

Abstract
The IL-1β-induced increase in intestinal epithelial tight junction (TJ) permeability has been postulated to be an important mechanism contributing to intestinal inflammation of Crohn's disease and other inflammatory conditions of the gut. The intra-cellular and molecular mechanisms that mediate the IL-1β-induced increase in intestinal TJ permeability remain unclear. The purpose of this study was to elucidate the mechanisms that mediate the IL-1β-induced increase in intestinal TJ permeability. Specifically, the role of myosin L chain kinase (MLCK) was investigated. IL-1β caused a progressive increase in MLCK protein expression. The time course of IL-1β-induced increase in MLCK level correlated linearly with increase in Caco-2 TJ permeability. Inhibition of the IL-1β-induced increase in MLCK protein expression prevented the increase in Caco-2 TJ permeability. Inhibition of the IL-1β-induced increase in MLCK activity also prevented the increase in Caco-2 TJ permeability. Additionally, knock-down of MLCK protein expression by small interference RNA prevented the IL-1β-induced increase in Caco-2 TJ permeability. The IL-1β-induced increase in MLCK protein expression was preceded by an increase in MLCK mRNA expression. The IL-1β-induced increase in MLCK mRNA transcription and subsequent increase in MLCK protein expression and Caco-2 TJ permeability was mediated by activation of NF-κB. In conclusion, our data indicate that the IL-1β increase in Caco-2 TJ permeability was mediated by an increase in MLCK expression and activity. Our findings also indicate that the IL-1β-induced increase in MLCK protein expression and Caco-2 TJ permeability was mediated by an NF-κB-dependent increase in MLCK gene transcription.
The defective intestinal epithelial tight junction (TJ)3 barrier has been implicated to play a key pathogenic role in the intestinal inflammation of Crohn's disease (CD) and other inflammatory conditions of the gut (1, 2). The defective intestinal TJ barrier allows an increase in the paracellular permeation of toxic luminal Ags that lead to intestinal and systemic inflammation. Intestinal epithelial TJs are the apical – most junctional complexes and act as a functional and structural barrier against paracellular permeation of hydrophilic luminal substances (2–5). Accumulating evidence indicate that immune system plays an important role in modulating intestinal TJ barrier function. Immune cells including neutrophils, dendritic cells, and monocytes have been directly implicated in inducing disturbance of TJ barrier function (6, 7). Additionally, proinflammatory cytokines, including TNF-α, IFN-γ, IL-1β, and IL-8 have been shown to cause a functional opening of intestinal TJ barrier (8–11).
It has been postulated (9–18) that proinflammatory cytokine-induced opening of the intestinal TJ barrier is an important mechanism contributing to the TJ barrier defect present in various inflammatory conditions of the gut. IL-1β is a prototypical multifunctional cytokine having wide-ranging biological activities (19–22). IL-1β is markedly elevated in CD and other inflammatory conditions of the gut and has been shown (22–24) to play a central role in the inflammatory process. A direct correlation between increasing levels of IL-1β and increasing severity of intestinal inflammation in CD has been demonstrated (25, 26). There is an imbalance between the level of IL-1β and its naturally occurring antagonist IL-1 receptor antagonist (IL-1ra) in CD patients, leading to the hypothesis that an excess of proinflammatory and deficiency of anti-inflammatory forms of IL-1 are important pathogenic factors in inflammatory bowel diseases (27, 28). There is also an increase incidence of IL-1β gene polymorphism in CD patients that determine the severity of intestinal inflammation in the affected patients (29, 30). Thus, excess IL-1β expression appears to be an important pathogenic factor contributing to intestinal inflammation of CD. IL-1ra has also been found to be effective in the treatment of immune-mediated inflammation in mice and is currently being developed for clinical usage in CD (31, 32).
Recent studies from our laboratory and others have shown (8, 33, 34) that IL-1β causes an increase in intestinal TJ permeability. The IL-1β-induced increase in intestinal TJ permeability has been postulated to play an important role in promoting intestinal inflammation by allowing increased paracellular permeation of luminal Ags (8). However, the intracellular mechanisms that mediate the IL-1β-induced increase in intestinal TJ permeability remain unclear.
Previous studies (17, 35–39) have shown that myosin L chain kinase (MLCK) plays a central role in the regulation of intestinal TJ permeability. Studies from our laboratory and others have shown that MLCK mediates pharmacologic (cytochalasins, ethanol, and low extracellular Ca++ solution) (37, 39–41), physiologic (Na+- glucose co-transport) (42), and microbial pathogen (enteropathogenic E. coli and C. difficile) (15, 43, 44) induced increase in intestinal TJ permeability. The activation of MLCK catalyzes the phosphorylation of myosin L chain (MLC); which in turn induces a contraction of peri-junctional actin-myosin filaments and opening of the TJ barrier (35, 45, 46). Inhibition of MLCK activation prevents the increase in intestinal TJ permeability (13, 38, 46). In CD patients, MLCK protein expression has been shown to be significantly increased in intestinal tissues (12, 47). The level of increase in intestinal epithelial MLCK protein expression correlated directly with the level of activity of CD, suggesting the possibility that the elevated intestinal MLCK protein expression in CD patients may contribute to the inflammatory process (12, 46). In this study, we examined the possibility that enterocyte MLCK plays an important role in mediating IL-1β modulation of intestinal epithelial TJ barrier.
Our data indicate that the IL-1β-induced increase in Caco-2 TJ permeability was mediated by an increase in MLCK protein expression and activity. Our studies also provide additional insight into the intracellular mechanisms that mediate the IL-1β-induced increase in MLCK protein expression and subsequent opening of the TJ barrier.
Materials and Methods
Chemicals
Cell culture medium (DMEM), trypsin, FBS, and related reagents were purchased from Life Technologies. Glutamine, penicillin, streptomycin, and PBS were purchased from Life Technologies-BRL. Anti-MLCK, NF-κB p65, and anti-β-actin Abs were obtained from Sigma-Aldrich. Anti-phospho-MLC S19 was purchased from StressGen. Biotinylated MLC peptide was ordered and prepared by AnaSpe. HRP-conjugated secondary Abs for Western blot analysis were purchased from Invitrogen. Cy-3 Abs for immunostaining were purchased from Jackson ImmunoResearch Laboratories. siRNA of MLCK, small interference RNA (siRNA) of NF-κB p65, and transfection reagents were from Dharmacon. Cycloheximide and actinomycin-D were purchased from Sigma-Aldrich. All other chemicals were of reagent grade and were purchased from Sigma-aldrich, VWR, or Fisher Scientific.
Cell cultures
Caco-2 cells (passage 20) were purchased from the American Type Culture Collection and maintained at 37°C in a culture medium composed of DMEM with 4.5 mg/ml glucose, 50 U/ml penicillin, 50 U/ml streptomycin, 4 mM glutamine, 25 mM HEPES, and 10% FBS. The cells were kept at 37°C in a 5% CO2 environment. Culture medium was changed every 2 days. Caco-2 cells were subcultured after partial digestion with 0.25% trypsin and 0.9 mM EDTA in Ca2+- and Mg2+-free PBS.
Determination of epithelial monolayer resistance and paracellular permeability
An epithelial voltohmeter (World Precision Instruments) was used for measurements of the transepithelial electrical resistance (TER) of the filter-grown Caco-2 intestinal monolayers as previously reported (48, 49). To study the time course effects of IL-1β on TER, Caco-2 monolayers were treated with increasing doses ranging 1–100 ng/ml over a 72-h time period. The effect of IL-1β on Caco-2 paracellular permeability was determined using an established paracellular marker inulin. For determination of mucosal-to-serosal flux rates of inulin, Caco-2-plated filters having epithelial resistance of 400–500 Ωcm2 were used. Known concentrations of permeability marker (2 μM) and its radioactive tracer were added to the apical solution. Low concentrations of permeability marker were used to ensure that negligible osmotic or concentration gradient was introduced.
Assessment of protein expression by Western blot analysis
To study the time course effect of IL-1β on MLCK and phospho-MLC protein expression, Caco-2 monolayers were treated with IL-1β (10 ng/ml) for varying time periods. At the end of the experimental period, Caco-2 monolayers were immediately rinsed with ice-cold PBS, and cells were lysed with lysis buffer (50 mM Tris · HCl, pH 7.5, 150 mM NaCl, 500 μM NaF, 2 mM EDTA, 100 μM vanadate, 100 μM PMSF, 1 μg/ml leupeptin, 1 μg/ml pepstatin A, 40 mM paranitrophenyl phosphate, 1 μg/ml aprotinin, and 1% Triton X-100) and scraped, and the cell lysates were placed in Microfuge tubes. Cell lysates were centrifuged to yield a clear lysate. Supernatant was collected, and protein measurement was performed using Bio-Rad Protein Assay kit (Bio-Rad Laboratories). Laemmli gel loading buffer was added to the lysate containing 10–20 μg of protein and boiled for 7 min, after which proteins were separated on an SDS-PAGE gel. Proteins from the gel were transferred to the membrane (Trans-Blot Transfer Medium, Nitrocellulose Membrane; Bio-Rad Laboratories) overnight. The membrane was incubated for 2 h in blocking solution (5% dry milk in TBS-Tween 20 buffer). The membrane was incubated with appropriate primary Abs in blocking solution. After being washed in TBS-1% Tween buffer, the membrane was incubated in appropriate secondary Abs and developed using the Santa Cruz Western Blotting Luminol Reagents (Santa Cruz Biotechnology) on the Kodak BioMax MS film (Fisher Scientific).
ELISA-based MLCK in vitro kinase activity
Biotinylated MLC was diluted in PBS and coated on streptavidin 96-well plates at 37°C for 1 h. The plates were washed 3 times with PBS, incubated with blocking solution (1 mg/ml BSA in PBS) at 37°C for 1 h, and then washed 3 times with PBS. The kinase reaction buffer (90 μl), provided by the manufacturer (MBL International) and the treated samples (10 μl) were added to the wells, and the kinase reaction was performed at 37°C for 30–60 min. The reaction was stopped by removing the reaction mixtures and washing the plates 3 times with washing buffer (20 mM Tris-HCl at pH 7.4, 0.5 M NaCl, and 0.05% Tween 20). The washed plates were incubated with the anti-phospho-MLC-S19 Ab (5 ng/ml) at room temperature for 1h. The plates were washed four times with washing buffer, and goat anti-rabbit IgG Ab (diluted at 1:2,000 in washing buffer) was added to the wells, and the plates were incubated at 37°C for 1 h. The plates were then washed four times and incubated with 100 μl substrate solution (tetramethylbenzidine) at 37°C for 5–15 min. A stop solution containing 0.5 N H2SO4 (100 μl) was added to stop the reaction. The absorbance at 450 nm was determined using the SpectrraMax 190 (Molecular Devices).
RNA isolation and reverse transcription
Caco-2 cells/filter (5 × 105) were seeded into six-well transwell permeable inserts and grown to confluency. Filter-grown Caco-2 cells were then treated with appropriate experimental reagents for desired time periods. At the end of the experimental period, cells were washed twice with ice-cold PBS. Total RNA was isolated using Qiagen RNeasy Kit (Qiagen) according to the manufacturer's protocol. Total RNA concentration was determined by absorbance at 260/280 nm using SpectrraMax 190 (Molecular Devices). The reverse transcription (RT) was conducted using the Gene-Amp Gold RNA PCR core kit (Applied Biosystems). Two micrograms of total RNA from each sample were reverse transcribed into cDNA in a 40-μl reaction containing 1× RT-PCR buffer, 2.5 mM MgCl2, 250 μM of each dNTP, 20 U RNase inhibitor, 10 mM DTT, 1.25 μM random hexamer, and 30 U multiscribe RT. The RT reactions were performed in a thermocycler (PTC-100; MJ Research) at 25°C for 10 min, 42°C for 30 min, and 95°C for 5 min.
Quantification of gene expression using real-time PCR
The real-time PCRs were conducted using ABI prism 7900 sequence detection system and Taqman universal PCR master mix kit (Applied Biosystems, Branchburg, NJ) as previously described (36, 46). Each real-time PCR contained 10 μl RT reaction mix, 25 μl 2× TaqMan universal PCR master mix, 0.2 μM probe, and 0.6 μM primers. Primer and probe design for the real-time PCR was made with Primer Express version 2 from Applied Biosystems. [The primers used in this study are as follows: MLCK specific primer pairs consisted of 5′AGGAAGGCAGCATTGAG GTTT-3′ (forward), 5–-GCTTTCAGCAGGCAGAGGTAA-3′ (reverse); probe specific for MLCK consisted of FAM 5′-TGAAGATGCTG GCTCC-3′ TAMRA; the internal control GAPDH-specific primer pairs consisted of 5′ CCACCCATGGCAAATTCC-3′ (forward), 5′-TGG GATTTCCATTGATGACCAG-3′ (reverse); probe specific for GAPDH consisted of JOE 5′TGGCACCGTCAAGGCTGAGAACG-3′ TAMRA]. All runs were performed according to the default PCR protocol (50°C for 2 min, 95°C for 10 min, 40 cycles of 95°C for 15 s, and 60°C for 1 min). For each sample, real-time PCR were performed in triplicate, and the average threshold cycle was calculated. A standard curve was generated to convert the threshold cycle to copy numbers. Expression of MLCK mRNA was normalized with GAPDH mRNA expression. The average copy number of MLCK mRNA expression in control samples was set to 1.0. The relative expression of MLCK mRNA in treated samples was determined as a fold increase compared with control samples.
Immunostaining of NF-κB p65 protein
Cellular localization of NF-κB p65 was assessed by immunofluorescent Ab labeling. At the end of the experimental period, filter-grown Caco-2 monolayers were washed twice in cold PBS and were fixed with 2% paraformaldehyde for 20 min. After being permeabilized with 0.1% Triton X-100 in PBS at room temperature for 20 min, Caco-2 monolayers were then incubated in blocking solution composed of BSA and normal donkey serum in PBS for 1 h. Cells were then labeled with primary Abs in blocking solution overnight at 4°C. After being washed with PBS, the cells were incubated in Cy-3-conjugated secondary Ab for 1h at room temperature. Mowiol was used to mount the filters onto the coverslips. Immunolocalizations of NF-κB p65 was visualized using a Nikon fluorescence microscope (Nikon) equipped with a Hamamatsu digital camera (Hamamatsu Photonics). Images were processed with Wasabi software (Hamamatsu Photonics Deutschland).
siRNA of MLCK or NF-κB p65
Targeted siRNA MLCK and NF-κB p65 were obtained from Dharmacon. Caco-2 monolayers were transiently transfected using DharmaFect transfection reagent (Lafayette). Briefly, cells/filter (5 × 105) were seeded into a twelve-well transwell plate and grown to confluency. Caco-2 monolayers were then washed with PBS twice and 1.0 ml Opti-MEM medium was added to the apical compartment of each filter and 1.5 ml were added to the basolateral compartment of each filter. Five nanograms of the siRNA of interest and 2 μl of DharmaFect reagent were preincubated in Opti-MEM. After 5 min of incubation, two solutions were mixed and incubated for another 20 min, and the mixture was added to the apical compartment of each filter. The IL-1β experiments were conducted 96 h after transfection. The efficiency of silencing NF-κB p65 and MLCK was confirmed by Western blot analysis.
Statistical analysis
Results are expressed as means ± SE. Statistical significance of differences between mean values was assessed with Student's t-tests for unpaired data. All reported significance levels represent two-tailed p values. A p value of <0.05 was used to indicate statistical significance. All experiments were repeated a minimum of three times to ensure reproducibility.
Results
IL-1β-induced increase in Caco-2 TJ permeability is mediated by an increase in MLCK protein expression
In the following studies, we examined the possibility that the IL-1β-induced increase in Caco-2 TJ permeability was regulated by an increase in MLCK protein expression. The time course effect of IL-1β on Caco-2 MLCK protein expression is shown in Fig. 1A. IL-1β (10 ng/ml) caused a progressive time-dependent increase in MLCK expression. The time course of IL-1β-induced increase in MLCK protein level closely paralleled the IL-1β-induced drop in Caco-2 TER (Fig. 1B) and increase in epithelial permeability to paracellular marker inulin (Fig. 1C). The comparison of IL-1β effect on MLCK protein expression vs the drop in Caco-2 TER indicated a direct linear relationship with a correlation coefficient of r = 0.99 (Fig. 1D). Similarly, IL-1β effect on MLCK protein level correlated linearly with an increase in paracellular permeability with a correlation coefficient of r = 0.97 (Fig. 1E). These results indicated that there was a direct linear relationship between IL-1β-induced increase in MLCK protein expression and increase in Caco-2 TJ permeability, suggesting a cause-and-effect relationship.
FIGURE 1.

Time course effect of IL-1β on Caco-2 myosin L chain kinase (MLCK) protein expression, transepithelial resistance (TER), and paracellular permeability. The Caco-2 MLCK protein expression was determined by Western blot analysis as described in Materials and Methods. The effect of IL-1β (10 ng/ml) on Caco-2 TER and mucosal-to-serosal flux of paracellular marker inulin (2 μM) were measured sequentially over the 48-h experimental period as described in Materials and Methods. A, Time course effect of IL-1β on Caco-2 MLCK protein expression (β-actin was used as an internal control for protein loading). B, T ime course effect of IL-1β on Caco-2 TER (means ± SE, n = 4). C, Time course effect of IL-1β on mucosal-to-serosal inulin flux (means ± SE, n = 4). *, p < 0.05 vs control. D, Graph of IL-1β effect on MLCK protein expression and Caco-2 TER (r = 0.99). E, Graph of IL-1β effect on MLCK protein expression and inulin flux (r = 0.97).
The possible causal relationship between IL-1β-induced increase in MLCK protein expression and increase in Caco-2 TJ permeability was investigated in the following studies. In these studies, the IL-1β-induced increase in MLCK protein expression was inhibited by potent protein synthesis inhibitor cycloheximide. As shown in Fig. 2A, cycloheximide inhibited the IL-1β-induced increase in MLCK expression. The cycloheximide inhibition of MLCK protein expression resulted in an inhibition of IL-1β-induced drop in Caco-2 TER (Fig. 2B). These findings suggested that the IL-1β-induced increase in MLCK protein expression was required for the drop in Caco-2 TER.
FIGURE 2.

The effect of protein synthesis inhibitor, cycloheximide (CHX) on IL-1β-induced increase in MLCK protein expression and drop in Caco-2 TER. A, Cycloheximide (5 μM) pretreatment prevented the IL-1β-induced up-regulation in MLCK protein expression as assessed by Western blot analysis. B, Cycloheximide prevented the IL-1β-induced drop in Caco-2 TER (means ± SE, n = 4). *, p < 0.001 vs control; **, p < 0.001 vs IL-1β treatment.
To further delineate the role of MLCK in IL-1β-induced increase in Caco-2 TJ permeability, IL-1β effect on Caco-2 MLCK activity was determined by in vitro kinase assay. In these studies, Caco-2 MLCK was immunoprecipitated following IL-1β treatment, and the kinase activity measured via an in vitro kinase assay. IL-1β treatment of filter-grown Caco-2 monolayers resulted in a progressive time-dependent increase in Caco-2 MLCK activity (Fig. 3A). The time course of IL-1β-induced increase in MLCK activity mirrored the IL-1β-induced increase in MLCK protein expression (Fig. 1A), indicating that the increase in Caco-2 MLCK activity was related to the increase in MLCK protein level. To confirm that IL-1β-induced increase in MLCK activity leads to an increase in MLC phosphorylation in Caco-2 cells, IL-1β effect on Caco-2 MLC phosphorylation was determined by immunoblot analysis. IL-1β treatment resulted in a progressive time-dependent increase in phosphorylated-MLC level in Caco-2 cells (Fig. 3B), indicating that the increase in MLCK activity correlates with an increase in Caco-2 MLC phosphorylation. The cycloheximide inhibition of MLCK protein synthesis also prevented the IL-1β-induced increase in MLCK activity (Fig. 3C), confirming that the increase in MLCK protein expression was responsible for the increase in MLCK activity. It is important to note that cycloheximide alone did not affect MLCK activity in quiescent Caco-2 cells (Fig. 3C), indicating that cycloheximide effect was due to inhibition of MLCK protein synthesis.
FIGURE 3.

Effect of IL-1β on Caco-2 MLCK activity. A, Time course effect of IL-1β on MLCK activity of filter-grown Caco-2 monolayers was determined by ELISA-based in vitro kinase activity measurements as described in Materials and Methods. After appropriate time period, MLCK activity was assessed by measuring the in vitro phosphorylation of MLC (S-19). IL-1β treatment resulted in a time-dependent increase in Caco-2 MLCK activity. *, p < 0.05 vs control. B, Time course effect of IL-1β on phosphorylation of MLC (P-MLC) in filter-grown Caco-2 monolayers over the 48-h experimental period as determined by Western blot analysis. C, Pretreatment with cycloheximide (5 μM) inhibited the IL-1β-induced increase in MLCK kinase activity, cycloheximide alone did not affect MLCK enzyme activity. *, p < 0.001 vs control; **, p < 0.001 vs IL-1β treatment.
Next, the effect of specific MLCK inhibitors ML-7 and ML-9 on IL-1β increase in Caco-2 TJ permeability was determined. The MLCK inhibitors ML-7 (10 μM) and ML-9 (20 μM), at doses shown to specifically inhibit MLCK activity (40, 49), prevented the IL-1β-induced increase in MLCK activity (Fig. 4A). The ML-7 and ML-9 inhibition of MLCK activity prevented the IL-1β-induced drop in Caco-2 TER (Fig. 4B). These data indicated that IL-1β-induced increase in Caco-2 MLCK activity was required for the drop in Caco-2 TER.
FIGURE 4.

Effect of MLCK inhibitors ML-7 and ML-9 on IL-1β-induced increase in Caco-2 MLCK activity and drop in Caco-2 TER. Filter-grown Caco-2 monolayers were treated with IL-1β (10 ng/ml) for a 48-h experimental period. ML-7 (10 μM) and ML-9 (20 μM) significantly prevented the IL-1β (10 ng/ml) induced increase in MLCK activity (in vitro kinase assay) and drop in Caco-2 TER (n = 4) (B). *, p < 0.001 vs control; **, p < 0.001 vs IL-1β treatment.
siRNA-induced MLCK knock-down prevents the IL-1β-induced increase in Caco-2 TJ permeability
To further validate the role of MLCK protein in IL-1β-induced increase in Caco-2 TJ permeability, MLCK protein expression was selectively knocked-down using MLCK siRNA. The MLCK siRNA transfection of Caco-2 monolayers resulted in a near complete depletion of MLCK expression as confirmed by immunoblot analysis of MLCK (Fig. 5A). SiRNA-induced depletion of MLCK protein expression prevented the IL-1β-induced drop in Caco-2 TER (Fig. 5B), confirming that MLCK expression was required for the IL-1β-induced modulation of Caco-2 TJ barrier function.
FIGURE 5.
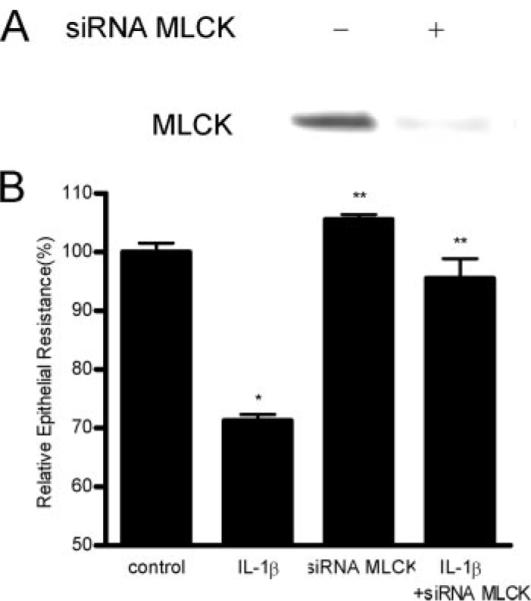
Effect of siRNA induced MLCK knock-down on IL-1β-induced drop on Caco-2 TER. Caco-2 monolayers were transfected with MLCK siRNA for a 96-h time period as described in Materials and Methods. A, MLCK siRNA transfection resulted in a near complete depletion in MLCK protein expression as determined by Western blot analysis. B, MLCK siRNA transfection prevented the IL-1β-induced drop in Caco-2 TER (n = 4). *, p < 0.001 vs control; **, p < 0.001 vs IL-1β treatment.
IL-1β-induced increase in MLCK protein expression is mediated by an increase in transcript expression
In the following studies, we examined the role of MLCK mRNA transcription in IL-1β-induced increase in MLCK expression or increase in Caco-2 TJ permeability. IL-1β effect on MLCK mRNA expression was determined by real-time PCR. IL-1β treatment resulted in a time-dependent increase in MLCK mRNA expression in Caco-2 monolayers (Fig. 6A). The inhibition of MLCK transcription by potent transcription inhibitor actinomycin-D (100 ng/ml) prevented the IL-1β increase in Caco-2 MLCK mRNA (Fig. 6B) and protein expression (Fig. 6C). Actinomycin-D also prevented the IL-1β-induced drop in Caco-2 TER (Fig. 6D). These findings suggested that IL-1β increase in MLCK protein expression and decrease in Caco-2 TER were mediated in part by an increase in MLCK transcript expression.
FIGURE 6.

IL-1β effect on Caco-2 MLCK mRNA expression. A, Time course effect of IL-1β on Caco-2 MLCK mRNA expression was determined by real-time PCR as described in the Materials and Methods. MLCK mRNA level was expressed relative to the control level which was assigned a value of 1. The average copy number of MLCK mRNA in controls was 4.63 × 1011. *, p < 0.001 vs control. B, Actinomycin-D (100 ng/ml) effect on IL-1β-induced increase in MLCK mRNA expression (6 h of IL-1β treatment). Actinomycin-D significantly inhibited the IL-1β increase in MLCK mRNA expression. *, p < 0.05 vs control; **, p < 0.05 vs IL-1β treatment. C, Actinomycin-D significantly prevented the IL-1β-induced up-regulation of MLCK protein expression as determined by Western blot analysis. D, Actinomycin-D also prevented the IL-1β-induced drop in Caco-2 TER (n = 4). *, p < 0.001 vs control; **, p < 0.001 vs IL-1β treatment.
IL-1β-induced increase in MLCK gene expression is mediated by nuclear transcription factor NF-κB
Previous studies have shown that activation of nuclear transcription factor NF-κB was required for the IL-1β-induced increase in Caco-2 TJ permeability (8). However, the mechanisms that mediated the NF-κB modulation of Caco-2 TJ permeability were not elucidated. In the present study, we examined the possibility that NF-κB mediated the IL-1β-induced increase in MLCK gene transcription. As shown in our previous study, IL-1β caused a rapid activation and nuclear translocation of NF-κB (Fig. 7A). The inhibition of NF-κB activation by known NF-κB inhibitor, pyrrolidine dithiocarbamate (PDTC), prevented the IL-1β-induced increase in MLCK mRNA expression (Fig. 7B). PDTC (100 μM) also prevented the IL-1β-induced increase in MLCK protein expression (Fig. 7C), suggesting that NF-κB activation was required for the IL-1β increase in MLCK mRNA and protein expression. In addition, inhibition of NF-κB activation by PDTC significantly prevented the IL-1β-induced drop in Caco-2 TER (Fig. 7D).
FIGURE 7.

IL-1β effect on NF-κB activation. A, IL-1β effect on NF-κB activation as assessed by NF-κB p65 nuclear translocation. NF-κB p65 cytoplasmic-to-nuclear translocation was determined by immunofluorescent staining with anti-NF-κB p65 Ab. Filter-grown Caco-2 monolayers were treated with 10 ng/ml IL-1β for 30 min. B, Effect of NF-κB inhibitor, PDTC (100 μM), on IL-1β-induced increase in MLCK mRNA expression. Caco-2 cells were pretreated with PDTC for 1 h before IL-1β treatment (6-h experimental period). MLCK mRNA level was determined by realtime PCR (n = 6). *, p < 0.05 vs control; **, p < 0.05 vs IL-1β treatment. C, Effect of PDTC (100 μM) on IL-1β up-regulation of MLCK protein expression. Filter-grown Caco-2 cells were treated with PDTC 1 h before IL-1β treatment for 48 h. MLCK protein expression was assessed by Western blot analysis. D, Effect of PDTC (100 μM) on IL-1β-induced drop in Caco-2 TER. *, p < 0.001 vs control; **, p < 0.001 vs IL-1β treatment.
To further validate the role of NF-κB in mediating the IL-1β increase in MLCK gene and protein expression, NF-κB p65 expression was silenced by siRNA transfection. NF-κB p65 siRNA transfection resulted in a near-complete silencing of NF-κB p65 expression in filter-grown Caco-2 cells (Fig. 8A). siRNA induced NF-κB p65 depletion inhibited the IL-1β-induced increase in MLCK protein expression (Fig. 8B). Moreover, NF-κB depletion completely prevented the IL-1β-induced drop in Caco-2 TER (Fig. 8C). Together, these data indicated that the IL-1β-induced increase in MLCK gene and protein expression was regulated by NF-κB activation; and inhibition of NF-κB induced increase in MLCK mRNA transcription prevented the IL-1β-induced increase in Caco-2 TJ permeability.
FIGURE 8.

Effect of siRNA induced knock-down of NF-κB p65 on IL-1β-induced increase in MLCK expression and drop in Caco-2 TER and. Filter-grown Caco-2 monolayers were transfected with NF-κB p65 siRNA for a 96-h time period as described in Materials and Methods. A, NF-κB p65 siRNA transfection resulted in a near complete knock-down of NF-κB p65 expression. B, siRNA NF-κB p65 transfection prevented the IL-1β-induced increase in MLCK protein expression. C, NF-κB p65 siRNA transfection prevented the IL-1β-induced drop in Caco-2 TER. *, p < 0.001 vs control; **, p < 0.001 vs IL-1β treatment.
Discussion
IL-1β is a proto-typical pro-inflammatory cytokine that promotes inflammatory response in various intestinal and systemic disorders (19, 21, 22, 50–52). Defective intestinal TJ barrier has been shown to be an important pathogenic mechanism contributing to intestinal inflammation of CD and other inflammatory conditions of the gut (2). Recent studies have shown that, in addition to its direct immune modulating actions, IL-1β causes a disruption of intestinal TJ barrier leading to an increase in paracellular permeation of luminal Ags (8, 33, 53, 54). The intracellular processes that mediate the IL-1β effect on intestinal TJ barrier remain unclear. Delineating the cellular and molecular processes that mediate the IL-1β modulation of intestinal TJ barrier is very important in understanding the cellular mechanisms that lead to intestinal TJ barrier defect during inflammatory conditions and in designing potential therapeutic strategies to induce re-tightening of the TJ barrier during inflammatory states.
Previous studies have shown that MLCK plays a central role in the regulation of intestinal epithelial TJ barrier function (41, 49, 55, 56). MLCK activation has been shown to be a central mechanism that regulates both pharmacologic (39–41) and physiologic (42) induced opening of intestinal TJ barrier. The cytochalasin, ethanol, or low extracellular Ca++ induced increase in Caco-2 TJ permeability required activation of MLCK; and inhibition of MLCK activity with MLCK inhibitors ML-7 and ML-9 prevented the increase in TJ permeability (37, 39–41, 57). Additionally, Na+-glucose co-transporter activation induced increase in Caco-2 TJ permeability also required MLCK activation (42). Similarly, in live animal studies, immune (T cell), stress, or bacterial endotoxin mediated increase in mice intestinal permeability was associated with an increase in MLCK expression and activity, and inhibition of MLCK activity prevented the increase in intestinal permeability and subsequent development of intestinal inflammation (58, 59). The pathophysiologic importance of MLCK protein expression in intestinal TJ barrier defect in CD was also suggested by a recent study showing that MLCK protein expression was markedly increased in intestinal tissue from CD patients (12). The degree of MLCK protein elevation directly correlated with the level of intestinal inflammation (12). Thus, accumulating evidence suggest that elevation in MLCK protein expression is likely to play an important causal role in intestinal TJ barrier defect during inflammatory conditions.
Our data indicated that IL-1β causes a progressive time-dependent increase in Caco-2 MLCK protein expression. The time course of IL-1β-induced increase in MLCK protein level directly correlated with a drop in Caco-2 TER and increase in paracellular permeability (Fig. 1), suggesting a direct cause-and-effect relationship between IL-1β-induced increase in Caco-2 MLCK protein level and increase in Caco-2 TJ permeability. The inhibition of MLCK protein synthesis with potent protein synthesis inhibitor cycloheximide prevented the IL-1β-induced drop in Caco-2 TER (Fig. 2B). The IL-1β-induced increase in Caco-2 MLCK protein level was also accompanied by a propotional increase in Caco-2 MLCK activity and increase in MLC phosphorylation in Caco-2 monolayers (Fig. 3). And, the inhibition of IL-1β-induced increase in MLCK enzyme activity with specific MLCK inhibitors ML-7 and ML-9 completely inhibited the IL-1β-induced drop in Caco-2 TER (Fig. 4B), indicating that IL-1β-induced increase in MLCK activity was also required for the IL-1β-induced increase in Caco-2 TJ permeability. Our data also indicated that the cycloheximide inhibition of the IL-1β-induced increase in MLCK protein expression completely inhibits the IL-1β-induced increase in MLCK activity (Fig. 3C). Because cycloheximide did not affect the basal Caco-2 MLCK activity, these data suggested that the IL-1β-induced increase in Caco-2 MLCK activity was due to an increase in MLCK protein level and not an increase in the specific activity of the enzyme. The requirement of MLCK protein in IL-1β modulation of Caco-2 TJ barrier was further validated by siRNA targeted knocked-down of MLCK protein expression. The siRNA induced silencing of Caco-2 MLCK protein expression also inhibited the IL-1β-induced drop in Caco-2 TER (Fig. 5B), further validating the role of MLCK protein in IL-1β-induced disruption of Caco-2 TJ barrier function.
The mechanism of MLCK modulation of intestinal TJ barrier function has been extensively studied. Previous studies have shown that cytochalasins and Na+-glucose co-transporter induced increase in intestinal TJ permeability in vitro and in vivo was triggered by MLCK-induced increase in MLC phosphorylation (42, 56, 57). MLC phosphorylation then leads to an energy-dependent contraction of peri-junctional acto-myosin filaments, and mechanical tension induced retraction of junctional membrane and TJ proteins, and functional opening of the TJ barrier (40, 57, 60). The over-expression of constitutively active MLCK has also been shown to cause MLC phosphorylation and functional opening of the TJ barrier in MDCK cells (45). Similarly, when constitutively active MLCK was expressed in Caco-2 cells, there was an increase in phosphorylation of MLC, re-organization of the peri-junctional F-actin, and increase in TJ permeability (35). In the present study, our data suggested that the IL-1β-induced increase in MLCK activity was due to an increase in MLCK protein level and not specific enzyme activity. The intracellular signaling pathways that modulate MLCK activity have been studied in various cell types (61). The signaling pathways that have been shown to up-regulate MLCK activity include Ca++/Calmodulin, PKC, ρ-kinase, MAP kinase and PI3 kinase (62–64). The signaling pathways that are known to down-regulate MLCK activity include MLC phosphatase, PKA, and PKG (65–67). Moreover, activation of signaling pathways that leads to an increase in MLCK activity have been shown to cause an increase in TJ permeability in various endothelial and epithelial cell types (18, 68, 69). Thus, specific signaling pathways that regulate MLCK activity may be targeted to modulate TJ barrier function.
In this study, we also examined the intracellular processes that mediated the IL-1β-induced increase in MLCK protein expression. Our data indicated that the IL-1β-induced increase in MLCK protein expression was due to an increase in MLCK gene transcription. IL-1β-induced increase in MLCK protein expression was preceded by an increase in MLCK mRNA expression (Fig. 6A); and the inhibition of IL-1β-induced increase in MLCK mRNA expression prevented the IL-1β increase in MLCK protein expression and subsequent drop in Caco-2 TER (Fig. 6, C and D). Previously, we showed that IL-1β causes an increase in NF-κB activation in Caco-2 cells; and that NF-κB activation was required for the IL-1β-induced increase in Caco-2 TJ permeability (8). However, the mechanisms that mediated the NF-κB modulation of Caco-2 TJ permeability were not delineated. Our present data indicated that IL-1β causes a rapid activation of NF-κB (Fig. 7A); and inhibition of NF-κB activation with known inhibitor, PDTC, prevented the IL-1β-induced increase in MLCK mRNA expression, MLCK protein expression, and Caco-2 TJ permeability (Fig. 7, B, C, and D). These data suggested that NF-κB modulation of Caco-2 TJ barrier was mediated in part by an increase in MLCK mRNA and protein expression. The role of NF-κB in mediating the IL-1β-induced increase in MLCK protein expression and Caco-2 TJ permeability was further validated by NF-κB p65 silencing studies using siRNA. The siRNA silencing of NF-κB p65 expression in Caco-2 cells completely inhibited the IL-1β-induced increase in MLCK protein expression (Fig. 8B) and the drop in Caco-2 TER (Fig. 8C), confirming the regulatory role of NF-κB in mediating the IL-1β-induced increase in MLCK mRNA transcription and protein expression.
Together, our results indicated that IL-1β causes a rapid activation of NF-κB. (The proposed scheme of IL-1β-induced increase in TJ permeability is outlined in Fig. 9). Activated NF-κB translocates to the nucleus and induces transcription of MLCK mRNA. The increase in MLCK mRNA then leads to an increase in MLCK protein expression and activity and opening of Caco-2 TJ barrier. The molecular processes that mediate the IL-1β-induced increase in MLCK mRNA remain unclear. Recent study by Graham et al. indicated that NF-κB binding motifs were present on the MLCK promoter region and could serve as a regulatory site to induce MLCK gene activation (70). Based on our present data and published studies showing the existence of NF-κB binding sites on MLCK promoter region, we speculate that IL-1β-induced increase in MLCK mRNA expression may be due to NF-κB induced regulation of MLCK promoter activity. We have recently initiated studies to examine the role of NF-κB in the regulation of MLCK promoter activity and delineate the molecular determinants that mediate the regulation of MLCK promoter activity.
FIGURE 9.

Proposed scheme of the IL-1β-induced increase in intestinal epithelial tight junction (TJ) permeability.
In conclusion, our data show for the first time that the IL-1β-induced increase in Caco-2 TJ permeability was mediated by an increase in MLCK protein expression and activity. The IL-1β-induced increase in MLCK protein expression was mediated by an increase in MLCK mRNA transcription. Our results indicated that IL-1β causes a rapid activation of NF-κB; the activated NF-κB translocates to the nucleus where it stimulates MLCK gene transcription; MLCK mRNA transcription leads to an increase in MLCK protein expression and MLCK activity; the increase in MLCK activity then leads to the opening of Caco-2 TJ barrier (Fig. 9). Thus, our data provide important new insight into the intracellular mechanisms that mediate the IL-1β modulation of the intestinal epithelial TJ barrier.
Acknowledgments
This research project was supported by a Veterans Affairs Merit Review grant from the Veterans Affairs Research Service and National Institute of Diabetes and Digestive and Kidney Diseases Grant R01-DK-64165-01 (to T.Y.M.).
Footnotes
Disclosures The authors have no financial conflict of interest.
Abbreviations used in this paper: TJ, tight junction; CD, Crohn's disease; siRNA, small interference RNA; MLCK, that myosin light chain kinase; TER, transepithelial electrical resistance.
References
- 1.Hollander D. Crohn's disease: a permeability disorder of the tight junction? Gut. 1988;29:1621–1624. doi: 10.1136/gut.29.12.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ma TY. Intestinal epithelial barrier dysfunction in Crohn's disease. Proc. Soc. Exp. Biol. Med. 1997;214:318–327. doi: 10.3181/00379727-214-44099. [DOI] [PubMed] [Google Scholar]
- 3.Berkes J, Viswanathan VK, Savkovic SD, Hecht G. Intestinal epithelial responses to enteric pathogens: effects on the tight junction barrier, ion transport, and inflammation. Gut. 2003;52:439–451. doi: 10.1136/gut.52.3.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hollander D. Intestinal permeability, leaky gut, and intestinal disorders. Curr. Gastroenterol. Rep. 1999;1:410–416. doi: 10.1007/s11894-999-0023-5. [DOI] [PubMed] [Google Scholar]
- 5.Anderson JM, Van Itallie CM. Tight junctions and the molecular basis for regulation of paracellular permeability. Am. J. Physiol. 1995;269:G467–G475. doi: 10.1152/ajpgi.1995.269.4.G467. [DOI] [PubMed] [Google Scholar]
- 6.Dimitrijevic OB, Stamatovic SM, Keep RF, Andjelkovic AV. Effects of the chemokine CCL2 on blood-brain barrier permeability during ischemia-reperfusion injury. J. Cereb. Blood Flow Metab. 2006;26:797–810. doi: 10.1038/sj.jcbfm.9600229. [DOI] [PubMed] [Google Scholar]
- 7.Martin-Padura I, Lostaglio S, Schneemann M, Williams L, Romano M, Fruscella P, Panzeri C, Stoppacciaro A, Ruco L, Villa A, et al. Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J. Cell Biol. 1998;142:117–127. doi: 10.1083/jcb.142.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Al-Sadi RM, Ma TY. IL-1β causes an increase in intestinal epithelial tight junction permeability. J. Immunol. 2007;178:4641–4649. doi: 10.4049/jimmunol.178.7.4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chiba H, Kojima T, Osanai M, Sawada N. The significance of interferon-γ-triggered internalization of tight-junction proteins in inflammatory bowel disease. Sci. STKE. 2006;2006:pe1. doi: 10.1126/stke.3162006pe1. [DOI] [PubMed] [Google Scholar]
- 10.Wang F, Graham WV, Wang Y, Witkowski ED, Schwarz BT, Turner JR. Interferon-γ and tumor necrosis factor-α synergize to induce intestinal epithelial barrier dysfunction by up-regulating myosin light chain kinase expression. Am. J. Pathol. 2005;166:409–419. doi: 10.1016/s0002-9440(10)62264-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang F, Schwarz BT, Graham WV, Wang Y, Su L, Clayburgh DR, Abraham C, Turner JR. IFN-γ-induced TNFR2 expression is required for TNF-dependent intestinal epithelial barrier dysfunction. Gastroenterology. 2006;131:1153–1163. doi: 10.1053/j.gastro.2006.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weber CR, Turner JR. Inflammatory bowel disease: is it really just another break in the wall? Gut. 2007;56:6–8. doi: 10.1136/gut.2006.104182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clayburgh DR, Shen L, Turner JR. A porous defense: the leaky epithelial barrier in intestinal disease. Lab. Invest. 2004;84:282–291. doi: 10.1038/labinvest.3700050. [DOI] [PubMed] [Google Scholar]
- 14.Chen ML, Ge Z, Fox JG, Schauer DB. Disruption of tight junctions and induction of proinflammatory cytokine responses in colonic epithelial cells by Campylobacter jejuni. Infect. Immun. 2006;74:6581–6589. doi: 10.1128/IAI.00958-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Savkovic SD, Ramaswamy A, Koutsouris A, Hecht G. EPEC-activated ERK1/2 participate in inflammatory response but not tight junction barrier disruption. Am. J. Physiol. 2001;281:G890–G898. doi: 10.1152/ajpgi.2001.281.4.G890. [DOI] [PubMed] [Google Scholar]
- 16.Watson CJ, Hoare CJ, Garrod DR, Carlson GL, Warhurst G. Interferon-γ selectively increases epithelial permeability to large molecules by activating different populations of paracellular pores. J. Cell Sci. 2005;118:5221–5230. doi: 10.1242/jcs.02630. [DOI] [PubMed] [Google Scholar]
- 17.Boivin MA, Ye D, Kennedy JC, Al-Sadi R, Shepela C, Ma TY. Mechanism of glucocorticoid regulation of the intestinal tight junction barrier. Am. J. Physiol. 2007;292:G590–G598. doi: 10.1152/ajpgi.00252.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Utech M, Ivanov AI, Samarin SN, Bruewer M, Turner JR, Mrsny RJ, Parkos CA, Nusrat A. Mechanism of IFN-γ-induced endocytosis of tight junction proteins: myosin II-dependent vacuolarization of the apical plasma membrane. Mol. Biol. Cell. 2005;16:5040–5052. doi: 10.1091/mbc.E05-03-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Monteleone G, Fina D, Caruso R, Pallone F. New mediators of immunity and inflammation in inflammatory bowel disease. Curr. Opin. Gastroenterol. 2006;22:361–364. doi: 10.1097/01.mog.0000231808.10773.8e. [DOI] [PubMed] [Google Scholar]
- 20.Kreydiyyeh SI, Al-Sadi R. Interleukin-1β increases urine flow rate and inhibits protein expression of Na+/K+-ATPase in the rat jejunum and kidney. J. Interferon Cytokine Res. 2002;22:1041–1048. doi: 10.1089/107999002760624279. [DOI] [PubMed] [Google Scholar]
- 21.O'Neill LA. The interleukin-1 receptor/Toll-like receptor superfamily: signal transduction during inflammation and host defense. Sci. STKE. 2000;2000:re1. doi: 10.1126/stke.442000re1. [DOI] [PubMed] [Google Scholar]
- 22.Dunne A, O'Neill LA. The interleukin-1 receptor/Toll-like receptor superfamily: signal transduction during inflammation and host defense. Sci. STKE. 2003;2003:re3. doi: 10.1126/stke.2003.171.re3. [DOI] [PubMed] [Google Scholar]
- 23.Dinarello CA. The interleukin-1 family: 10 years of discovery. FASEB J. 1994;8:1314–1325. [PubMed] [Google Scholar]
- 24.Stylianou E, Saklatvala J. Interleukin-1. Int. J. Biochem. Cell. Biol. 1998;30:1075–1079. doi: 10.1016/s1357-2725(98)00081-8. [DOI] [PubMed] [Google Scholar]
- 25.Carty E, De Brabander M, Feakins RM, Rampton DS. Measurement of in vivo rectal mucosal cytokine and eicosanoid production in ulcerative colitis using filter paper. Gut. 2000;46:487–492. doi: 10.1136/gut.46.4.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reinecker HC, Steffen M, Witthoeft T, Pflueger I, Schreiber S, MacDermott RP, Raedler A. Enhanced secretion of tumour necrosis factor-α, IL-6, and IL-1 β by isolated lamina propria mononuclear cells from patients with ulcerative colitis and Crohn's disease. Clin. Exp. Immunol. 1993;94:174–181. doi: 10.1111/j.1365-2249.1993.tb05997.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cominelli F, Pizarro TT. Interleukin-1 and interleukin-1 receptor antagonist in inflammatory bowel disease. Aliment. Pharmacol. Ther. 1996;10(Suppl. 2):49–54. doi: 10.1046/j.1365-2036.1996.22164020.x. [DOI] [PubMed] [Google Scholar]
- 28.Mittal RD, Bid HK, Ghoshal UC. IL-1 receptor antagonist (IL-1Ra) gene polymorphism in patients with inflammatory bowel disease in India. Scand. J. Gastroenterol. 2005;40:827–831. doi: 10.1080/00365520510015629. [DOI] [PubMed] [Google Scholar]
- 29.Heresbach D, Alizadeh M, Dabadie A, Le Berre N, Colombel JF, Yaouanq J, Bretagne JF, Semana G. Significance of interleukin-1β and interleukin-1 receptor antagonist genetic polymorphism in inflammatory bowel diseases. Am. J. Gastroenterol. 1997;92:1164–1169. [PubMed] [Google Scholar]
- 30.Nemetz A, Nosti-Escanilla MP, Molnar T, Kope A, Kovacs A, Feher J, Tulassay Z, Nagy F, Garcia-Gonzalez MA, Pena AS. IL1B gene polymorphisms influence the course and severity of inflammatory bowel disease. Immunogenetics. 1999;49:527–531. doi: 10.1007/s002510050530. [DOI] [PubMed] [Google Scholar]
- 31.Barksby HE, Lea SR, Preshaw PM, Taylor JJ. The expanding family of interleukin-1 cytokines and their role in destructive inflammatory disorders. Clin. Exp. Immunol. 2007;149:217–225. doi: 10.1111/j.1365-2249.2007.03441.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dinarello CA. Anti-cytokine therapeutics and infections. Vaccine. 2003;21(Suppl. 2):S24–S34. doi: 10.1016/s0264-410x(03)00196-8. [DOI] [PubMed] [Google Scholar]
- 33.Brun P, Castagliuolo I, Leo VD, Buda A, Pinzani M, Palu G, Martines D. Increased intestinal permeability in obese mice: new evidence in the pathogenesis of nonalcoholic steatohepatitis. Am. J. Physiol. 2007;292:G518–G525. doi: 10.1152/ajpgi.00024.2006. [DOI] [PubMed] [Google Scholar]
- 34.Sun Z, Olanders K, Lasson A, Dib M, Annborn M, Andersson K, Wang X, Andersson R. Effective treatment of gut barrier dysfunction using an antioxidant, a PAF inhibitor, and monoclonal antibodies against the adhesion molecule PECAM-1. J. Surg. Res. 2002;105:220–233. doi: 10.1006/jsre.2001.6342. [DOI] [PubMed] [Google Scholar]
- 35.Shen L, Black ED, Witkowski ED, Lencer WI, Guerriero V, Schneeberger EE, Turner JR. Myosin light chain phosphorylation regulates barrier function by remodeling tight junction structure. J. Cell Sci. 2006;119:2095–2106. doi: 10.1242/jcs.02915. [DOI] [PubMed] [Google Scholar]
- 36.Ye D, Ma I, Ma TY. Molecular mechanism of tumor necrosis factor-α modulation of intestinal epithelial tight junction barrier. Am. J. Physiol. 2006;290:G496–G504. doi: 10.1152/ajpgi.00318.2005. [DOI] [PubMed] [Google Scholar]
- 37.Ma TY, Tran D, Hoa N, Nguyen D, Merryfield M, Tarnawski A. Mechanism of extracellular calcium regulation of intestinal epithelial tight junction permeability: role of cytoskeletal involvement. Microsc. Res. Tech. 2000;51:156–168. doi: 10.1002/1097-0029(20001015)51:2<156::AID-JEMT7>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 38.Clayburgh DR, Rosen S, Witkowski ED, Wang F, Blair S, Dudek S, Garcia JG, Alverdy JC, Turner JR. A differentiation-dependent splice variant of myosin light chain kinase, MLCK1, regulates epithelial tight junction permeability. J. Biol. Chem. 2004;279:55506–55513. doi: 10.1074/jbc.M408822200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ma TY, Hollander D, Tran LT, Nguyen D, Hoa N, Bhalla D. Cytoskeletal regulation of Caco-2 intestinal monolayer paracellular permeability. J. Cell. Physiol. 1995;164:533–545. doi: 10.1002/jcp.1041640311. [DOI] [PubMed] [Google Scholar]
- 40.Ma TY, Hoa NT, Tran DD, Bui V, Pedram A, Mills S, Merryfield M. Cytochalasin B modulation of Caco-2 tight junction barrier: role of myosin light chain kinase. Am. J. Physiol. 2000;279:G875–G885. doi: 10.1152/ajpgi.2000.279.5.G875. [DOI] [PubMed] [Google Scholar]
- 41.Ma TY, Nguyen D, Bui V, Nguyen H, Hoa N. Ethanol modulation of intestinal epithelial tight junction barrier. Am. J. Physiol. 1999;276:G965–G974. doi: 10.1152/ajpgi.1999.276.4.G965. [DOI] [PubMed] [Google Scholar]
- 42.Turner JR, Black ED, Ward J, Tse CM, Uchwat FA, Alli HA, Donowitz M, Madara JL, Angle JM. Transepithelial resistance can be regulated by the intestinal brush-border Na+/H+ exchanger NHE3. Am. J. Physiol. 2000;279:C1918–C1924. doi: 10.1152/ajpcell.2000.279.6.C1918. [DOI] [PubMed] [Google Scholar]
- 43.Hecht G, Pothoulakis C, LaMont JT, Madara JL. Clostridium difficile toxin A perturbs cytoskeletal structure and tight junction permeability of cultured human intestinal epithelial monolayers. J. Clin. Invest. 1988;82:1516–1524. doi: 10.1172/JCI113760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Spitz J, Yuhan R, Koutsouris A, Blatt C, Alverdy J, Hecht G. Enteropathogenic Escherichia coli adherence to intestinal epithelial monolayers diminishes barrier function. Am. J. Physiol. 1995;268:G374–G379. doi: 10.1152/ajpgi.1995.268.2.G374. [DOI] [PubMed] [Google Scholar]
- 45.Hecht G, Pestic L, Nikcevic G, Koutsouris A, Tripuraneni J, Lorimer DD, Nowak G, Guerriero V, Jr., Elson EL, Lanerolle PD. Expression of the catalytic domain of myosin light chain kinase increases paracellular permeability. Am. J. Physiol. 1996;271:C1678–C1684. doi: 10.1152/ajpcell.1996.271.5.C1678. [DOI] [PubMed] [Google Scholar]
- 46.Ma TY, Boivin MA, Ye D, Pedram A, Said HM. Mechanism of TNF-α modulation of Caco-2 intestinal epithelial tight junction barrier: role of myosin light-chain kinase protein expression. Am. J. Physiol. 2005;288:G422–G430. doi: 10.1152/ajpgi.00412.2004. [DOI] [PubMed] [Google Scholar]
- 47.Blair SA, Kane SV, Clayburgh DR, Turner JR. Epithelial myosin light chain kinase expression and activity are upregulated in inflammatory bowel disease. Lab. Invest. 2006;86:191–201. doi: 10.1038/labinvest.3700373. [DOI] [PubMed] [Google Scholar]
- 48.Dokladny K, Moseley PL, Ma TY. Physiologically relevant increase in temperature causes an increase in intestinal epithelial tight junction permeability. Am. J. Physiol. 2006;290:G204–G212. doi: 10.1152/ajpgi.00401.2005. [DOI] [PubMed] [Google Scholar]
- 49.Ma TY, Iwamoto GK, Hoa NT, Akotia V, Pedram A, Boivin MA, Said HM. TNF-α-induced increase in intestinal epithelial tight junction permeability requires NF-κB activation. Am. J. Physiol. 286:G367–G376. doi: 10.1152/ajpgi.00173.2003. [DOI] [PubMed] [Google Scholar]
- 50.O'Neill LA, Dinarello CA. The IL-1 receptor/toll-like receptor superfamily: crucial receptors for inflammation and host defense. Immunol. Today. 2000;21:206–209. doi: 10.1016/s0167-5699(00)01611-x. [DOI] [PubMed] [Google Scholar]
- 51.Nakamura M, Saito H, Kasanuki J, Tamura Y, Yoshida S. Cytokine production in patients with inflammatory bowel disease. Gut. 1992;33:933–937. doi: 10.1136/gut.33.7.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ligumsky M, Simon PL, Karmeli F, Rachmilewitz D. Role of interleukin 1 in inflammatory bowel disease: enhanced production during active disease. Gut. 1990;31:686–689. doi: 10.1136/gut.31.6.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hardin J, Kroeker K, Chung B, Gall DG. Effect of proinflammatory interleukins on jejunal nutrient transport. Gut. 2000;47:184–191. doi: 10.1136/gut.47.2.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Matysiak-Budnik T, Thomas-Collignon A, Megraud F, Heyman M. Alterations of epithelial permeability by Helicobacter and IL-1β in vitro: protective effect of rebamipide. Dig. Dis. Sci. 2001;46:1558–1566. doi: 10.1023/a:1010664626431. [DOI] [PubMed] [Google Scholar]
- 55.McKenzie JA, Ridley AJ. Roles of ρ/ROCK and MLCK in TNF-α-induced changes in endothelial morphology and permeability. J. Cell. Physiol. 2007;213:221–228. doi: 10.1002/jcp.21114. [DOI] [PubMed] [Google Scholar]
- 56.Turner JR. “Putting the squeeze” on the tight junction: understanding cytoskeletal regulation. Semin. Cell. Dev. Biol. 2000;11:301–308. doi: 10.1006/scdb.2000.0180. [DOI] [PubMed] [Google Scholar]
- 57.Madara JL, Barenberg D, Carlson S. Effects of cytochalasin D on occluding junctions of intestinal absorptive cells: further evidence that the cytoskeleton may influence paracellular permeability and junctional charge selectivity. J. Cell Biol. 1986;102:2125–2136. doi: 10.1083/jcb.102.6.2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ferrier L, Mazelin L, Cenac N, Desreumaux P, Janin A, Emilie D, Colombel JF, Garcia-Villar R, Fioramonti J, Bueno L. Stress-induced disruption of colonic epithelial barrier: role of interferon-γ and myosin light chain kinase in mice. Gastroenterology. 2003;125:795–804. doi: 10.1016/s0016-5085(03)01057-6. [DOI] [PubMed] [Google Scholar]
- 59.Cenac N, Chin AC, Garcia-Villar R, Salvador-Cartier C, Ferrier L, Vergnolle N, Buret AG, Fioramonti J, Bueno L. PAR2 activation alters colonic paracellular permeability in mice via IFN-γ-dependent and -independent pathways. J. Physiol. 2004;558:913–925. doi: 10.1113/jphysiol.2004.061721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Madara JL, Stafford J, Barenberg D, Carlson S. Functional coupling of tight junctions and microfilaments in T84 monolayers. Am. J. Physiol. 1988;254:G416–G423. doi: 10.1152/ajpgi.1988.254.3.G416. [DOI] [PubMed] [Google Scholar]
- 61.Prahalad P, Calvo I, Waechter H, Matthews JB, Zuk A, Matlin KS. Regulation of MDCK cell-substratum adhesion by RhoA and myosin light chain kinase after ATP depletion. Am. J. Physiol. 2004;286:C693–C707. doi: 10.1152/ajpcell.00124.2003. [DOI] [PubMed] [Google Scholar]
- 62.Murthy KS. Signaling for contraction and relaxation in smooth muscle of the gut. Annu. Rev. Physiol. 2006;68:345–374. doi: 10.1146/annurev.physiol.68.040504.094707. [DOI] [PubMed] [Google Scholar]
- 63.Sward K, Mita M, Wilson DP, Deng JT, Susnjar M, Walsh MP. The role of RhoA and ρ-associated kinase in vascular smooth muscle contraction. Curr. Hypertens. Rep. 2003;5:66–72. doi: 10.1007/s11906-003-0013-1. [DOI] [PubMed] [Google Scholar]
- 64.Walsh MP, Susnjar M, Deng J, Sutherland C, Kiss E, Wilson DP. Phosphorylation of the protein phosphatase type 1 inhibitor protein CPI-17 by protein kinase C. Methods Mol. Biol. 2007;365:209–223. doi: 10.1385/1-59745-267-X:209. [DOI] [PubMed] [Google Scholar]
- 65.Wu X, Somlyo AV, Somlyo AP. Cyclic GMP-dependent stimulation reverses G-protein-coupled inhibition of smooth muscle myosin light chain phosphate. Biochem. Biophys. Res. Commun. 1996;220:658–663. doi: 10.1006/bbrc.1996.0460. [DOI] [PubMed] [Google Scholar]
- 66.Khromov AS, Wang H, Choudhury N, McDuffie M, Herring BP, Nakamoto R, Owens GK, Somlyo AP, Somlyo AV. Smooth muscle of telokin-deficient mice exhibits increased sensitivity to Ca2+ and decreased cGMP-induced relaxation. Proc. Natl. Acad. Sci. USA. 2006;103:2440–2445. doi: 10.1073/pnas.0508566103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sobieszek A, Andruchov OY, Grabarek Z, Kulikova N, Liebetrau C, Matusovsky OS. Modulation of myosin filament activation by telokin in smooth muscle liberation of myosin kinase and phosphatase from supramolecular complexes. Biophys. Chem. 2005;113:25–40. doi: 10.1016/j.bpc.2004.07.038. [DOI] [PubMed] [Google Scholar]
- 68.Norwood N, Moore TM, Dean DA, Bhattacharjee R, Li M, Stevens T. Store-operated calcium entry and increased endothelial cell permeability. Am. J. Physiol. 2000;279:L815–L824. doi: 10.1152/ajplung.2000.279.5.L815. [DOI] [PubMed] [Google Scholar]
- 69.Yuan SY, Wu MH, Ustinova EE, Guo M, Tinsley JH, De Lanerolle P, Xu W. Myosin light chain phosphorylation in neutrophil-stimulated coronary microvascular leakage. Circ. Res. 2002;90:1214–1221. doi: 10.1161/01.res.0000020402.73609.f1. [DOI] [PubMed] [Google Scholar]
- 70.Graham WV, Wang F, Clayburgh DR, Cheng JX, Yoon B, Wang Y, Lin A, Turner JR. Tumor necrosis factor-induced long myosin light chain kinase transcription is regulated by differentiation-dependent signaling events: characterization of the human long myosin light chain kinase promoter. J. Biol. Chem. 2006;281:26205–26215. doi: 10.1074/jbc.M602164200. [DOI] [PubMed] [Google Scholar]