


Abstract
Arguably, the most bizarre mitochondrial DNA (mtDNA) is that of the euglenozoan eukaryote Diplonema papillatum. The genome consists of numerous small circular chromosomes none of which appears to encode a complete gene. For instance, the cox1 coding sequence is spread out over nine different chromosomes in non-overlapping pieces (modules), which are transcribed separately and joined to a contiguous mRNA by trans-splicing. Here, we examine how many genes are encoded by Diplonema mtDNA and whether all are fragmented and their transcripts trans-spliced. Module identification is challenging due to the sequence divergence of Diplonema mitochondrial genes. By employing most sensitive protein profile search algorithms and comparing genomic with cDNA sequence, we recognize a total of 11 typical mitochondrial genes. The 10 protein-coding genes are systematically chopped up into three to 12 modules of 60–350 bp length. The corresponding mRNAs are all trans-spliced. Identification of ribosomal RNAs is most difficult. So far, we only detect the 3′-module of the large subunit ribosomal RNA (rRNA); it does not trans-splice with other pieces. The small subunit rRNA gene remains elusive. Our results open new intriguing questions about the biochemistry and evolution of mitochondrial trans-splicing in Diplonema.
INTRODUCTION
The mitochondrial genome generally consists of a single chromosome (in multiple copies), on which reside from as few as five genes in the apicomplexan parasite Plasmodium to up to approximately 100 in jakobid flagellates. Mitochondrial genes are involved in central biological processes, notably respiration, oxidative phosphorylation and translation, and occasionally transcription, RNA maturation and protein import. Mitochondrial introns, which in some organisms make up more than half of a genome, belong to either group I or II [for reviews, see (1,2)].
Studies of mitochondria as well as chloroplasts have uncovered several novel modes of gene expression that were also detected later in the nucleus. One such example is trans-splicing of exons that are encoded in distant genomic regions or even on different strands and transcribed separately (3). Another intriguing feature first discovered in organelles and specifically in mitochondria is RNA editing, a process that corrects ‘errors’ in a gene at the level of its transcript. RNA editing in mitochondria proceeds by several fundamentally different molecular mechanisms (reviewed in ref. 4). C-to-U substitution editing in plant mitochondria involves nucleotide modification; tRNA editing, found in mitochondria of diverse eukaryotes, replaces mispaired nucleotides with pairing ones; and global insertion editing in slime mould mitochondria proceeds in a transcription-dependent fashion. Finally, editing in mitochondria of kinetoplastid flagellates consists of uridine insertions and deletions, a process templated by short guide RNAs (gRNAs) that are encoded on hundreds of small DNA minicircles. Mitochondria of kinetoplastids clearly stand out as most eccentric due to their unusual genome architecture and unconventional gene expression mechanism (5). It is unknown how these particularities arose and whether they are shared by the kinetoplastids’ sistergroup, the diplonemids.
The first study of mitochondrial genes in the free-living flagellate Diplonema papillatum was published >10 years ago (6). The authors described the 3′-half of the cox1 cDNA (specifying cytochrome c oxidase subunit 1) and provided evidence for a mitochondrial DNA (mtDNA) of unusual architecture—probably consisting of numerous small circles. We demonstrated later that Diplonema has a multipartite mitochondrial genome with circular chromosomes of two size classes, 6 kb (Class A) and 7 kb (Class B) (7); one representative each of Classes A and B chromosomes has been completely sequenced (8). To our surprise, the cox1 gene turned out to be discontinuous and split up into nine pieces (modules), each of which is located on a different chromosome. After separate transcription of these modules, the contiguous cox1 mRNA is generated by a trans-splicing process. Furthermore, we found one case of apparent RNA editing in cox1 involving the addition of Us between two gene modules, reminiscent of U-insertion/deletion editing in kinetoplastids (8).
Here, we report about chromosome structure, set of genes and pattern of gene fragmentation in Diplonema mtDNA. Delineation of gene pieces and gene function annotation was a challenging task, because Diplonema mitochondrial genes are highly divergent with sequences very different from any known organism including kinetoplastids.
MATERIALS AND METHODS
Sequences deposited in public-domain databases
Accession numbers of sequences deposited in GenBank are listed in Table 1.
Table 1.
Sequences deposited in the public domain
| Sequence | Length (bp) | Description | GenBank acc. no. | References |
|---|---|---|---|---|
| Chromosome A (A3207) | 5852 | Carries cox1 Module 9 | EU12356 | Marande and Burger (8) |
| Chromosome A (A3208) | 5802 | Carries cox1 Module 9 | HQ288823 | This report |
| Chromosome A (A4001) | 5794 | Carries nad7 Module 6 | HQ288824 | This report |
| Chromosome B (B3209) | 7182 | Carries cox1 Module 4 | EU12357 | Marande and Burger (8) |
| cox1 cassettes | set of sequences | Comprise cox1 Modules 1–9 | HQ288825-33 | This report |
| cob cDNA (dp4278rian41) | 1214 | Sequences include 5′-UTR and A-tail; all module junctions are annotated | HQ288819 | This report |
| cox1 cDNA (dp4030riaC21) | 2280 | EU123538 | This report: updated version of Marande and Burger (8) | |
| cox2 cDNA (dp0321iah548) | 924 | HQ288820 | This report | |
| cox3 cDNA (dp0301iaF27) | 1012 | HQ288821 | This report | |
| nad7 cDNA (dp4285ian41) | 1251 | HQ288822 | This report |
Strain, culture and mtDNA extraction
Diplonema papillatum (ATCC 50162) was obtained from the American Type Culture Collection. The organism was cultivated axenically at 22°C in artificial seawater enriched with 1% fetal horse serum (Wisent) and 0.1% tryptone. Mitochondrial DNA was extracted from the organelle fraction separated by differential centrifugation and a sucrose gradient. Alternatively, mtDNA was extracted by TRIzol (Invitrogen), whereby the small mitochondrial circles cofractionate with RNA (7,9).
PCR and RT-PCR
We used the polymerase chain reaction (PCR) kit of TAKARA (Bio Inc.) as recommended, by adding 1% Dimethylsulfoxide (DMSO) and 1M Betaine to the reaction mix. The primer pairs that allow specific amplification of A and B class chromosomes are listed in Supplementary Table S1. Several cDNAs from the cDNA library (see below) were truncated, likely due to the G+C-rich templates, which cause premature termination of the reverse transcriptase reaction. To complete cDNAs, we performed (regular) RT–PCR and nested RT–PCR using poly(A) RNA as template. For regular RT–PCR, first strand (cDNA) synthesis was primed with an exon-specific oligonucleotide using the Powerscript reverse transcriptase of the Creator SMART cDNA library construction kit (TAKARA). Subsequent PCR amplification was conducted for 35 cycles using the first-strand primer plus the SMART IV primer that anneals with the most 5′-end of the transcript. Nested RT–PCR was employed when sequence information was available to design a primer upstream of the first-strand primer and included the following, additional step. One-fiftieth of the product was employed as template for a second PCR amplification (35 cycles) using the SMART IV primer plus the ‘upstream primer’.
Library construction and DNA sequencing
Mitochondrial genomic clones were obtained by several different approaches: (i) cloning of restriction fragments including entire linearized Class B chromosomes (XhoI) and smaller fragments (XhoI for Class A chromosomes and BamHI for both classes); (ii) cloning of PCR-amplified chromosomes using module-specific primers facing ‘outwards’ (Figure 1A); (iii) cloning of PCR-amplified cassettes using primers annealing in the constant regions adjacent to cassettes; and (iv) cloning of mechanically broken whole genome random fragments. For detailed protocols, see (10). The genomic clone 3.0 and two subclones of it (3.6, 3.9) that include a 4-kb BamHI fragment of Diplonema mtDNA were initially generated by S. Yasuhira in L. Simpson’s laboratory and were kindly provided to us by D. Maslov; clone 3.0 was identified in Simpson’s group through hybridization with labelled cox1 cDNA. Prior to cloning, we size-fractionated blunt-ended DNA fragments on agarose gel and cloned them into the EcoRV-site of vector pBFL6cat (derived from pBluescript, Stratagene). Whole chromosomes were sequenced by primer walking. Incomplete cassettes were completed by PCR amplification. For primers used for sequencing, PCR and RT–PCR, see Supplementary Table S1. The cDNA library was constructed from poly(A) RNA using the components of the SMART kit (see above) including the vector pDNRlib. The method has been described in detail elsewhere (11). Mitochondrial cDNAs were distinguished from nuclear cDNAs by sequence identity with mtDNA sequence (cassettes or constant regions). Plasmid DNA was extracted with the QIAprep 96 Turbo Miniprep Kit (Qiagen). For sequencing reactions, we used the Sanger-dye terminator technology (kit ABI PRISM Big Dye terminator version 3.0/3.1, Perkin-Elmer). Reactions were run on an MJ BaseStation or an ABI 3730 capillary sequencer. We generated in total 675 kb high-quality raw sequence and 250 kb assembled sequence by the Sanger technology. Mitochondrial genome sequences were also present in the set of reads generated from total DNA by the 454 technology (GS FLX/Titanium) at the Institute of Molecular Genetics in Prague. A total of approximately 183 000 readings were clustered at 99% with Cd-hit (12) yielding ∼26 500 clusters, among which ∼4600 were mitochondrial as identified by Blast (e-value <1.0e-10) against the sequences of complete chromosomes, A3207 and B3209. Cluster-representing readings constitute 1.480 Mbp high-quality raw sequence and 377 kb assembled sequence. Sanger readings (abi and scf format) were assembled with Phred, Phrap (–q 0.9) and Consed (13) using wrapper scripts developed in-house. Fasta-formatted sequence files with integrated annotations (Masterfile format, http://megasun.bch.umontreal.ca/ogmp/ogmpid.html) were generated with Cosmea, a program developed in-house. To allow Phred assembly of 454 readings (ssf-files), we transformed these readings into Fasta files and the corresponding quality files and then generated pseudo-scf files. About 450 readings were also assembled by Mira (http://www.chevreux.org/projects_mira.html) (14). All in-house developed tools are described at the URL given above and available on request.
Figure 1.

Mitochondrial chromosome architecture in D. papillatum. (A) The constant region is identical within Classes A and B chromosomes. The portion shared by Classes A- and B chromosomes is indicated in dark grey. The cassette includes the coding region (gene module) and two unique module-flanking regions. (B) repeat structure of two representative chromosomes, A3207 (Class A) and B3209 (Class B). Triangles denoted a–h symbolize distinct repeat motifs that are all arranged in the same orientation. The replication origin inferred from the GC skew is indicated by a hollow arrow. For GenBank accession numbers, see Table 1.
Basic sequence analysis
We used the in-house developed tools Flip for translation of nucleotide sequence into protein sequence, and Pepper for codon usage analysis and extracting various genetic elements from the Masterfile. Sequence similarity searches were performed locally with Blast (15) and Fasta (16), and remotely with Blast and psiBlast (17) against NCBI’s databases. Multiple sequence alignments were obtained with Muscle (18). Alignments were visualized and edited with the Genetic Data Environment (GDE) (19). For the analysis of sequence repeats, we used Dotter v3.1 (http://www.cgb.ki.se/cgb/groups/sonnhammer/Dotter.html) (20), and the cumulative GC skew [(G–C)/(G+C)] was calculated using the University of Pittsburgh bioinformatics software and web tools collection at the URL http://bioinformatics2.pitt.edu/index.html.
HMM search
To identify the protein-coding content of small gene fragments, we generated hidden Markov model (HMM) profiles of typical mitochondrion-encoded proteins and portions thereof, using HMMER3 (21). The proteins and species used for generating the profiles are listed in Supplementary Table S2. Profiles were searched against all open reading frames >10 amino acids long in Diplonema mtDNA and cDNA sequences. HMM predictions were evaluated and validated by several criteria. (i) The HMMER e-value must be <0.005 for profiles of protein fragments. (ii) To eliminate fortuitous hits outside of cassettes, the nucleotide sequence that gives rise to the matching open reading frame from Diplonema must have low similarity (<50%) to the constant regions of chromosomes A3207 and B3209. (iii) To test whether hits come from mitochondrion- or rather nucleus-encoded open reading frames (ORFs), we looked for Classes A and B constant regions adjacent to the matching reading frame (>80% identity). (iv) The sequence divergence of the Diplonema protein fragment must be less than that of the corresponding regions of trypanosome proteins, because phylogenetic inferences show that the free-living diplonemids are less derived than parasitic trypanosomes (22). This latter criterion was verified by inspecting the multiple protein alignment by eye.
RESULTS
Sequencing of Diplonema mtDNA
We chose three sequencing strategies: (i) sequencing of entire, individual chromosomes or regions thereof (the cassettes, see below) amplified by PCR; (ii) whole genome (shotgun) sequencing of mtDNA (Sanger technology); and (iii) massive parallel sequencing (454 technology) of total genomic DNA. In the 454 data set, mitochondrial readings were identified by Blast against the sequence of two representative chromosomes, A3207 and B3209 (see ‘Materials and Methods’ section). Together these data confirm that all mitochondrial chromosomes consist of two unequal parts. The large constant region (95% of the total length) is nearly identical in sequence between members of the same chromosome class, and about 1/3 of this region is shared between Classes A and B chromosomes. The second—minor—part of Diplonema mitochondrial chromosomes is the ‘cassette’ (5% of the total length), which distinguishes individual chromosomes from one another. Only this portion of the chromosome includes coding sequence (Figure 1A).
Structure of mitochondrial chromosomes
We analysed in detail three complete Diplonema Class A chromosomes that were cloned and sequenced individually (A3207, A3208 and A4001); A3207 and A3208 carry the same cassette. Further, we analysed a population of Class B chromosomes carrying the same cassette that were sequenced collectively and assembled to one consensus sequence (B3209). In sum, within a class, constant regions are 97–98% identical in sequence over their entire length with variations consisting of nucleotide substitutions and small insertions/deletions. Classes A and B chromosomes share 2.6 kb of their constant region with 97% sequence identity (Supplementary Table S3).
Constant regions carry a number of dispersed direct and tandem repeats (>40 bp). Most conspicuous are the tandem repeat arrays of Class B chromosomes (Figure 1B and Supplementary Figures S1A, S1B). A pronounced cumulative guanine-cytosine (GC) skew minimum (23) suggests a replication origin located in the portion of the constant region that is shared between Classes A and B chromosomes (Supplementary Figures S2A, S2B). Experimental mapping will be required to confirm and pinpoint more precisely the predicted replication origin.
Cassettes and gene modules
To date we found 75 distinct cassettes whereof 4/5 reside on Class A and 1/5 on Class B chromosomes. Cassettes are on average 300 bp long, ranging from 190 to 470 bp, and enclose gene modules that are framed by unique non-coding sequence (module-flanking regions, see Figure 1A). Modules occur in different orientations with regard to the constant region and there is no correlation between a coding region’s orientation and the chromosome class on which it resides (Table 2).
Table 2.
Cassette structure for completely determined genes and cDNAs
| Gene | Module no. | Length of cassettea (upstream flanking/module/ downstream flanking) | Chromosome class (chromosome id) | Strand |
|---|---|---|---|---|
| cob | 1 | 276 (24/198/54) | A | + |
| 2 | 286 (34/154/96) | A | – | |
| 3 | 290 (53/138/99) | A | + | |
| 4 | 294 (42/198/54) | A | – | |
| 5 | 317 (15/279/22) | A | – | |
| 6 | 206 (39/123/44) | B | – | |
| cox1 | 1 | 282 (52/195/35) | B | + |
| 2 | 222 (34/124/63) | A | + | |
| 3 | 321 (26/263/32) | A | + | |
| 4 | 310 (43/226/47) | B (B3209) | + | |
| 5 | 266 (63/179/24) | A | + | |
| 6 | 284 (35/169/80) | A | + | |
| 7 | 241 (116/89/36) | A | + | |
| 8 | 251 (118/110/23) | A | + | |
| 9 | 311 (11/248/52) | A (A3207; A3208) | – | |
| cox2 | 1 | 308 (41/237/30) | A | + |
| 2 | 248 (80/160/8) | B | + | |
| 3 | 288 (57/76/155) | A | + | |
| 4 | 284 (26/125/133) | A | – | |
| cox3 | 1 | 357 (4/344/9) | A | + |
| 2 | 333 (55/266/12) | A | + | |
| 3 | 304 (17/230/57) | A | – | |
| nad7 | 1 | 296 (40/221/35) | A | + |
| 2 | 274 (110/75/89) | A | – | |
| 3 | 289 (149/133/7) | A | + | |
| 4 | 186 (46/64/76) | B | – | |
| 5 | 284 (81/192/11) | A | + | |
| 6 | 295 (36/66/193) | A (A4001) | – | |
| 7 | 253 (31/169/53) | B | + | |
| 8 | 219 (34/182/3) | A | + | |
| 9 | 273 (66/79/128) | A | – |
aSizes of modules and flanking regions have been inferred by comparison of genomic and cDNA sequences. The chromosome class was inferred from the sequence of the constant region adjacent to the cassettes. Underscores highlight the minimum and maximum length of modules and module-flanking regions. In cases where module junctions are not precisely known (see text), the shortest length is indicated. For sequence accession numbers, see Table 1.
All genes encoded by Diplonema mtDNA appear to be fragmented. This was tested for cox1 (8), cox2, cob and nad7 (this report, data not shown) by PCR spanning ∼500-bp stretches of coding regions, using total cellular DNA.
Initial Blast and Fasta comparisons (15,16) with sequences in public databases identified unambiguously only ∼10% of the gene fragments (7 out of 75), i.e. those corresponding to highly conserved gene regions. To detect more genes, we conducted HMM searches (21) with profiles generated from taxonomically diverse sets of typical mitochondrial proteins and portions thereof. In silico predictions were evaluated based on E-values and several other criteria as described in the ‘Materials and Methods’ section.
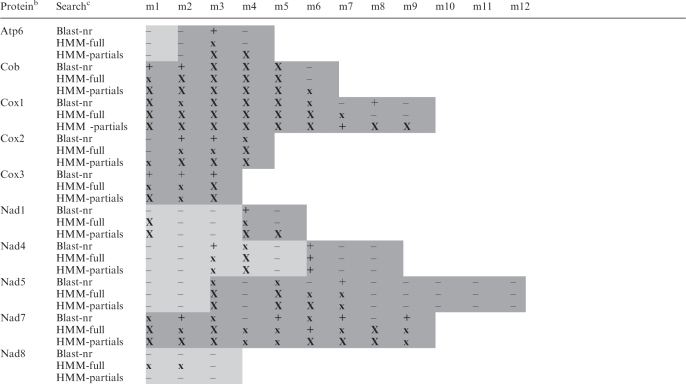
HMM profile searches assigned ∼60% of gene fragments (44 out of 75) to mitochondrial proteins (Table 3). The notoriously poorly conserved nad8 gene (coding for nicotinamide adenine dinucleotide (NADH) dehydrogenase subunit 8) is at the detection limit as illustrated in the multiple protein alignment of the predicted Diplonema nad8 with homologs of other eukaryotes (Supplementary Figure S3). To provide supporting evidence for predicted gene assignments and identify more modules, we generated cDNA data as described below.
Table 3.
In silico assignment of gene modules in mtDNA sequencea
 |
|---|
aShading indicates modules that a given gene includes. Dark shading, modules for which cDNA data were obtained. X, identified module displaying a strong hit (Blast: ≤1.0e-12; HMM-full: ≤1.0e-8; HMM-partials: ≤1.0e-5). x, identified module displaying a moderate hit (Blast: between <1.0e-12 and >1.0e-5; HMM-full: >1.0e-8; HMM-partials: >1.0e-5). +, identified module displaying a marginal hit with an e-value in the range of numerous false positives (Blast: ≥1.0e-5; HMM-full: >2.0e-2; HMM-partials: >1.0e-2); true positives have been validated by the criteria described in the ‘Materials and Methods’ section. –, non-identified module by similarity search with top hits of unrelated proteins. Modules which could not be identified by the three in silico methods are likely not missing from the genomic data set (see ‘Materials and Methods’ section for coverage), but rather unrecognizable. Note that many modules have been assigned by database searches with the conceptually translated contiguous cDNA sequence, while no significant results were obtained with individual gene modules.
bProteins encoded by mtDNA. For full product names, see Table 4.
cBlast-nr, remote blastp search of proteins <14 residues long, conceptually translated in all six frames from Diplonema cassette sequences, against the non-redundant nucleotide database in GenBank. Hits to published Diplonema sequences were not considered in this table. HMM-full, search with a profile HMM, which was constructed from complete homologous proteins from non-diplonemid species, against proteins conceptually translated in all six frames from Diplonema cassette-sequences. HMM-partials, profile HMMs were constructed from protein sub-regions corresponding to modules in Diplonema mtDNA.
Mitochondrial transcriptome and gene complement
Diplonema mitochondrial transcripts are poly-adenylated just like the transcripts of its nuclear genes. The cDNA library constructed from total cellular poly(A) RNA contained ∼5% clones of identifiable mitochondrial origin. About 1/3 of cDNAs were full length, the others were truncated lacking the 5′-portion and are being completed individually by RT–PCR (see ‘Materials and Methods’ section for details).
Gene assignment to cDNA sequences was by and large possible through Blast and Fasta due to the extended length of coding sequence. Assigned cDNAs, in turn, allowed annotation of several genomic modules. For example, Module 2 of atp6, initially unrecognized in genomic sequence, was identified as such in the cDNA, because it is attached to the well conserved region of Module 3.
The mitochondrial origin of cDNAs was corroborated by the existence of corresponding modules in mtDNA or the finding of incompletely processed modules bounded by stretches of Classes A and B constant regions. One cDNA (GenBank Acc. no. EC843366.1) showed significant sequence similarity to rps12 and was initially considered to represent a mitochondrial transcript, but it lacked support for mitochondrial origin by the above criteria. Indeed, thorough analysis revealed a much better match with nucleus- than mitochondrion-encoded counterparts from trypanosomes. Therefore, this gene of Diplonema is now considered nucleus- and not mitochondrion encoded.
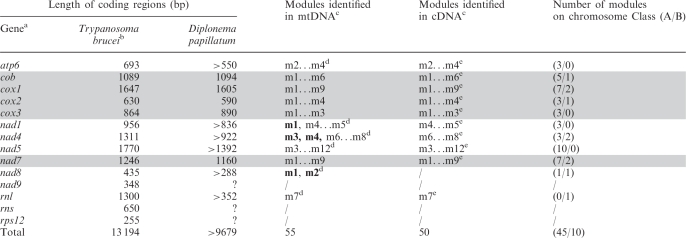
The gene set inferred from genomic and cDNA sequence is fairly conventional (Table 4). Currently, we have cDNA sequences from 11 distinct mitochondrial genes, five of them complete. Protein-coding genes specify apocytochrome b (cob), three cytochrome oxidase subunits (cox1-3), five subunits of NADH dehydrogenase (nad1, 4, 5, 7, 8) and one ATP synthase subunit (atp6). Sequences of the five complete cDNAs (cob, cox1-3, nad7) with annotated module junctions are deposited in GenBank (Table 1). All but one stretch of cDNA-coding regions (nad5-module 6) could be mapped to available genomic sequences, whereas several modules detected in mtDNA (mostly those corresponding to the transcripts’ 5′-end) are yet missing in our cDNA data.
Table 4.
Gene content and structure of D. papillatum mtDNA
 |
|---|
aGenes and corresponding gene products are: atp6, ATP synthase subunit 6; cob, cytochrome b apoprotein; cox1-cox3, cytochrome c oxidase subunits; nad1-nad9, NADH dehydrogenase subunits; rps12, ribosomal protein S12; rnl, LSU rRNA; rns, SSU rRNA. Completely sequenced genes and cDNAs are highlighted by grey background.
bThe gene set of Trypanosoma brucei serves for comparison. Data are inferred from the following GenBank accession nos. (protein GI). cox1: AAB59223 (343538); cox3: AAA32122 (343596); cob: CAA24915 (578828); atp6: AAA97428 (343544); nad1, rps2, cox2: M94286 (343546); nad4: AAB59224 (343539); nad5: AAB59225.1 (343540); nad7: M55645 (343542); nad8 AA91499 (552291); nad9: AAA03749 (162166).
cm1…m12, all modules from m1 to m12. Modules in boldface indicate those so far only found in genomic DNA. Modules exclusively found in the 454 data set are nad5-m3 and nad8-m2. For incomplete genes, module numbers have been estimated from multiple protein alignments assuming an average module length of 170 bp.
dTentative module numbers, see footnote c.
ePoly-adenylated 3′-terminal module.
Every protein-coding gene in Diplonema mtDNA is trans-spliced to form a conventional mRNA. These mRNAs have no trailer at their 3′-end, rather, the poly(A) tail starts immediately downstream of the stop codon, and sometimes, the stop codon is completed by polyadenylation as is the case, for example, in animal (24) and dinoflagellate mitochondria (25). The transcripts’ non-coding 5′-end (5′-UTR) varies in length by 28–600 nt.
Genes coding for structural RNAs are scarce in Diplonema mtDNA. Transfer RNA genes appear to be absent. Of the two canonical ribosomal RNA (rRNA) genes, only one has been detected so far, i.e. rnl that encodes the large subunit (LSU) rRNA. The 300-nt long cDNA corresponds to the 3′-terminal portion of bacterial LSUrRNAs including the highly conserved sarcin/ricin loop domain, is polyadenylated and highly overrepresented in the cDNA library. Interestingly, the cDNA has the same length [not counting the poly(A)-tail] as the genomic module, which indicates that this rnl transcript is not trans-spliced, but instead, LSUrRNA (the gene product itself) is fragmented in Diplonema mitochondria. The gene (rns) encoding the small subunit (SSU) rRNA remains unidentified; the gene and its product are probably fragmented as well. Mitochondrial rRNAs in pieces have been observed in several other protists, fungi and animals, but not in Euglenozoa (26–29).
Finally, cDNA data confirm that Diplonema mitochondria use a non-standard genetic code that is common in this organelle, with TGA specifying tryptophan. GTG is most likely a start codon in addition to ATG (e.g. in cox2, cox3, and nad7). The strongest evidence for a GTG-initiation codon comes from nad7, whose first ATG occurs only downstream of an otherwise conserved region of the protein (52 triplets downstream of the designated GTG-start). Overall codon frequency of the five completely sequenced protein-coding genes shows an unusual strong bias against ATT, TTA, CAA and CTT (Supplementary Table S4).
Structure of mitochondrial genes
Transcript data not only allowed identification of additional coding regions, but also allowed to pinpoint exactly the gene modules within cassettes. Modules are 60–350 bp long (average 170 bp), generally centred within the cassettes and flanked by 0 (not shown) to 200 bp (see Table 2). The complete set of cassette sequences that comprise cox1 modules is deposited in GenBank (see Table 1).
The sequences of genomic modules align precisely with the corresponding cDNA sequence in a non-overlapping fashion as depicted for cox1 in Figure 2. In some instances, the exact junction is uncertain (±4; e.g. in cox2), because of identical sequence in the flanking regions of neighbouring modules. These uncertainties will be clarified eventually by sequencing more individual, fully processed module transcripts that contain only coding sequence. Whereas in essence, the concatenate of genomic modules is identical with the cDNA sequence, there is one flagrant exception, six non-encoded Us inserted between Modules 4 and 5 of cox1 (8). This remains so far the only bona fide editing site in Diplonema mitochondria.
Figure 2.

Genomic cox1 modules aligned according to cox1 cDNA. Parts of module-flanking regions are shown as well. m1–m9, gene Modules 1–9. Six Ts are present at the junction of Modules 4 and 5 in cDNA, but not in the corresponding genomic modules or their flanking regions. Bold uppercase, sequence of genomic modules corresponding to that of cDNA. Lowercase, sequence of module-flanking regions in mtDNA. Five module junctions (1/2, 2/3, 5/6, 7/8, 8/9) cannot be inferred accurately from the genomic sequence alone; all junctions except 7/8 (highlighted in grey) have been mapped by sequencing transcript intermediates (not shown).
In the current data set, the number of modules per gene is proportional to the gene length and ranges from 3 to 12 (see Table 4). Nearly five times more modules reside on Class A than on Class B chromosomes, and this ratio is similar across all genes. There is no correspondence between the chromosome class and the ordinal number of the module encoded by the chromosome (see Table 2).
DISCUSSION
Genome architecture and gene structure
In Diplonema mitochondria, the single gene modules encoded on 6–7 kb chromosomes are on average 170 bp long, yielding a coding versus non-coding ratio of only ∼3%. The number of distinct mitochondrial chromosomes is at least 75 (this is the number of currently identified distinct cassettes), and thus the estimated genome size is >500 kb, which is extremely large for mtDNA. The currently largest known mtDNA is that of muskmelon with 2400 kb (30), probably comprising accumulated chloroplast sequences and repeats as seen in other Cucurbitacean mtDNAs (31).
The above-discussed genome sizes refer to unique sequence, not considering redundancy through multiple copies of distinct chromosomes as typically found in mitochondrial genomes. Evidence for multiple copies in Diplonema mitochondria comes from DNA sequencing, where we observed chromosomes with the same cassette having polymorphisms in their constant regions (see Supplementary Table S3, comparison A3207 and A3208). Multiple chromosome copies are also in concordance with the extraordinary large amount of organelle DNA seen by DAPI staining of Diplonema whole cells (7).
In Cryptobia helicis (Bodonidae, Kinetoplastida), quantitative analysis estimated 14 copies of maxicircles and a total of 8400 minicircles (32). The high copy number was proposed to assure that random segregation during mitochondrial division provides both daughter organelles with a complete chromosome set, a reasoning that also rationalizes the high copy number of Diplonema mitochondrial chromosomes. A more sophisticated approach to equitable chromosome segregation is realized in trypanosomes (the second family within Kinetoplastida) by physical catenation of the molecules into a single network (for a review, see 33).
Diplonema is not the only organism with multipartite mtDNA. A few chromosomes make up mtDNA of certain fungi and animals (for a review, see 26), whereas chromosome numbers in thousands occur in kinetoplastids (34), in a unicellular relative of animals Amoebidium parasiticum (35) and potentially also in dinoflagellates (25). Notable also are multicircle chloroplast genomes of dinoflagellates (36). These multipartite organelle genomes encode one to several genes per chromosome. Diplonema is unique in that all its mitochondrial chromosomes sequenced so far contain one and only one ‘piece’ of a (known or putative) gene.
Expression of fragmented genes
In Diplonema, all mitochondrion-encoded genes appear to be fragmented, and lack contiguous copies elsewhere in the cell. Fragmented genes occur in various organisms and genomes, and are expressed in different ways. In the nucleus of ciliates, gene fragments are joined by DNA splicing prior to transcription (37). Alternatively, gene fragments can be expressed as such, leading to fragmented gene products. For instance, the mitochondrial rRNAs of the green alga Chlamydomonas consist of up to eight pieces (all encoded by a single chromosome) that are believed to associate by hydrogen bonds only (38). Examples of fragmented mitochondrial proteins include NADH dehydrogenase subunit 1 of the ciliate Tetrahymena and cytochrome c oxidase subunit 2 of the green alga Scenedesmus (reviewed in 26). The third scenario is that gene pieces are joined at the RNA level. Prevalent in organelles is trans-splicing mediated by group-II introns (3). Recently, also group I intron-mediated trans-splicing was discovered and reported first in mitochondria of the animal Trichoplax (39) and later also in mitochondria of a protist and a land plant (40,41). Spliced-leader (SL) trans-splicing occurs in the nucleus of diplonemids, kinetoplastids, rotifers, dinoflagellates and animals. Here, a non-translated transcript is attached covalently to the 5′-end of all the coding transcripts (3). Finally, trans-splicing of nuclear pre-mRNAs occurs sporadically in animals including human (reviewed in 42). All trans-splicing events reported in the literature—spliceosomal, Group I- and Group II mediated—involve at most four separate transcripts (43), much less than we see in Diplonema mitochondria.
In Diplonema mitochondria, we rule out DNA splicing, since contiguous genes could not be amplified by PCR of total DNA spanning more than one module. Likewise, fragmented proteins can be excluded, because the corresponding cDNAs are contiguous and of expected length. Further, trans-splicing mediated by Groups I, II, archaeal or spliceosomal introns is most unlikely for Diplonema mitochondria, because the regions adjacent to cox1 modules lack the hallmarks of these introns (see cox1 cassette sequences, Table 1). Also lacking are catalytic RNA signatures such as hammerhead, which has been proposed to direct self-ligation of viroidal RNA (44). Interestingly, an equally enigmatic trans-splicing event has been recently discovered in cox3 (encoding cytochrome c oxidase subunit 3) of the dinoflagellate Karlodinium (reviewed in 25), but it remains to be seen whether in this species mitochondrial trans-splicing is the rule or rather the exception. This finding adds one more to the long list of plesiomorphic characters shared between the phylogenetically most distant euglenozoans and dinoflagellates (45).
Commonalities and differences of diplonemid and kinetoplastid mtDNAs
Diplonema and the sistergroup kinetoplastids both have a multipartite mtDNA. In addition, Diplonema mitochondrial chromosomes resemble trypanosomatid minicircles, in that both contain a major constant (core) region with high sequence conservation within their chromosome class (46). Yet, with more than 500 kb unique sequence, the mitochondrial genome of Diplonema is significantly larger than that of trypanosomes and related kinetoplastids, whose mtDNA sizes range from 100 to 200 kb when summing up the maxicircle of 20–40 kb plus up to 100 distinct minicircles of typically 1–2 kb (reviewed in ref. 7).
All genes currently identified in mtDNA of Diplonema are also present in mtDNA of kinetoplastids, but only 4/5 vice versa (see Table 4). One of the genes expected but not found in Diplonema mtDNA is nad9 encoding subunit 9 of NADH dehydrogenase. Since nad9 is globally well conserved in sequence but short, it may be present but not encountered in the genomic and cDNA libraries of Diplonema. Alternatively, it may have migrated to the nucleus. Another gene present in mtDNA of kinetoplastids but apparently missing in Diplonema is rps12, coding for the mitochondrial ribosomal protein S12. However, the S12 sequence is highly variable across taxa so that it is equally probable that the gene is absent from, or present but unrecognized, in Diplonema mtDNA. Among the approximately 20 hypothetical (unassigned) modules, about 15 should be the missing fragments of yet partially sequenced genes. Another five would be sufficient to account for the expected modules of SSUrRNA—an essential component of mitochondrial ribosomes and otherwise present in all mitochondrial genomes—and in addition one or very few small genes such as nad9.
Transfer RNA genes appear to be absent from mtDNA of both Diplonema and kinetoplastids. It was shown experimentally in kinetoplastids (and certain other eukaryotes) that tRNAs are imported into mitochondria from the nucleus (47). The nuclear genes encoding mitochondrial tRNAs are believed to have initially resided on mtDNA and later been transferred to the nucleus. Some tRNA genes may also have been lost and substituted by genuine nuclear genes. This is likely the case for tRNA-Trp in Trypanosoma brucei, where a single nucleus-encoded gene serves both cytoplasmic and mitochondrial translation. The tRNA imported into mitochondria undergoes C to U modification in the first position of the anticodon. Editing changes the anticodon from CCA (only decoding TGG-Trp) to UCA (decoding both TGG-Trp and TGA-Trp)—a retrofit to comply with the mitochondrial genetic code (48). It would be interesting to see if the same situation applies to Diplonema as its mitochondrial translation code also includes TGA-Trp codons (see Supplementary Table S4).
Mitochondrial mRNAs of kinetoplastids and Diplonema are decorated with an A-tail, which is rather the exception in this organelle. Although, poly(A) is obviously a trait of stable mitochondrial transcripts in kinetoplastids and diplonemids as well as in animals (49) and dinoflagellates (25), it is instead an RNA degradation signal in plant mitochondria and bacteria (50). Yet, in kinetoplastids the situation is more complicated. Here, mRNA stability together with RNA editing depend on the precise length of the poly(A) tail (51).
Finally, both kinetoplastids and Diplonema feature U-based mitochondrial RNA editing: U-insertion/deletion editing to various extents in kinetoplastids (5), and U-insertion RNA editing, so far at a single site, in Diplonema. Given the scarcity of occurrence, we suggest that mitochondrial RNA editing in Diplonema is a primitive form of that in kinetoplastids. Whether or not guide RNAs direct also the process in Diplonema is under investigation.
Conclusion and outlook
The mitochondrial genome of Diplonema is exceptional in every aspect: genome architecture, gene structure and gene expression. Notably, other diplonemid species seem to display the same unusual features (52), providing data for promising comparative studies. An intriguing question bears on the genetic makeup of mtDNA in the common ancestor of kinetoplastids and diplonemids. The answer may be found in euglenids, a euglenozoan group that emerged before the divergence of kinetoplastids and diplonemids. The euglenid mitochondrial genome is virtually unexplored.
ACCESSION NUMBER
GenBank acc. nos. HQ288819-33.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Canadian Institute for Health Research (MOP-79309 to G.B.); Czech Ministry of Education, Youth and Sports (1M0520 and 6007665801 to C.V. and J.L.); and Grant Agency of the Czech Republic 204/09/1667 (to J.L.); Premium Academiae award (to J.L.). Funding for open access charge: Canadian Institute for Health Research.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
We thank J. Paces and J. Ridl (IMG, Prague) for performing 454 DNA sequencing and data management, and S. Prigent (Université de Lyon, France) for in silico gene assignment by HMM in the context of a Master’s internship project supervised by GB. M.W. Gray and M. Schnare (Dalhousie University, Halifax) kindly assisted in the identification of LSUrRNA, and B. Franz Lang (Université de Montréal) contributed through helpful discussions and suggestions to the manuscript. We also acknowledge N. Beck (Université de Montréal) for software development and maintenance, L. Simpson (UCLA) and his former trainees D. Maslov and S. Yasuhira, for providing mitochondrial genomic clones, and finally I. Plante, Y. Zhu, and J.H. Song (Université de Montréal) for excellent technical assistance. G.B. designed and supervised the study, conducted data management and sequence analyses, and wrote the manuscript. W.M. conducted mtDNA and cDNA sequencing and comparative sequence analysis and wrote part of the manuscript in the context of his thesis supervised by G.B. S.T. prepared Diplonema DNA for 454 sequencing, Diplonema RNA for cDNA sequencing, constructed the cDNA library, and wrote parts of the manuscript. C.V. and his group performed 454 sequencing of mitochondrial (and nuclear) DNA. J.L. co-supervised WM and served as a hub between the two sequencing endeavors. All authors contributed to the final manuscript version.
REFERENCES
- 1.Lang BF, Gray MW, Burger G. Mitochondrial genome evolution and the origin of eukaryotes. Annu. Rev. Genet. 1999;33:351–397. doi: 10.1146/annurev.genet.33.1.351. [DOI] [PubMed] [Google Scholar]
- 2.Gray MW, Lang BF, Burger G. Mitochondria of protists. Annu. Rev. Genet. 2004;38:477–524. doi: 10.1146/annurev.genet.37.110801.142526. [DOI] [PubMed] [Google Scholar]
- 3.Bonen L. Trans-splicing of pre-mRNA in plants, animals, and protists. FASEB J. 1993;7:40–46. doi: 10.1096/fasebj.7.1.8422973. [DOI] [PubMed] [Google Scholar]
- 4.Gott JM, Emeson RB. Functions and mechanisms of RNA editing. Annu. Rev. Genet. 2000;34:499–531. doi: 10.1146/annurev.genet.34.1.499. [DOI] [PubMed] [Google Scholar]
- 5.Stuart KD, Schnaufer A, Ernst NL, Panigrahi AK. Complex management: RNA editing in trypanosomes. Trends Biochem. Sci. 2005;30:97–105. doi: 10.1016/j.tibs.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 6.Maslov DA, Yasuhira S, Simpson L. Phylogenetic affinities of Diplonema within the Euglenozoa as inferred from the SSU rRNA gene and partial COI protein sequences. Protist. 1999;150:33–42. doi: 10.1016/S1434-4610(99)70007-6. [DOI] [PubMed] [Google Scholar]
- 7.Marande W, Lukeš J, Burger G. Unique mitochondrial genome structure in diplonemids, the sister group of kinetoplastids. Eukaryot Cell. 2005;4:1137–1146. doi: 10.1128/EC.4.6.1137-1146.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marande W, Burger G. Mitochondrial DNA as a genomic jigsaw puzzle. Science. 2007;318:415. doi: 10.1126/science.1148033. [DOI] [PubMed] [Google Scholar]
- 9.Lang BF, Burger G. Purification of mitochondrial and plastid DNA. Nat. Protoc. 2007;2:652–660. doi: 10.1038/nprot.2007.58. [DOI] [PubMed] [Google Scholar]
- 10.Burger G, Lavrov DV, Forget L, Lang BF. Sequencing complete mitochondrial and plastid genomes. Nat. Protoc. 2007;2:603–614. doi: 10.1038/nprot.2007.59. [DOI] [PubMed] [Google Scholar]
- 11.Rodriguez-Ezpeleta N, Teijeiro S, Forget L, Burger G, Lang BF. EST databases and Web tools for EST projects. In: Parkinson J, editor. Methods in Molecular Biology: Expressed Sequence Tags (ESTs) Vol. 533. Totowa, NJ: Humana Press; 2009. [Google Scholar]
- 12.Huang Y, Niu B, Gao Y, Fu L, Li W. CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics. 2010;26:680–682. doi: 10.1093/bioinformatics/btq003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gordon D. Curr. Protoc. Bioinformatics. 2003. Viewing and editing assembled sequences using Consed. Chapter 11, Unit 11.12. [DOI] [PubMed] [Google Scholar]
- 14.Chevreux B, Pfisterer T, Drescher B, Driesel AJ, Muller WE, Wetter T, Suhai S. Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res. 2004;14:1147–1159. doi: 10.1101/gr.1917404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 16.Pearson WR. Flexible sequence similarity searching with the FASTA3 program package. Methods Mol. Biol. 2000;132:185–219. doi: 10.1385/1-59259-192-2:185. [DOI] [PubMed] [Google Scholar]
- 17.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith SW, Overbeek R, Woese CR, Gilbert W, Gillevet PM. The genetic data environment an expandable GUI for multiple sequence analysis. Comput. Appl. Biosci. 1994;10:671–675. doi: 10.1093/bioinformatics/10.6.671. [DOI] [PubMed] [Google Scholar]
- 20.Sonnhammer EL, Durbin R. A dot-matrix program with dynamic threshold control suited for genomic DNA and protein sequence analysis. Gene. 1995;167:GC1–10. doi: 10.1016/0378-1119(95)00714-8. [DOI] [PubMed] [Google Scholar]
- 21.Eddy SR. A new generation of homology search tools based on probabilistic inference. Genome Inform. 2009;23:205–211. [PubMed] [Google Scholar]
- 22.Simpson AG, Gill EE, Callahan HA, Litaker RW, Roger AJ. Early evolution within kinetoplastids (Euglenozoa), and the late emergence of trypanosomatids. Protist. 2004;155:407–422. doi: 10.1078/1434461042650389. [DOI] [PubMed] [Google Scholar]
- 23.Lobry JR. Asymmetric substitution patterns in the two DNA strands of bacteria. Mol. Biol. Evol. 1996;13:660–665. doi: 10.1093/oxfordjournals.molbev.a025626. [DOI] [PubMed] [Google Scholar]
- 24.Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- 25.Waller RF, Jackson CJ. Dinoflagellate mitochondrial genomes: stretching the rules of molecular biology. Bioessays. 2009;31:237–245. doi: 10.1002/bies.200800164. [DOI] [PubMed] [Google Scholar]
- 26.Burger G, Gray MW, Lang BF. Mitochondrial genomes - anything goes. Trends Genet. 2003;19:709–716. doi: 10.1016/j.tig.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 27.Kamikawa R, Inagaki Y, Sako Y. Fragmentation of mitochondrial large subunit rRNA in the dinoflagellate Alexandrium catenella and the evolution of rRNA structure in alveolate mitochondria. Protist. 2007;158:239–245. doi: 10.1016/j.protis.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 28.Dellaporta SL, Xu A, Sagasser S, Jakob W, Moreno MA, Buss LW, Schierwater B. Mitochondrial genome of Trichoplax adhaerens supports placozoa as the basal lower metazoan phylum. Proc. Natl Acad. Sci. USA. 2006;103:8751–8756. doi: 10.1073/pnas.0602076103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bullerwell CE, Forget L, Lang BF. Evolution of monoblepharidalean fungi based on complete mitochondrial genome sequences. Nucleic Acids Res. 2003;31:1614–1623. doi: 10.1093/nar/gkg264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ward BL, Anderson RS, Bendich AJ. The mitochondrial genome is large and variable in a family of plants (Cucurbitaceae) Cell. 1981;25:793–803. doi: 10.1016/0092-8674(81)90187-2. [DOI] [PubMed] [Google Scholar]
- 31.Alverson AJ, Wei X, Rice DW, Stern DB, Barry K, Palmer JD. Insights into the evolution of mitochondrial genome size from complete sequences of Citrullus lanatus and Cucurbita pepo (Cucurbitaceae) Mol. Biol. Evol. 2010 doi: 10.1093/molbev/msq029. doi:10.1093/molbev/msq029 [Epub ahead of print, 29 January 2010] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lukeš J, Jirku M, Avliyakulov N, Benada O. Pankinetoplast DNA structure in a primitive bodonid flagellate, Cryptobia helicis. EMBO J. 1998;17:838–846. doi: 10.1093/emboj/17.3.838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu B, Liu Y, Motyka SA, Agbo EE, Englund PT. Fellowship of the rings: the replication of kinetoplast DNA. Trends Parasitol. 2005;21:363–369. doi: 10.1016/j.pt.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 34.Simpson L. The mitochondrial genome of kinetoplastid protozoa: genomic organization, transcription, replication, and evolution. Annu. Rev. Microbiol. 1987;41:363–382. doi: 10.1146/annurev.mi.41.100187.002051. [DOI] [PubMed] [Google Scholar]
- 35.Burger G, Forget L, Zhu Y, Gray MW, Lang BF. Unique mitochondrial genome architecture in unicellular relatives of animals. Proc. Natl Acad. Sci. USA. 2003;100:892–897. doi: 10.1073/pnas.0336115100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Z, Cavalier-Smith T, Green BR. Evolution of dinoflagellate unigenic minicircles and the partially concerted divergence of their putative replicon origins. Mol. Biol. Evol. 2002;19:489–500. doi: 10.1093/oxfordjournals.molbev.a004104. [DOI] [PubMed] [Google Scholar]
- 37.Landweber LF, Kuo TC, Curtis EA. Evolution and assembly of an extremely scrambled gene. Proc. Natl Acad. Sci. USA. 2000;97:3298–3303. doi: 10.1073/pnas.040574697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boer PH, Gray MW. Scrambled ribosomal RNA gene pieces in Chlamydomonas reinhardtii mitochondrial DNA. Cell. 1988;55:399–411. doi: 10.1016/0092-8674(88)90026-8. [DOI] [PubMed] [Google Scholar]
- 39.Burger G, Yan Y, Javadi P, Lang BF. Group I-intron trans-splicing and mRNA editing in the mitochondria of placozoan animals. Trends Genet. 2009;25:381–386. doi: 10.1016/j.tig.2009.07.003. [DOI] [PubMed] [Google Scholar]
- 40.Grewe F, Viehoever P, Weisshaar B, Knoop V. A trans-splicing group I intron and tRNA-hyperediting in the mitochondrial genome of the lycophyte Isoetes engelmannii. Nucleic Acids Res. 2009;37:5093–5104. doi: 10.1093/nar/gkp532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pombert JF, Keeling PJ. The mitochondrial genome of the entomoparasitic green alga Helicosporidium. PLoS One. 2010;5:e8954. doi: 10.1371/journal.pone.0008954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Herai RH, Yamagishi ME. Detection of human interchromosomal trans-splicing in sequence databanks. Brief Bioinform. 2009;11:198–209. doi: 10.1093/bib/bbp041. [DOI] [PubMed] [Google Scholar]
- 43.Glanz S, Kuck U. Trans-splicing of organelle introns–a detour to continuous RNAs. Bioessays. 2009;31:921–934. doi: 10.1002/bies.200900036. [DOI] [PubMed] [Google Scholar]
- 44.Lafontaine D, Beaudry D, Marquis P, Perreault JP. Intra- and intermolecular nonenzymatic ligations occur within transcripts derived from the peach latent mosaic viroid. Virology. 1995;212:705–709. doi: 10.1006/viro.1995.1528. [DOI] [PubMed] [Google Scholar]
- 45.Lukeš J, Leander BS, Keeling PJ. Cascades of convergent evolution: the corresponding evolutionary histories of euglenozoans and dinoflagellates. Proc. Natl Acad. Sci. USA. 2009;106 (Suppl. 1):9963–9970. doi: 10.1073/pnas.0901004106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Simpson L. The genomic organization of guide RNA genes in kinetoplastid protozoa: several conundrums and their solutions. Mol. Biochem. Parasitol. 1997;86:133–141. doi: 10.1016/s0166-6851(97)00037-6. [DOI] [PubMed] [Google Scholar]
- 47.Alfonzo JD, Soll D. Mitochondrial tRNA import–the challenge to understand has just begun. Biol. Chem. 2009;390:717–722. doi: 10.1515/BC.2009.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alfonzo JD, Blanc V, Estevez AM, Rubio MA, Simpson L. C to U editing of the anticodon of imported mitochondrial tRNA(Trp) allows decoding of the UGA stop codon in Leishmania tarentolae. EMBO J. 1999;18:7056–7062. doi: 10.1093/emboj/18.24.7056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ojala D, Attardi G. Identification and partial characterization of multiple discrete polyadenylic acid containing RNA components coded for by HeLa cell mitochondrial DNA. J. Mol. Biol. 1974;88:205–219. doi: 10.1016/0022-2836(74)90305-2. [DOI] [PubMed] [Google Scholar]
- 50.Jacobson A, Peltz SW. Interrelationships of the pathways of mRNA decay and translation in eukaryotic cells. Annu. Rev. Biochem. 1996;65:693–739. doi: 10.1146/annurev.bi.65.070196.003401. [DOI] [PubMed] [Google Scholar]
- 51.Etheridge RD, Aphasizheva I, Gershon PD, Aphasizhev R. 3′ adenylation determines mRNA abundance and monitors completion of RNA editing in T. brucei mitochondria. EMBO J. 2008;27:1596–1608. doi: 10.1038/emboj.2008.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roy J, Faktorova D, Lukeš J, Burger G. Unusual mitochondrial genome structures throughout the Euglenozoa. Protist. 2007;158:385–396. doi: 10.1016/j.protis.2007.03.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.