP79/150 in hippocampal neurons. A, Cultured rat embryonic day 18 (E18) hippocampal neurons were transfected with control vector or human AKAP79 using the Nucleofector System. Biotinylation assays were performed at DIV14; surface Kv4.2 was labeled with NHS-SS-Biotin and probed with mouse anti-Kv4.2 (1:2000). Surface level of endogenous Kv4.2 is significantly decreased in neurons overexpressing AKAP79 compared with the control. β-Actin is labeled as negative surface control. Human AKAP79 is detected in AKAP79-transfected rat neurons only. GAPDH served as a loading control. AKAP79 decreases surface Kv4.2 by ∼30% (n = 5, p < 0.05). B, On-cell Western assay of surface endogenous Kv4.2 in hippocampal cultured neurons. The top row is transfected with control vector, and the second row is after AKAP79 overexpression. Lines 1–2 are treated with Ht31 control (Ht31C) and lines 3–4 are treated with Ht31. Intensity levels of surface endogenous Kv4.2 (green) were significantly decreased in wells of neurons overexpressing AKAP79 compared with the control, but not in the wells treated with Ht31. The intensity levels of total β-actin (red) were not different in each well, demonstrating that equal numbers of neurons are found in each well. C, Pooled data showing a significant reduction of surface Kv4.2 by AKAP79 without Ht31 treatment. Error bars represent SEM (n = 6, *p < 0.05). D, Electrophysiological recording results from hippocampal cultured neurons show that, for those groups treated with Ht31 control peptide, overexpressed AKAP79 (n = 11) induced a significant decrease of Kv4.2-mediated current density compared with control (n = 14). However, this current density reduction by AKAP overexpression was not found in neurons treated with Ht31 peptide (n = 12). We note that current density in hippocampal neurons overexpressing AKAP79 after Ht31 (D) does not reach the same magnitude as found for control neurons after Ht31 treatment (Fig. 3D). Evidence from biotinylation experiments (A) shows no change in total Kv4.2 expression in neurons overexpressing AKAP79. Together, these findings suggest that while steady-state total Kv4.2 levels persist in AKAP79-overexpressing neurons, the fraction of channels able to remain stably expressed in the membrane is decreased both in control and in the presence of Ht31 upon AKAP79 overexpression. Error bars represent SEM. Calibration: 200 pA, 200 ms.
Next, we used a previously characterized AKAP150shRNAi (siAKAP150, (Hoshi et al., 2005)) to examine the effect of chronic knockdown of endogenous AKAP150 on endogenous A-type K+ currents in hippocampal neurons. Consistent with our overexpression results, knockdown of AKAP150 increased Kv4.2 surface expression (Fig. 5A; siAKAP150 = 1.33 ± 0.08 normalized to control; n = 5; p < 0.05) and increased A-type K+ density (Fig. 5B; siCtrl = 269.3 ± 23.3 pA/pF, n = 15; siAKAP150 = 446.6 ± 49.3 pA/pF, n = 12; p < 0.05) in hippocampal neurons.
Figure 5.
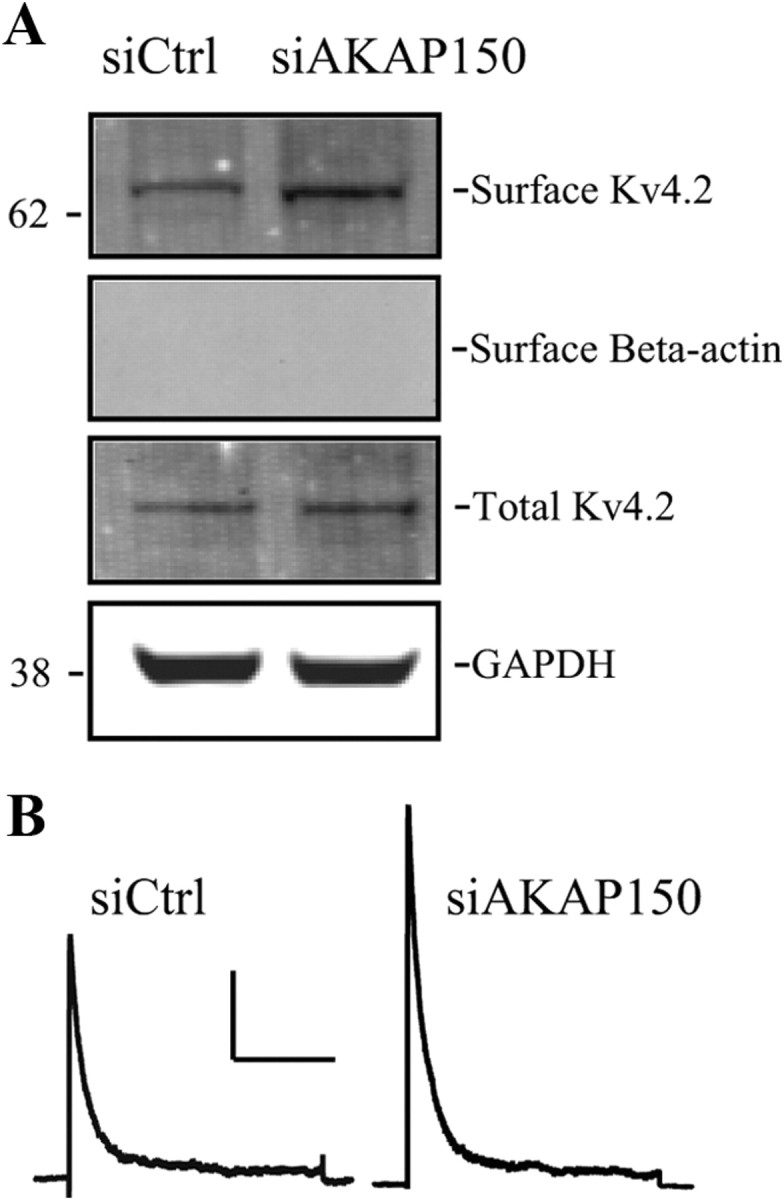
Knockdown of endogenous rat AKAP150 increases Kv4.2 surface expression in hippocampal neurons. A, Cultured rat embryonic day 18 (E18) hippocampal neurons were transfected with control siRNA (siCtrl) or siAKAP150 by using the Nucleofector System. Biotinylation assay shows that surface level of endogenous Kv4.2 is significantly increased by knock-down of endogenous AKAP150 by siAKAP150 compared with siCtrl. GAPDH served as a loading control. β-Actin is labeled as negative surface control. Surface Kv4.2 level is increased ∼30% by siAKAP150 knock-down (n = 5, p < 0.05). B, As with the biotinylation results, electrophysiological recordings from cultured hippocampal neurons showed peak current density of Kv4.2-mediated currents also significantly increased (∼1.5-fold) by knock-down endogenous AKAP150 with siAKAP150 (n = 12) compared with siCtrl (n = 15). Calibration: 200 pA, 200 ms.
PKA phosphorylation of Kv4.2 site S552 is required for regulation by AKAP79
To examine phosphorylation of Kv4.2 by AKAP79 we coexpressed the two proteins in COS7 cells. In a biotinylation assay, we found that mutation of site Ser552 to prevent its phosphorylation by PKA prevented the decrease in Kv4.2 expression levels observed with AKAP79 expression (Fig. 6A,B; n = 5; p > 0.1). Electrophysiological recordings from COS7 cells showed that coexpression of Kv4.2S552A and AKAP79 resulted in enhanced A-type K+ current density compared with wild-type Kv4.2 with AKAP79 (Fig. 6C; n = 10 for Kv4.2 + AKAP79, n = 10 for Kv4.2S552A + AKAP79; p < 0.05). Application of the Ht31 to prevent PKA anchoring by AKAP79 enhanced A-current density in cells expressing wild-type Kv4.2 (Fig. 6C; n = 10 for Kv4.2 + AKAP79 + Ht31C; n = 10 for Kv4.2 + AKAP79 + Ht31; p < 0.05) but not those expressing Kv4.2S552A (Fig. 6C; n = 10 for Kv4.2S552A + AKAP79 + Ht31C; n = 13 for Kv4.2S552A + AKAP79 + Ht31; p > 0.1).
Figure 6.
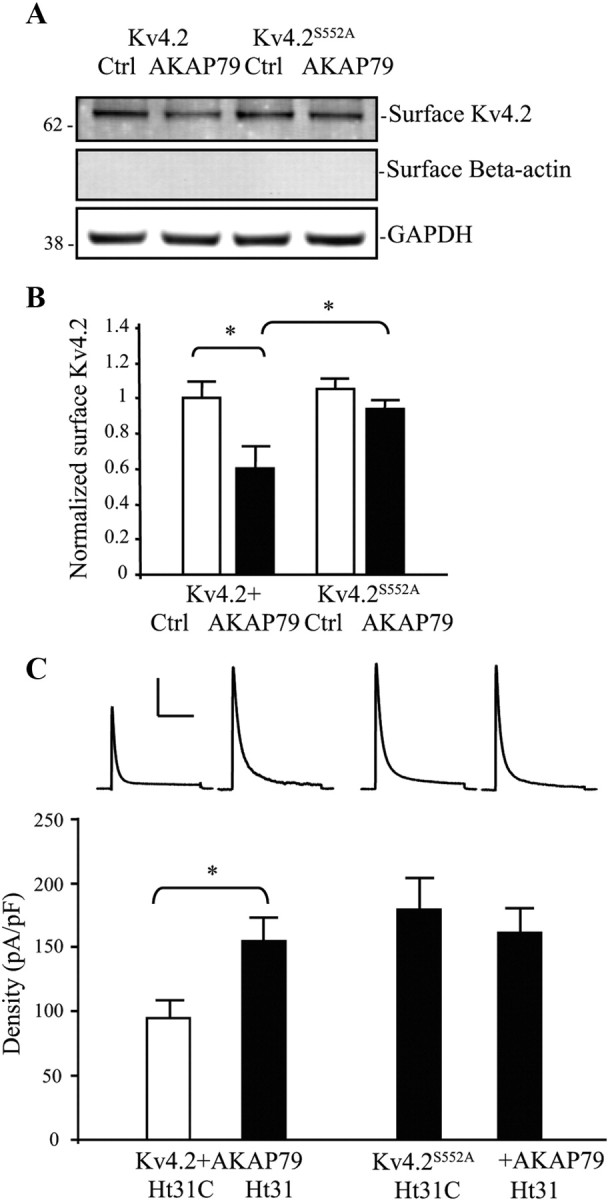
AKAP79 regulation of Kv4.2 trafficking requires Kv4.2 PKA phosphorylation site S552. A, COS7 cells cotransfected Kv4.2 or Kv4.2S552A and either control or AKAP79. Surface proteins were labeled with NHS-SS-Biotin and probed with mouse anti-Kv4.2 (1:2000). Cells coexpressing Kv4.2 and AKAP79 showed decreased surface expression of Kv4.2 compared with control. Surface expression of the Kv4.2 phospho-mutant S552A (Kv4.2S552A) was not changed by AKAP79 coexpression. GAPDH served as a loading control. β-Actin is labeled as negative surface control. B, Pooled data normalized to total Kv4.2 expression show that AKAP79 coexpression decreased surface Kv4.2 expression by ∼40% (n = 5, p < 0.05), and without a significant effect on Kv4.2S552A (n = 5, *p < 0.05). Error bars represent SEM. C, In electrophysiological recordings from COS7 cells, peak current density of Kv4.2-mediated currents increased (∼1.5-fold) by coexpression of AKAP79 with the phospho-mutant Kv4.2S552A (n = 10) compared with Kv4.2 (n = 10). Ht31 application (15 min) significantly increased Kv4.2-mediated peak current density in the neurons cotransfected with Kv4.2 and AKAP79 (n = 10, p < 0.05), but this peak current density enhancement by Ht31 was not found in cells expressing Kv4.2S552A and AKAP79 (n = 13, p < 0.05). Error bars represent SEM. Calibration: 200 pA, 200 ms.
These results indicate that AKAP79 directly impacts Kv4.2 expression levels through PKA anchoring. Additionally, the finding that PKA phosphorylation of Kv4.2 at site S552 underlies reduced Kv4.2 surface expression in the presence of AKAP79 is consistent with our previous finding that PKA phosphorylation of S552 mediates activity-dependent Kv4.2 trafficking (Hammond et al., 2008). If so, it follows that CaN dephosphorylation of Kv4.2 through the AKAP79 complex could lead to increased Kv4.2 surface expression. To examine this possibility, we coexpressed Kv4.2 and AKAP79 in COS7 cells and applied the CaN inhibitor FK506. Surface Kv4.2 was significantly decreased in these experiments indicating a basal level of CaN activity acts to enhance surface expression of Kv4.2-AKAP79 complexes (Fig. 7A,B; n = 5; p < 0.05). FK506 also counter-acted the enhanced Kv4.2 surface expression found after Ht31 (Fig. 7A,B; n = 5; p < 0.05) suggesting pools of both phosphorylated and dephosphorylated Kv4.2 under basal (control) conditions.
Figure 7.

Calcineurin regulation of Kv4.2 surface expression in hippocampal neurons and COS7 cells. A, COS7 cells are cotransfected with Kv4.2 and AKAP79, and treated with CaN inhibitor FK506 (4 μm, 30 min) followed with Ht31 control peptide or Ht31 (10 μm, 15 min). Surface proteins were labeled with NHS-SS-Biotin and probed with mouse anti-Kv4.2 (1:2000). Cells with FK506 treatment had 20% decrease of surface Kv4.2 expression in the Ht31 control group. In contrast, the cells treated with both FK506 and Ht31 show no significant changes compared with the Ht31 control. GAPDH served as a loading control. β-Actin is labeled as negative surface control. B, Pooled data normalized to total Kv4.2 expression shows that FK506 decreases surface Kv4.2 expression by ∼40% (n = 5, p < 0.05) and no difference between control and FK506 + Ht31 treatment (n = 5, p > 0.05). Error bars represent SEM. C, In electrophysiological recordings from cultured hippocampal neurons, peak A-current density was significantly decreased by FK506 (4 μm, 20 min) treatment (n = 9) compared with control peptide alone (n = 14, p < 0.05). This decrease was also found in neurons cotreated with FK506 and Ht31 peptide (n = 13) compared with Ht31 treatment alone (n = 11). As with the biochemical results, cells treated with both FK506 and Ht31 show no significant changes compared with the Ht31 control peptide in the absence of FK506. Error bars represent SEM. Calibration: 200 pA, 200 ms. p < 0.05.
We next applied intracellular FK506 to prevent CaN activation in neurons and directly recorded A-type K+ currents in nucleated patch recordings. Results showed that A-current density was significantly reduced compared with control (Fig. 7C; n = 14 for −FK506; n = 9 for +FK506; p < 0.05). Coapplication of FK506 with Ht31 lead to A-current densities similar to that found for neurons expressing siAKAP150 where both PKA and CaN′s effects on Kv4.2 channels would be diminished (Fig. 5) but not as high as for control neurons in the presence of Ht31 (Fig. 3), where only PKA's influence of the channel is affected. These results suggest dual PKA and CaN regulation of the phosphorylation state of Kv4.2 Ser552 in neurons under basal conditions, affecting A-current magnitude. Rapamycin, which shares common target proteins with FK506 but does not inhibit CaN was used as negative control. Rapamycin did not alter A-current density in nucleated patches (rapamycin = 247.8 ± 41.1 pA/pF; n = 10; p > 0.1).
Neuronal firing patterns and membrane excitability are regulated by PKA anchoring
Given the strong affect of AKAP PKA anchoring on A-type K+ current magnitude, we expected a corresponding effect on neuronal excitability (Kim et al., 2005). Intracellular application of Ht31 peptide resulted in significantly decreased neuronal excitability in recordings from hippocampal neurons (Fig. 8). Ht31 produced a decrease in the number of spikes fired in response to a +200 pA current injection (Fig. 8A,C). At least part of the reduced firing was due to an increase in the amount of time before first AP onset (Fig. 8B,E). In addition, after Ht31, AP threshold was increased and after-hyperpolarization strength enhanced (Fig. 8D,F). Finally Ht31 caused a significant decrease in cellular input resistance (Table 1). Neither control peptide nor rapamycin affected firing or membrane properties (data not shown; n = 10 for Ht31C; n = 10 for rapamycin; p > 0.1). Decreasing Kv4.2 expression by preventing CaN activation lead to corresponding increases in excitability (Fig. 8; n = 10 for Ctrl; n = 9 for FK506; p < 0.05).
Figure 8.
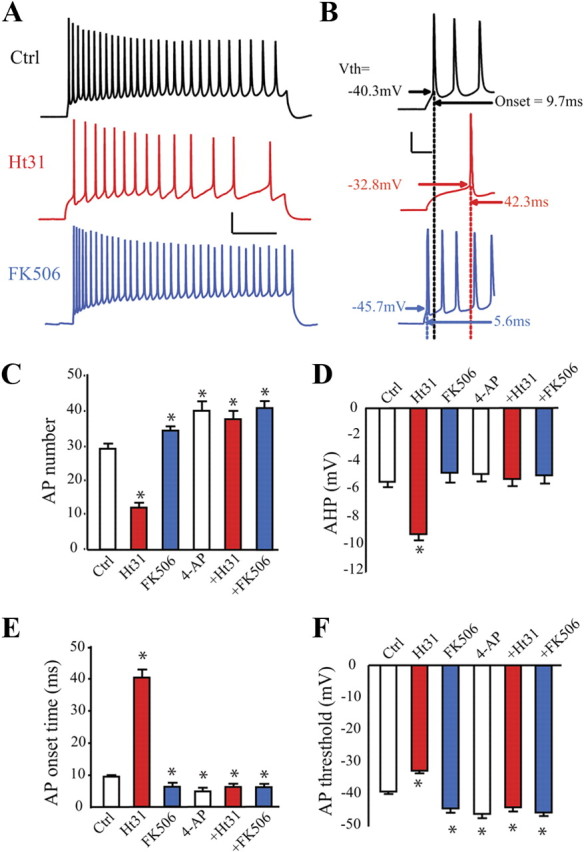
PKA anchoring controls hippocampal neuron excitability. A, Traces in response to a 1000 ms, 200 pA current injection for control hippocampal neurons (black trace), neurons treated with Ht31 peptide (red trace) or the calcineurin (protein phosphatase 2B) inhibitor FK506 (blue trace). Calibration: 20 mV, 200 ms. B, Expanded traces from A to more clearly depict AP onset time and threshold. Scale bar: 20 mV, 20 ms. C, The number of APs recorded from control neurons (no drug treatment, open bar), from neurons treated with intracellular Ht31 peptide (red bar), with FK506 (blue bar), with 5 mm intracellular 4-AP (second open bar), with 4-AP + Ht31 (second red bar), or with 4-AP + FK506 (second blue bar) over the course of 1 s for a 200 pA current injection. After 15 min treatment, AP number is significantly decreased by Ht31 application (n = 10 for Ctrl, n = 8 for Ht31), increased by FK506 (n = 9), 4-AP (n = 12), 4-AP + Ht31 (n = 13) and 4-AP + FK506 (n = 12) application. D–F, Pooled data showing first AP onset time, AHP data and AP threshold for control neurons (open bars, n = 10), or neurons treated with Ht31 peptide (red bars, n = 8), FK506 (blue bars, n = 9), 4-AP (second open bar, n = 12), 4-AP + Ht31 (second red bar, n = 13), or 4-AP + FK506 (second blue bar, n = 12). *p < 0.05.
Table 1.
PKA anchoring affects hippocampal neuron resting membrane properties
| Peak potential (mV) | Steady-state potential (mV) | “Sag” potential (mV) | Input resistance (MΩ) | |
|---|---|---|---|---|
| Ctrl (n = 10) | −104.5 ± 3.1 | −94.5 ± 3.3 | 9.8 ± 0.3 | 149.2 ± 5.8 |
| Ht31 (n = 8) | −83.7 ± 2.8* | −74.4 ± 2.6* | 9.3 ± 0.9 | 82.5 ± 2.6* |
| FK506 (n = 9) | −119.7 ± 4.3* | −110.2 ± 4.2* | 9.5 ± 0.3 | 198.6 ± 8.7* |
| 4-AP (n = 12) | −128.1 ± 6.6* | −118.7 ± 6.7* | 9.4 ± 0.9 | 245.7 ± 6.5* |
| 4-AP + Ht31 (n = 13) | −124.1 ± 3.8* | −114.4 ± 3.8* | 9.7 ± 0.5 | 210.9 ± 6.7* |
| 4-AP + FK506 (n = 12) | −127.8 ± 3.7* | −118.2 ± 4.8* | 9.6 ± 0.3 | 232.3 ± 10.4* |
Subthreshold voltage transients were generated in response to a +200 pA, 1000 ms current injection for control neurons, or neurons treated with Ht31 peptide, FK506, 4-AP, 4-AP + Ht31, or 4-AP + FK506. Compared with control neurons, both peak and steady-state responses are reduced in the Ht31 treatment group (n = 10 for Ctrl, n = 8 for Ht31 peptide) and enhanced in the FK506, 4-AP, 4-AP + Ht31, and 4-AP + FK506 groups, affecting cellular input resistance. The difference potential or “sag” between peak and steady-state voltages did not differ between experimental groups, indicating that the hyperpolarization-activated nonspecific cation current, Ih, was not affected.
*p < 0.05.
The profile of excitability changes presented in Figure 8 is consistent with changes in A-type K+ current (Kim et al., 2005; Jung and Hoffman, 2009) and suggests that AKAP79/150 regulation of Kv4.2 phosphorylation state fundamentally impacts neuronal excitability. To confirm that AKAP PKA anchoring contributes to the intrinsic excitability of hippocampal neurons through regulation of A-type K+ currents, we treated neurons with the intracellular K+ channel blocker, 4-aminopyridine (4-AP) (5 mm), alone or together with Ht31 or FK506 in the pipette. Results showed that, compared with control, intracellular 4-AP significantly increased neuronal excitability (Fig. 8C–F, Table 1; n = 12 for 4-AP; n = 13 for 4-AP + Ht31, n = 12 for 4-AP + FK506; p < 0.05). However, with 4-AP treatment, neither Ht31 nor FK506 altered neuronal excitability (Fig. 8C–F, Table 1; p > 0.1), demonstrating that AKAP79/150 likely affects excitability through regulation of 4-AP-sensitive Kv4.2 surface expression.
Discussion
The detailed molecular mechanisms underlying ion channel trafficking are of great interest because of their contribution to plasticity-induced changes in neuronal function (Shah et al., 2010). We have presented several lines of evidence indicating a functional interaction between Kv4.2 with the PKA-anchoring protein AKAP79/150. Using a peptide (Ht31), which blocks PKA anchoring to AKAP79, we found increased surface expression of Kv4.2 indicating that PKA anchoring by AKAP79 normally controls Kv4.2 surface expression. Protein phosphatase 2B (calcineurin) block led to a decrease in surface Kv4.2 expression, indicating a balance in the phosphorylation state of Kv4.2 in basal conditions. Corresponding effects on neuronal excitability show the Kv4.2-AKAP79 interaction is physiologically important, providing a mechanism for rapid, dynamic regulation of intrinsic neuronal excitability of hippocampal neurons.
The sum of our co-IP experiments indicates a direct interaction between Kv4.2 and AKAP79. The MAGUK-binding site of AKAP79 (153–315) is necessary for this interaction along with Kv4.2 amino acid between 417 and 490. Neither PKA activation nor MAGUK proteins are required although the C terminus of AKAP79 including the PKA-binding domain appears to be a minor site for Kv4.2 interaction. To our knowledge this is the first example of direct binding of another AKAP partner at this MAGUK-binding site. It will be interesting to see whether Kv4.2 and MAGUK proteins compete for binding at this site, although our finding that AKAP79 can co-IP Kv4.2ΔPDZ and PSD-95 from the same cells suggest that these proteins may form a ternary complex. However, if there is competition at this binding site, then Kv4.2 may act to sequester AKAP79 from synapses to prevent its interaction with MAGUKs and therefore GluR1 (GRIA1) AMPA receptors. This would be yet another case where Kv4.2 and AMPARs are anti-regulated. Chemically and synaptically evoked LTP both induce GluR1 AMPAR insertion, which is coincident with Kv4.2 internalization (Shi et al., 1999; Kim et al., 2007). Additionally, the Ht31 peptide, which we found to increase Kv4.2 expression, acts to decrease GluR1 surface expression (Tavalin et al., 2002; Snyder et al., 2005), suggesting possible coregulation of Kv4.2 and AMPAR trafficking during LTP.
Coregulation of Kv4.2 by PKA and calcineurin
In the course of these experiments, we noticed that disrupting PKA anchoring with Ht31 peptide produced a larger effect on A-type K+ currents in control neurons compared with neurons expressing either enhanced (AKAP79 overexpression) or downregulated levels of AKAP79/150 (siAKAP150). AKAP79 also anchors the protein phosphatase 2B (calcineurin, CaN) and previous studies of GluR1 AMPA receptors and L-type Ca2+ channels have shown that AKAP-anchored PKA and CaN have opposing actions in regulation of channel phosphorylation, activity or trafficking (Colledge et al., 2000; Dell'Acqua et al., 2002; Tavalin et al., 2002; Snyder et al., 2005; Smith et al., 2006; Oliveria et al., 2007; Bhattacharyya et al., 2009). We report here a comparable antagonistic coregulation of Kv4.2 surface expression by PKA and CaN. While CaN block with FK506 and prevention of AKAP PKA anchoring with Ht31 are both able to modulate Kv4.2 surface expression (Figs. 3, 7) and hippocampal firing properties (Fig. 8), experiments using simultaneous application of FK506 and Ht31 resulted in A-current densities similar to control (Fig. 7), suggesting pools of both phosphorylated and unphosphorylated Kv4.2 under basal conditions. However, our overexpression (Fig. 4) and knockdown (Fig. 5) experiments (where AKAP anchoring of both PKA and CaN are affected) resulted in Kv4.2 expression and A-current densities consistent with a stronger effect of interrupting PKA anchoring. In agreement, Ht31 produced a larger effect on hippocampal neuron excitability than did FK506 (Fig. 8). Because AKAP79/150 interacts with Kv4.2 and plays a dominant role in localizing PKA in dendrites and dendritic spines (Smith et al., 2006; Tunquist et al., 2008; Weisenhaus et al., 2010), these Ht31 results indicate that AKAP79/150 is likely to be the primary AKAP responsible for Kv4.2 regulation in hippocampal neurons.
Role of KChIP auxiliary proteins in regulating Kv4.2-AKAP channel expression
Contrary to these current findings in neurons suggesting a net inhibitory effect of AKAP79/150 on surface Kv4.2 expression (Figs. 4, 5), previous results using heterologous expression systems showed that AKAP79/150 acts to increase Kv4.2 surface expression (Lin et al., 2010). However, the increase in Kv4.2 expression brought by AKAP79 coexpression required the presence of KChIP4a auxiliary subunits as AKAP79 + Kv4.2 by itself caused a decrease in surface Kv4.2 expression (Fig. 6). Kv4.2 surface augmentation was specific to KChIP4a as Kv4.2 subunits coexpressed with KChIPs 1 and 2 are not affected by AKAP79 expression (data not shown). Therefore, AKAP79 can have opposing affects on Kv4.2 surface expression, depending on which auxiliary subunits are present. In heterologous systems, AKAP79 has a direct effect of decreasing functional Kv4.2 expression (Fig. 6).
In addition, the increase in A-current density in the presence of Ht31 (Fig. 3) was not accompanied by a change in the rate of current inactivation or in the voltage dependence of channel opening in neurons (data not shown) suggesting that the composition of accessory subunits of the A-channel complex was not different between control and those enhanced by Ht31 application (Hoffman and Johnston, 1998; Schrader et al., 2002; Jerng et al., 2004; Maffie and Rudy, 2008; Lin et al., 2010). Together, these observations, along with the fact that we observe AKAP-dependent regulation of Kv4.2 surface expression, A-current density and associated firing properties in neurons, suggest that there are populations of Kv4.2 channels in disassociated hippocampal neurons that are either not in complex with KChIP subunits or that their regulation observed in heterologous systems is not consistent with that found in neurons.
Physiological function of AKAP-Kv4.2 interactions
AKAP scaffolding of PKA, PKC and CaN have been shown to affect the expression and activity of other voltage-gated channels in neurons including L-type Ca2+ channels (PKA and CaN) (Oliveria et al., 2007), KCNQ channels producing the M-current (PKC) (Hoshi et al., 2003; Bal et al., 2010) and voltage-gated Na+ currents (PKA) (Cantrell et al., 1999). As for Kv4.2, activity-dependent regulation of these channels by AKAPs may also be expected to result in changes in neuronal excitability. In addition, activity-dependent Kv2.1 dephosphorylation by CaN induces a hyperpolarizing shift in the voltage-dependent activation of channels, causing a decrease in neuronal excitability (Park et al., 2006). However, the voltage-profile (i.e., mode of action and subthreshold effects) of the excitability changes reported here for disruption of PKA anchoring by Ht31 cannot be adequately explained by any of these previous results and match well excitability changes attributable to Kv4.2 channels (Kim et al., 2005). In particular, M-current modulation depends on AKAP79/150-anchored PKC but not anchored PKA (Hoshi et al., 2005) and disrupting PKA anchoring to AKAP15 increases neuronal Na+ currents (Cantrell et al., 1999). Although typically not thought to act subthreshold, AKAP-linked, neuronal L-type Ca2+ channels could, in part, contribute to the effects observed here. Disruption of PKA anchoring decreases, while inhibition of CaN activity increases, endogenous L-channel currents in hippocampal neurons through AKAP79/150 scaffolding (Oliveria et al., 2007). These changes in L-channel activity are in the right direction to favor decreased or increased excitability similar to the changes in A-current reported here. CaV1.3 L-channels activate at lower voltage thresholds than CaV1.2 L-channels (Catterall et al., 2005) and thus are more attractive candidates for contributing to the excitability changes measured. However, CaV1.2 is more abundantly expressed in hippocampal neurons and only CaV1.2 has been confirmed to interact directly with AKAP79/150 (Oliveria et al., 2007). Moreover, a recent report in adult CA1 hippocampal pyramidal neurons found that downregulation of CaV1.2 by enzymatic removal of the extracellular matrix molecule hyaluronic acid had no effect on input resistance, action potential generation, or the spike adaptation pattern in CA1 pyramidal cells, arguing against any effects on cell excitability (Kochlamazashvili et al., 2010). Finally, our data showing that 4-AP increased excitability and prevented additional regulation by HT31 and FK506 argues that changes in A-current density are best able to explain the observed AKAP effects on intrinsic excitability.
A role for AKAP proteins in regulating AMPAR-mediated currents, AMPAR trafficking, and synaptic plasticity has been well established (Snyder et al., 2005; Smith et al., 2006; Lu et al., 2007; Nie et al., 2007; Tunquist et al., 2008; Bhattacharyya et al., 2009; Weisenhaus et al., 2010). The induction and expression of synaptic plasticity is accompanied by changes neuronal excitability (intrinsic plasticity) (Bliss and Lomo, 1973; Watanabe et al., 2002; Frick et al., 2004; Xu et al., 2005; Disterhoft and Oh, 2006; Narayanan and Johnston, 2007; Campanac et al., 2008; Jung and Hoffman, 2009; Rosenkranz et al., 2009) and recent studies have found that ion channel modulation and trafficking contribute to plasticity-induced alterations in neuronal function (Zhang and Linden, 2003; Shah et al., 2010). Intrinsic plasticity has been hypothesized to act as an additional memory-storage mechanism (Zhang and Linden, 2003). If so, it is essential to understand how voltage- and/or calcium-gated channels are modulated and how this plasticity in their function contributes to learning and memory. If and how Kv4.2 trafficking by AKAP79/150 affects synaptic plasticity will require additional tools designed to specifically impact AKAP79/150-Kv4.2 interaction to distinguish results from AKAP effects on AMPAR expression.
Footnotes
This work was supported by the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development. We thank Begum Choudhury for excellent technical support.
References
- Bal M, Zhang J, Hernandez CC, Zaika O, Shapiro MS. Ca2+/calmodulin disrupts AKAP79/150 interactions with KCNQ (M-Type) K+ channels. J Neurosci. 2010;30:2311–2323. doi: 10.1523/JNEUROSCI.5175-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya S, Biou V, Xu W, Schlüter O, Malenka RC. A critical role for PSD-95/AKAP interactions in endocytosis of synaptic AMPA receptors. Nat Neurosci. 2009;12:172–181. doi: 10.1038/nn.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol. 1973;232:331–356. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanac E, Daoudal G, Ankri N, Debanne D. Downregulation of dendritic I(h) in CA1 pyramidal neurons after LTP. J Neurosci. 2008;28:8635–8643. doi: 10.1523/JNEUROSCI.1411-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantrell AR, Tibbs VC, Westenbroek RE, Scheuer T, Catterall WA. Dopaminergic modulation of voltage-gated Na+ current in rat hippocampal neurons requires anchoring of cAMP-dependent protein kinase. J Neurosci. 1999;19(RC21):1–6. doi: 10.1523/JNEUROSCI.19-17-j0003.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr DW, Hausken ZE, Fraser ID, Stofko-Hahn RE, Scott JD. Association of the type II cAMP-dependent protein kinase with a human thyroid RII-anchoring protein. Cloning and characterization of the RII-binding domain. J Biol Chem. 1992;267:13376–13382. [PubMed] [Google Scholar]
- Catterall WA, Perez-Reyes E, Snutch TP, Striessnig J. International Union of Pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol Rev. 2005;57:411–425. doi: 10.1124/pr.57.4.5. [DOI] [PubMed] [Google Scholar]
- Chen X, Yuan LL, Zhao C, Birnbaum SG, Frick A, Jung WE, Schwarz TL, Sweatt JD, Johnston D. Deletion of Kv4.2 gene eliminates dendritic A-type K+ current and enhances induction of long-term potentiation in hippocampal CA1 pyramidal neurons. J Neurosci. 2006;26:12143–12151. doi: 10.1523/JNEUROSCI.2667-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coghlan VM, Perrino BA, Howard M, Langeberg LK, Hicks JB, Gallatin WM, Scott JD. Association of protein kinase A and protein phosphatase 2B with a common anchoring protein. Science. 1995;267:108–111. doi: 10.1126/science.7528941. [DOI] [PubMed] [Google Scholar]
- Colledge M, Dean RA, Scott GK, Langeberg LK, Huganir RL, Scott JD. Targeting of PKA to glutamate receptors through a MAGUK-AKAP complex. Neuron. 2000;27:107–119. doi: 10.1016/s0896-6273(00)00013-1. [DOI] [PubMed] [Google Scholar]
- Dell'Acqua ML, Dodge KL, Tavalin SJ, Scott JD. Mapping the protein phosphatase-2B anchoring site on AKAP79. Binding and inhibition of phosphatase activity are mediated by residues 315–360. J Biol Chem. 2002;277:48796–48802. doi: 10.1074/jbc.M207833200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disterhoft JF, Oh MM. Learning, aging and intrinsic neuronal plasticity. Trends Neurosci. 2006;29:587–599. doi: 10.1016/j.tins.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Ehrlich I, Malinow R. Postsynaptic density 95 controls AMPA receptor incorporation during long-term potentiation and experience-driven synaptic plasticity. J Neurosci. 2004;24:916–927. doi: 10.1523/JNEUROSCI.4733-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Husseini AE, Schnell E, Chetkovich DM, Nicoll RA, Bredt DS. PSD-95 involvement in maturation of excitatory synapses. Science. 2000;290:1364–1368. [PubMed] [Google Scholar]
- Elias GM, Funke L, Stein V, Grant SG, Bredt DS, Nicoll RA. Synapse-specific and developmentally regulated targeting of AMPA receptors by a family of MAGUK scaffolding proteins. Neuron. 2006;52:307–320. doi: 10.1016/j.neuron.2006.09.012. [DOI] [PubMed] [Google Scholar]
- Fan Y, Fricker D, Brager DH, Chen X, Lu HC, Chitwood RA, Johnston D. Activity-dependent decrease of excitability in rat hippocampal neurons through increases in I(h) Nat Neurosci. 2005;8:1542–1551. doi: 10.1038/nn1568. [DOI] [PubMed] [Google Scholar]
- Frick A, Magee J, Johnston D. LTP is accompanied by an enhanced local excitability of pyramidal neuron dendrites. Nat Neurosci. 2004;7:126–135. doi: 10.1038/nn1178. [DOI] [PubMed] [Google Scholar]
- Gardoni F, Mauceri D, Marcello E, Sala C, Di Luca M, Jeromin A. SAP97 directs the localization of Kv4.2 to spines in hippocampal neurons: regulation by CaMKII. J Biol Chem. 2007;282:28691–28699. doi: 10.1074/jbc.M701899200. [DOI] [PubMed] [Google Scholar]
- Gomez LL, Alam S, Smith KE, Horne E, Dell'Acqua ML. Regulation of A-kinase anchoring protein 79/150-cAMP-dependent protein kinase postsynaptic targeting by NMDA receptor activation of calcineurin and remodeling of dendritic actin. J Neurosci. 2002;22:7027–7044. doi: 10.1523/JNEUROSCI.22-16-07027.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond RS, Lin L, Sidorov MS, Wikenheiser AM, Hoffman DA. Protein kinase a mediates activity-dependent Kv4.2 channel trafficking. J Neurosci. 2008;28:7513–7519. doi: 10.1523/JNEUROSCI.1951-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman DA, Johnston D. Downregulation of transient K+ channels in dendrites of hippocampal CA1 pyramidal neurons by activation of PKA and PKC. J Neurosci. 1998;18:3521–3528. doi: 10.1523/JNEUROSCI.18-10-03521.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman DA, Magee JC, Colbert CM, Johnston D. K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature. 1997;387:869–875. doi: 10.1038/43119. [DOI] [PubMed] [Google Scholar]
- Hoshi N, Zhang JS, Omaki M, Takeuchi T, Yokoyama S, Wanaverbecq N, Langeberg LK, Yoneda Y, Scott JD, Brown DA, Higashida H. AKAP150 signaling complex promotes suppression of the M-current by muscarinic agonists. Nat Neurosci. 2003;6:564–571. doi: 10.1038/nn1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi N, Langeberg LK, Scott JD. Distinct enzyme combinations in AKAP signalling complexes permit functional diversity. Nat Cell Biol. 2005;7:1066–1073. doi: 10.1038/ncb1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerng HH, Pfaffinger PJ, Covarrubias M. Molecular physiology and modulation of somatodendritic A-type potassium channels. Mol Cell Neurosci. 2004;27:343–369. doi: 10.1016/j.mcn.2004.06.011. [DOI] [PubMed] [Google Scholar]
- Jung SC, Hoffman DA. Biphasic somatic A-type K channel downregulation mediates intrinsic plasticity in hippocampal CA1 pyramidal neurons. PLoS One. 2009;4:e6549. doi: 10.1371/journal.pone.0006549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Hoffman DA. Potassium channels: newly found players in synaptic plasticity. Neuroscientist. 2008;14:276–286. doi: 10.1177/1073858408315041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Dittgen T, Nimmerjahn A, Waters J, Pawlak V, Helmchen F, Schlesinger S, Seeburg PH, Osten P. Sindbis vector SINrep(nsP2S726): a tool for rapid heterologous expression with attenuated cytotoxicity in neurons. J Neurosci Methods. 2004;133:81–90. doi: 10.1016/j.jneumeth.2003.09.029. [DOI] [PubMed] [Google Scholar]
- Kim J, Wei DS, Hoffman DA. Kv4 potassium channel subunits control action potential repolarization and frequency-dependent broadening in rat hippocampal CA1 pyramidal neurones. J Physiol. 2005;569:41–57. doi: 10.1113/jphysiol.2005.095042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Jung SC, Clemens AM, Petralia RS, Hoffman DA. Regulation of dendritic excitability by activity-dependent trafficking of the A-type K+ channel subunit Kv4.2 in hippocampal neurons. Neuron. 2007;54:933–947. doi: 10.1016/j.neuron.2007.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klauck TM, Faux MC, Labudda K, Langeberg LK, Jaken S, Scott JD. Coordination of three signaling enzymes by AKAP79, a mammalian scaffold protein. Science. 1996;271:1589–1592. doi: 10.1126/science.271.5255.1589. [DOI] [PubMed] [Google Scholar]
- Kochlamazashvili G, Henneberger C, Bukalo O, Dvoretskova E, Senkov O, Lievens PM, Westenbroek R, Engel AK, Catterall WA, Rusakov DA, Schachner M, Dityatev A. The extracellular matrix molecule hyaluronic acid regulates hippocampal synaptic plasticity by modulating postsynaptic L-type Ca(2+) channels. Neuron. 2010;67:116–128. doi: 10.1016/j.neuron.2010.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Sun W, Wikenheiser AM, Kung F, Hoffman DA. KChIP4a regulates Kv4.2 channel trafficking through PKA phosphorylation. Mol Cell Neurosci. 2010;43:315–325. doi: 10.1016/j.mcn.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Allen M, Halt AR, Weisenhaus M, Dallapiazza RF, Hall DD, Usachev YM, McKnight GS, Hell JW. Age-dependent requirement of AKAP150-anchored PKA and GluR2-lacking AMPA receptors in LTP. EMBO J. 2007;26:4879–4890. doi: 10.1038/sj.emboj.7601884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffie J, Rudy B. Weighing the evidence for a ternary protein complex mediating A-type K+ currents in neurons. J Physiol. 2008;586:5609–5623. doi: 10.1113/jphysiol.2008.161620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe AM, Makhinson M, He Y, Ramsay MF, Morris RG, Morrison JH, O'Dell TJ, Grant SG. Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature. 1998;396:433–439. doi: 10.1038/24790. [DOI] [PubMed] [Google Scholar]
- Narayanan R, Johnston D. Long-term potentiation in rat hippocampal neurons is accompanied by spatially widespread changes in intrinsic oscillatory dynamics and excitability. Neuron. 2007;56:1061–1075. doi: 10.1016/j.neuron.2007.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie T, McDonough CB, Huang T, Nguyen PV, Abel T. Genetic disruption of protein kinase A anchoring reveals a role for compartmentalized kinase signaling in theta-burst long-term potentiation and spatial memory. J Neurosci. 2007;27:10278–10288. doi: 10.1523/JNEUROSCI.1602-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveria SF, Gomez LL, Dell'Acqua ML. Imaging kinase–AKAP79–phosphatase scaffold complexes at the plasma membrane in living cells using FRET microscopy. J Cell Biol. 2003;160:101–112. doi: 10.1083/jcb.200209127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveria SF, Dell'Acqua ML, Sather WA. AKAP79/150 anchoring of calcineurin controls neuronal L-type Ca2+ channel activity and nuclear signaling. Neuron. 2007;55:261–275. doi: 10.1016/j.neuron.2007.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KS, Mohapatra DP, Misonou H, Trimmer JS. Graded regulation of the Kv2.1 potassium channel by variable phosphorylation. Science. 2006;313:976–979. doi: 10.1126/science.1124254. [DOI] [PubMed] [Google Scholar]
- Robertson HR, Gibson ES, Benke TA, Dell'Acqua ML. Regulation of postsynaptic structure and function by an A-kinase anchoring protein-membrane-associated guanylate kinase scaffolding complex. J Neurosci. 2009;29:7929–7943. doi: 10.1523/JNEUROSCI.6093-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenkranz JA, Frick A, Johnston D. Kinase-dependent modification of dendritic excitability after long-term potentiation. J Physiol. 2009;587:115–125. doi: 10.1113/jphysiol.2008.158816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumbaugh G, Sia GM, Garner CC, Huganir RL. Synapse-associated protein-97 isoform-specific regulation of surface AMPA receptors and synaptic function in cultured neurons. J Neurosci. 2003;23:4567–4576. doi: 10.1523/JNEUROSCI.23-11-04567.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlüter OM, Xu W, Malenka RC. Alternative N-terminal domains of PSD-95 and SAP97 govern activity-dependent regulation of synaptic AMPA receptor function. Neuron. 2006;51:99–111. doi: 10.1016/j.neuron.2006.05.016. [DOI] [PubMed] [Google Scholar]
- Schnell E, Sizemore M, Karimzadegan S, Chen L, Bredt DS, Nicoll RA. Direct interactions between PSD-95 and stargazin control synaptic AMPA receptor number. Proc Natl Acad Sci U S A. 2002;99:13902–13907. doi: 10.1073/pnas.172511199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader LA, Anderson AE, Mayne A, Pfaffinger PJ, Sweatt JD. PKA modulation of Kv4.2-encoded A-type potassium channels requires formation of a supramolecular complex. J Neurosci. 2002;22:10123–10133. doi: 10.1523/JNEUROSCI.22-23-10123.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah MM, Hammond RS, Hoffman DA. Dendritic ion channel trafficking and plasticity. Trends Neurosci. 2010;33:307–316. doi: 10.1016/j.tins.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi SH, Hayashi Y, Petralia RS, Zaman SH, Wenthold RJ, Svoboda K, Malinow R. Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science. 1999;284:1811–1816. doi: 10.1126/science.284.5421.1811. [DOI] [PubMed] [Google Scholar]
- Smith KE, Gibson ES, Dell'Acqua ML. cAMP-dependent protein kinase postsynaptic localization regulated by NMDA receptor activation through translocation of an A-kinase anchoring protein scaffold protein. J Neurosci. 2006;26:2391–2402. doi: 10.1523/JNEUROSCI.3092-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder EM, Colledge M, Crozier RA, Chen WS, Scott JD, Bear MF. Role for A kinase-anchoring proteins (AKAPS) in glutamate receptor trafficking and long term synaptic depression. J Biol Chem. 2005;280:16962–16968. doi: 10.1074/jbc.M409693200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavalin SJ, Colledge M, Hell JW, Langeberg LK, Huganir RL, Scott JD. Regulation of GluR1 by the A-kinase anchoring protein 79 (AKAP79) signaling complex shares properties with long-term depression. J Neurosci. 2002;22:3044–3051. doi: 10.1523/JNEUROSCI.22-08-03044.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunquist BJ, Hoshi N, Guire ES, Zhang F, Mullendorff K, Langeberg LK, Raber J, Scott JD. Loss of AKAP150 perturbs distinct neuronal processes in mice. Proc Natl Acad Sci U S A. 2008;105:12557–12562. doi: 10.1073/pnas.0805922105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Hoffman DA, Migliore M, Johnston D. Dendritic K+ channels contribute to spike-timing dependent long-term potentiation in hippocampal pyramidal neurons. Proc Natl Acad Sci U S A. 2002;99:8366–8371. doi: 10.1073/pnas.122210599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisenhaus M, Allen ML, Yang L, Lu Y, Nichols CB, Su T, Hell JW, McKnight GS. Mutations in AKAP5 disrupt dendritic signaling complexes and lead to electrophysiological and behavioral phenotypes in mice. PLoS One. 2010;5:e10325. doi: 10.1371/journal.pone.0010325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong W, Newell EW, Jugloff DG, Jones OT, Schlichter LC. Cell surface targeting and clustering interactions between heterologously expressed PSD-95 and the Shal voltage-gated potassium channel, Kv4.2. J Biol Chem. 2002;277:20423–20430. doi: 10.1074/jbc.M109412200. [DOI] [PubMed] [Google Scholar]
- Xu J, Kang N, Jiang L, Nedergaard M, Kang J. Activity-dependent long-term potentiation of intrinsic excitability in hippocampal CA1 pyramidal neurons. J Neurosci. 2005;25:1750–1760. doi: 10.1523/JNEUROSCI.4217-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Schlüter OM, Steiner P, Czervionke BL, Sabatini B, Malenka RC. Molecular dissociation of the role of PSD-95 in regulating synaptic strength and LTD. Neuron. 2008;57:248–262. doi: 10.1016/j.neuron.2007.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Linden DJ. The other side of the engram: experience-driven changes in neuronal intrinsic excitability. Nat Rev Neurosci. 2003;4:885–900. doi: 10.1038/nrn1248. [DOI] [PubMed] [Google Scholar]