

Abstract
ATP7A primarily functions to egress copper from cells, thereby supplying this cofactor to secreted copper-accepting enzymes. This ATPase has attracted significant attention since the discovery of its mutation leading to human Menkes disease and the demonstration of its distribution in various tissues. Recently, we reported that ATP7A is expressed in the human vasculature. In the present study, we investigated the cellular expression of ATP7A in atherosclerotic lesions of LDL receptor −/− mice. Subsequently, we examined the role of ATP7A in regulating the oxidation of LDL in a macrophage cell model. We observed that ATP7A is expressed in atherosclerotic murine aorta and colocalizes with macrophages. To investigate the function of ATP7A, we downregulated ATP7A expression in THP-1 derived macrophages using small interfering RNA (siRNA). ATP7A downregulation attenuated cell-mediated oxidation of LDL. Moreover, downregulation of ATP7A resulted in decreased expression and enzymatic activity of cytosolic phospholipase A2 α (cPLA2α), a key intracellular enzyme involved in cell-mediated LDL oxidation. In addition, cPLA2α promoter activity was decreased after downregulation of ATP7A, suggesting that ATP7A transcriptionally regulates cPLA2α expression. Finally, cPLA2α overexpression increased LDL oxidation, which was blocked by coadministration of ATP7A siRNA oligonucleotides. These findings suggest a novel mechanism linking ATP7A to cPLA2α and LDL oxidation, suggesting that this copper transporter could play a previously unrecognized role in the pathogenesis of atherosclerosis.
ATP7A, otherwise known as the Menkes disease gene, is located at Xq12-13, consists of 23 exons, and encodes a copper-transporting P-type ATPase. Its gene product, ATP7A, a single-chain, 178 kDa polypeptide, is localized to the trans-Golgi network in cells under basal conditions [see reviews in (1–3)]. ATP7A egresses copper from cells, and supplying copper to secreted copper-accepting enzymes. This P-type ATPase has attracted significant attention since the discovery of its mutation leading to human Menkes disease and the demonstration of its distribution in various tissues, including placenta, brain, testes, kidney, and lung (4–6). Although affected infants with Menkes disease appear healthy at birth and develop normally for 6–8 weeks, death by 3 years of age is typical due to hypotonia, seizures, and failure to thrive (7).
Recently, we reported that ATP7A is highly expressed in the human vasculature, including aortic smooth muscle cells, aortic endothelial cells, and umbilical vein endothelial cells. Moreover, we identified a novel vascular function of ATP7A to modulate the expression and activity of a copper-accepting enzyme, extracellular superoxide dismutase (8). This finding suggests that ATP7A is closely associated with its copper-accepting enzymes in the vasculature (2) and raises the possibility that the ATPase could regulate other important functions in the blood vessel wall. Moreover, we also recently reported that phorbol-12-myristate-13-acetate (PMA) induces ATP7A expression in THP-1 cells (9). Interestingly, ATP7A−/− mice develop spontaneous aortic aneurysms, yet very little lipid accumulates in their aortic wall (10), suggesting that ATP7A might play a previously unrecognized role in the regulation of atherosclerosis.
Oxidation of LDL is critical to the initiation and progression of atherosclerosis. Macrophages present in arterial lesions are thought to be the principal mediators of LDL oxidation in the arterial wall [see review in (11)]. Macrophage can modulate LDL oxidation through extracellular [ceruloplasmin (12), paraoxonase-1 (13), and myeloperoxidase (14)] and intracellular [12 and 15-lipoxygenase (LOX) (15, 16), NADPH oxidase (17), LDL receptor-related protein 1 (LRP1) (18), and cytosolic phospholipase A2 α (cPLA2α) (19)] mechanisms. Moreover, LDL oxidation by macrophages was recently reported to occur in the intracellular compartment within lysosomes (20).
In the present study, we investigated the cellular expression of ATP7A in murine atherosclerotic lesions. Subsequently, we examined the role of ATP7A in regulating LDL oxidation in a macrophage cell model. Our study also revealed a potential novel link between ATP7A, cPLA2α, and LDL oxidation. Based on these studies, we conclude that ATP7A is expressed in macrophages and may function to facilitate LDL oxidation.
MATERIALS AND METHODS
Animal studies
Eight-week-old LDL receptor (LDLR)−/− mice on a C57BL/6 background (Jackson Laboratories, Bar Harbor, ME) were maintained on a Western-type high-fat/-cholesterol diet for 12 weeks and then euthanized. The heart and aorta were perfused with phosphate-buffered saline. The aortic root and ascending aorta were carefully dissected and placed in phosphate-buffered saline containing 30% sucrose for 18 h, embedded in OCT medium, frozen, and sectioned with a cryostat at 10 µm intervals throughout the aortic sinus and the aortic arch. Following immunostaining with antibodies against ATP7A and MOMA (8, 21), sections were viewed under an Olympus fluorescence microscope with excitation at 455/490 nm, a band pass of 500 nm, and a 515 nm barrier filter. Equal exposure time was used for all the images.
Cell culture
The human monocytic cell line THP-1 was obtained from the American Type Culture Collection (Rockville, MD) and maintained in culture using RPMI 1640 medium (Hyclone, Logan, UT) supplemented with 10% FBS, penicillin (100 units/ml), and streptomycin (100 µg/ml). Cells were maintained in 5% CO2 tissue culture incubators at 37°C until ready for use. Cells were stimulated by 100 nM PMA (Sigma, St. Louis, MO) to induce differentiation into macrophages. The differentiated THP-1 cells, hereafter referred to as THP-1-derived macrophages, have previously been used as a physiologically robust macrophage model as evidenced by the expression of cytokines and adhesion molecules, differentiation markers, and phagocytic function characteristic of macrophages (22, 23). For experiments to assess LDL oxidation, THP-1-derived macrophages were incubated with native LDL (250 µg/ml protein) in Ham's F12 medium without FBS for the indicated time periods.
Human LDL preparation
LDL was prepared from the serum of human volunteers by discontinuous density gradient ultracentrifugation. In some experiments, LDL was purchased from Biomedical Technologies (Stoughton, MA). LDL was stored in the dark at 4°C and used within 1 month of isolation. Prior to each experiment, the LDL was dialyzed with Slide-A-Lyzer cassettes (10,000 molcular weight cut-off; Pierce, Rockford, IL) against 1 l of phosphate-buffered saline (pH 7.4, two changes) for 24 h at 4°C, then against 0.5 l of Ham's F12 medium for 12 h. After dialysis, LDL was sterilized by passing through a low protein binding filter (0.45 µm pore size, Nalgene).
Thiobarbituric acid assay
The thiobarbituric acid (TBA) assay was used to assess the extent of cell-mediated LDL oxidation as described previously (19). TBA reacts with malondialdehyde (MDA) and MDA-like derivatives to form TBA reactive species, which can be quantified by spectrophotometry at 535 nm. Data are presented as MDA equivalents (nM MDA/mg LDL protein).
Small interfering RNA
The ATP7A small interfering (siRNA) oligonucleotides (Ambion, Austin, TX) targeted exon 11 (sense, GCAACUAUUGUAACUCUUGtt; antisense, CAAGAGUUACAAUAGUUGCtt). Scrambled and GAPDH siRNA sequences were used as negative controls. All siRNAs were obtained in lyophilized, annealed form, resuspended in double-distilled water to a stock concentration of 20 µM, and stored at −20°C in 100 µl aliquots. Cells were transfected with siRNA oligonucleotides using oligofectamine transfection reagent (Invitrogen, Carlsbad, CA) or DMRIE-C reagent (Invitrogen) according to the manufacturer's instructions.
Western blotting
Cells were incubated for 30 min at 4°C in 1% Triton X-100 lysis buffer (50 mmol/L HEPES, 50 mmol/L NaCl, 5 mmol/L EDTA) with a tablet of protease inhibitors (Roche, Indianapolis, IN). Samples were then centrifuged at 18,000 g for 10 min, and proteins in the supernatant were separated using SDS-PAGE, transferred to nitrocellulose membranes (Bio-Rad, Hercules, CA), blocked, and incubated overnight at 4°C with ATP7A or cPLA2α primary antibodies (Santa Cruz) (8, 19). After incubation with HRP-conjugated secondary antibodies, proteins were detected by chemiluminescence (Bio-Rad). Equal gel loading was determined by Ponceau S staining of nitrocellulose membrane following transfer and by blotting with α-tubulin antibodies (Sigma).
RNA preparation
TRI Reagent (Sigma) was used to isolate total RNA following the manufacturer's instructions, with minor modifications. For total RNA isolation, after the ethanol precipitation step in the TRI Reagent extraction procedure, an additional cleanup was performed using RNeasy Mini kit (QIAGEN, Valencia, CA) to improve the purity of total RNA. In some cases, the quality of RNA was assessed using standard techniques, including examination of the 260 to 280 nm optical density absorbance ratio and detection of distinct 28S and 18S rRNA bands on ethidium bromide-stained agarose gels.
RT-PCR
cDNA was synthesized using a Retroscript First-Strand Synthesis Kit (Ambion) following the manufacturer's instructions. PCR was performed using the Mx3000PTM PCR system (Stratagene, La Jolla, CA) under the following conditions: denaturation at 94°C for 1 min, annealing at 55°C for 30 s, and extension at 72°C for 1 min. All RT-PCR experiments were performed in triplicate. Equal aliquots from 25 or 30 thermocycles were electrophoresed in 1.5% agarose gel and quantified by densitometry analysis (Kodak Digital 1D Science). The abundance of target mRNA was calculated in relation to the GAPDH mRNA in the same sample. cPLA2α primer sequence: forward: TGGCTCTGTGTGATCAGGAG, reverse: GAGCCAGAAAGACCAGCAAC. GAPDH primer sequence: forward: AACACAGTCCATGCCATCAC, reverse: TCCACCACCCTGTTGCTGTA.
cPLA2 activity assay
cPLA2 activity was determined using an assay kit (Cayman Chemical) with 2-deoxy-2-thioarachidonoylphosphatidylcholine as the substrate, as described previously (24). To exclude secretory PLA2 and calcium-independent PLA2, supernatants of cell homogenates were concentrated by Y30 filters (Millipore), followed by incubation with bromoenol lactone (Cayman Chemical), a calcium-independent PLA2 inhibitor. Samples (10 µl) were finally assayed in a 96-well plate, and the OD values were measured at 414 nm.
Cell transfection and reporter assays
A cPLA2α reporter construct containing 2.4 kb (−2,487 to +40 bp) of a 5′-flanking region of rat cPLA2α gene was ligated into the promoterless vector PA3-Luc (PA3-Luc/cPLA2α) (25, 26). The phRLTK vector (Promega) containing the Renilla luciferase gene was used as an internal control. Transfections of THP-1 cells were carried out using DMRIE-C reagent (Invitrogen) as previously described (27). The DMRIE-C reagent was first mixed with 5 µg of PA3-Luc/cPLA2α vector DNA, 0.5 µg of phRLTK vector, and 5 nM of control or ATP7A siRNA oligonucleotides to form DMRIE-C-DNA complexes. After cells were transfected with DMRIE-C-DNA complexes in 200 µl of serum-free Opti-MEM I (Invitrogen) for 4 h, 2 ml of growth medium containing 15% FBS and 100 nM PMA was added to each well for an additional 48 h incubation. Cells were then harvested, and cell lysates were extracted with passive lysis buffer (Promega), which was used to determine the luciferase activity. The luciferase activity was then normalized to Renilla luciferase activity and expressed as relative luciferase activity.
Transfection of cPLA2α plasmids
A human cPLA2α construct was cloned into the enhanced cyan fluorescent protein (ECFP) vector (Clotech) kindly provided by Dr. Christina C. Leslie, National Jewish Medical and Research Center (28, 29). Overexpression of cPLA2α using this plasmid mimics the function and cellular translocation of the endogenous enzyme (29–31). DMRIE-C reagent was used to transfect ECFP-cPLA2α vector DNA (4 µg) and siRNA oligonucleotides (5 nM) into THP-1 cells. Four hours after transfection, 2 ml of growth medium containing 15% FBS and 100 nM PMA was added into each well for an additional 48 h. Expression of ECFP-cPLA2α was monitored by fluorescence microscopy and further confirmed by Western blot.
Statistical analyses
Data are presented as mean ± SEM. Data were compared between groups of cells by t-testing when one comparison was performed or by ANOVA for multiple comparisons. When significance was indicated by ANOVA, the Tukey-Kramer post hoc test was used to specify between-group differences. Values of P < 0.05 were considered statistically significant.
RESULTS
Colocalization of ATP7A with macrophages in atherosclerotic lesions
In a previous report, we observed robust staining of ATP7A in murine aortic atherosclerotic lesions (8). In the current report, immunofluorescence was performed to determine the cell type(s) in which ATP7A was expressed in vivo. Figure 1 illustrates representative data showing that ATP7A expression is largely colocalized with expression of MOMA, a widely used mouse macrophage subpopulation marker, in intimal lesions of aortas from LDLR−/− mice. The specificity of our antibody was verified by demonstrating lack of ATP7A expression in fibroblasts isolated from a patient with Menkes disease (32), whereas expression was detected in fibroblasts isolated from a healthy control patient (data not shown). Therefore, these data suggest that ATP7A is expressed in macrophages within atherosclerotic lesions.
Fig. 1.
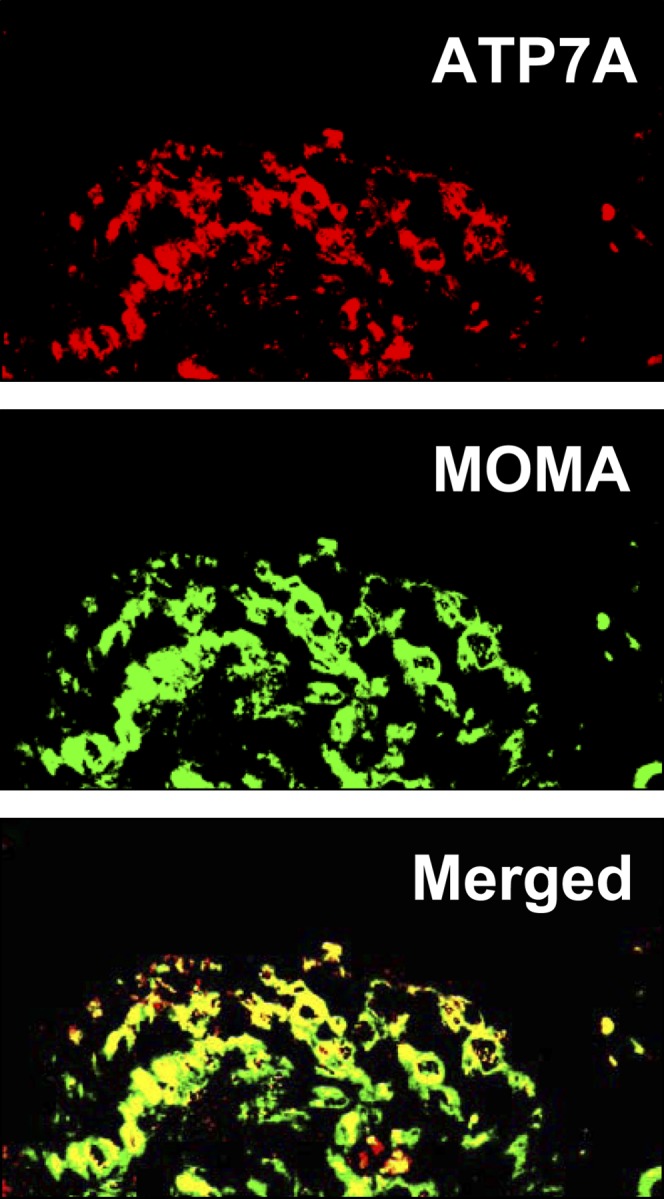
ATP7A is expressed in macrophages of aortic intimal lesions from LDLR−/− mice. Aortic sections from LDLR −/−mice maintained on a Western-type high-fat/-cholesterol diet for 12 weeks were immunostained with antibodies against ATP7A and MOMA as described in Materials and Methods. Overlay of ATP7A (red) and MOMA (green) signals shows colocalization (yellow signal) in aortic intimal lesions. Images are representative of three separate experiments.
Inhibition of LDL oxidation after downregulation of ATP7A in THP-1-derived macrophages
To begin to investigate the function of ATP7A in macrophages, siRNA technique was employed to downregulate the expression of ATP7A in THP-1-derived macrophages. THP-1 cells can be induced by PMA to differentiate into a macrophage-like cell, as defined by an increase in adherence and surface expression of macrophage specific markers. Following treatment with 100 nM PMA for 24 h to induce macrophage differentiation, the THP-1 cells were incubated with siRNA oligonucleotides to downregulate ATP7A expression. Figure 2A shows that after treatment with ATP7A- specific siRNA oligonucleotides for 24 h, ATP7A protein was downregulated by ≈9-fold compared with that of scrambled oligonucleotides and nonoligonucleotide treated cells. Moreover, this level of inhibition of ATP7A expression persisted for up to 4 d postincubation (data not shown).
Fig. 2.

A: Effect of siRNA on ATP7A protein expression in human THP-1-derived macrophages. THP-1-derived macrophages (5 × 105 cells/ml) were incubated with either 5 nM of ATP7A-specific (lanes 5 and 6) or control (lanes 3 and 4) siRNA oligonucleotides for 24 h. Control cells (lanes 1 and 2) were exposed to transfectant without siRNA oligonucleotides. The protein level of ATP7A was determined by Western blot, with α tubulin used as a loading control. Representative blots (upper panel) are from five individual experiments, and quantification is shown in the lower panel. *P < 0.01 versus ATP7A-downregulated macrophages. B: Effect of ATP7A downregulation on cell mediated LDL oxidation. After incubation with ATP7A or control siRNA oligonucleotides for 24 h, THP-1-derived macrophages were incubated with native LDL (250 µg/ml protein) in Ham's F12 medium without FBS for the indicated time periods. Following incubation, lipid oxidation was assessed by the TBA assay as described in the Materials and Methods. Each data point is the mean of triplicate measurements after subtraction of cell-free control. * P < 0.05 versus ATP7A-downregulated macrophages. ** P < 0.01 versus ATP7A-downregulated macrophages.
Next, we investigated the role of ATP7A in macrophage mediated LDL oxidation. Figure 2B demonstrates that the THP-1-derived macrophages oxidized human LDL (250 µg/ml) over a 48 h time course, which was significantly decreased following transfection with ATP7A- specific siRNA oligonucleotides compared with controls after 24, 36, or 48 h of incubation. As a negative control, LDL incubated in medium (RPMI 1640) alone (without cells) exhibited no detectable TBA reactive species (data not shown). Importantly, cell viability was similar between ATP7A-downregulated macrophages and control cells, as assessed by MTT assay (data not shown). These observations suggest that endogenous ATP7A contributes to LDL oxidation in THP-1-derived macrophages. In addition, compared with control oligonucleotides, treatment with ATP7A siRNA oligonucleotides substantially reduced O2•− levels (an average of 40%) in THP-1-derived macrophages (data not shown).
As ATP7A functions to egress copper from cells, and extracellular copper has potential to promote LDL oxidation, we separately examined the effects of conditioned medium versus cells on LDL oxidation. For these experiments, after downregulation of ATP7A for 48 h, the conditioned medium was harvested and incubated with native LDL for an additional 48 h. As shown in Fig. 3, the conditioned medium elicited much less LDL oxidation compared with that observed in the presence of cells. Moreover, a similar level of LDL oxidation was elicited by conditioned medium from ATP7A downregulated versus control cells. In contrast, cell-mediated LDL oxidation was reduced following downregulation of ATP7A. These findings suggest that downregulation of ATP7A does not suppress LDL oxidation simply by preventing copper egress into the incubation medium.
Fig. 3.

Comparison of LDL oxidation mediated by cells versus conditioned medium in THP-1-derived macrophages. After incubation with ATP7A or control siRNA oligonucleotides for 48 h, the conditioned media (left two bars) from THP-1-derived macrophages were harvested and then incubated with native LDL (250 µg/ml protein) in Ham's F12 medium without FBS for an additional 48 h. Afterwards, lipid oxidation was assessed by the TBA assay, while cell mediated LDL oxidation (right two bars) was performed as described in Fig. 2B. Each data point is the mean of triplicate measurements after subtraction of cell-free control. *P < 0.01 versus ATP7A-downregulated macrophages. ns, Not significant.
Downregulation of ATP7A leads to reduced cPLA2α expression and cPLA2 activity
Because our results suggested that downregulation of ATP7A in THP-1 macrophages inhibits cell-mediated LDL oxidation and because ablation of cPLA2α inhibits macrophage-mediated LDL oxidation (19), we examined whether ATP7A regulates cPLA2α expression and activity in macrophages. Figure 4 shows that expression (mRNA and protein) and activity of cPLA2α were decreased following downregulation of ATP7A by siRNA transfection. To examine the mechanisms leading to reduced cPLA2α expression, cells were transiently cotransfected with a plasmid encoding 2.4 kb of cPLA2α promoter along with control or ATP7A siRNA oligonucleotides. We coupled a relatively large region of the cPLA2α promoter upstream of the reporter gene to ensure a high likelihood that the DNA introduced contained relevant regulatory elements for transcriptional control. Downregulation of ATP7A decreased cPLA2α promoter activity by 3- to 4-fold (Fig. 4D), which suggests that endogenous ATP7A transcriptionally regulates expression of cPLA2α.
Fig. 4.

Effect of downregulation of ATP7A on mRNA levels (A), protein levels (B), enzymatic activity (C), and promoter activity (D) of cPLA2α in human THP-1-derived macrophages. A: Following treatment with ATP7A-specific or control siRNA oligonucleotides for 24 h, total RNA was isolated, and human cPLA2α and GAPDH expression was quantified by RT-PCR. The abundance of target mRNA was calculated in relation to the GAPDH mRNA in the same sample. The data are presented as fold change in mRNA expression compared with control cells (treated with control siRNA oligonucleotides). Each data point is the mean of triplicate measurements. *P < 0.05 versus ATP7A-downregulated macrophages. B and C: Following treatment with ATP7A-specific or control siRNA oligonucleotides for 48 h, cell lysates were prepared and the protein expression and activity of cPLA2α was assessed as described in Fig. 2. For Western blot, α-tubulin served as a loading control. Representative blots are from three separate experiments. *P < 0.05 versus ATP7A-downregulated macrophages. D: Cells were cotransfected with the cPLA2α-luciferase reporter plasmid (PA3-Luc/cPLA2α) along with control or ATP7A siRNA oligonucleotides. Forty-eight hours later, lysates were prepared, and luciferase activity (normalized to phRLTK activity) was determined as described in Materials and Methods. Results represent the mean of three independent experiments. *P < 0.01 versus ATP7A-downregulated cells.
In addition to cPLA2α, we also examined the expression of other genes reported to be involved in macrophage-mediated intracellular LDL oxidation, including 12 and 15 LOX and LRP1. In contrast to the expression of cPLA2α (Fig. 4B), mRNA levels of 12 LOX, 15 LOX, and LRP1 were not affected by downregulation of ATP7A (data not shown). These results suggest that endogenous ATP7A does not indiscriminately regulate expression of enzymes implicated in macrophage-mediated oxidation of LDL.
cPLA2α overexpression induces LDL oxidation, which is blocked by ATP7A downregulation
To further investigate whether cPLA2α has a causative role in ATP7A-mediated LDL oxidation, cPLA2α was overexpressed in ATP7A-downregulated THP-1 macrophages. Consistent with a previous finding (19) that chemical inhibitors of, and antisense oligonucleotides directed against, cPLA2 led to decreased macrophage-mediated LDL oxidation, we found that cPLA2α overexpression increased LDL oxidation in THP-1 cells (Fig. 5). Downregulation of ATP7A abolished the increased LDL oxidation induced by overexpression of cPLA2α. Taken together, these results suggest a role for cPLA2α in ATP7A-mediated LDL oxidation in THP-1 cells.
Fig. 5.

Effects of ATP7A downregulation on LDL oxidation in cells overexpressing cPLA2α. Cells were cotransfected with the ECGF-cPLA2α vector or control vector along with siRNA oligonucleotides targeted to ATP7A or control. Forty-eight hours later, cells were incubated with native LDL (250 µg/ml protein) in Ham's F12 medium without FBS for additional 48 h. Afterwards, lipid oxidation was assessed by the TBA assay. Each data point is the mean of triplicate measurements after subtraction of cell-free control. * P < 0.05 versus ATP7A-downregulated or control macrophages.
DISCUSSION
We previously reported that ATP7A is highly expressed in the vasculature, including vascular smooth muscle cells and endothelial cells, suggesting a previously unrecognized role in cardiovascular homeostasis (8, 33). Here, we report that ATP7A is robustly expressed in intimal macrophages of murine atherosclerotic aorta. Moreover, in a macrophage cell model, we demonstrate that the downregulation of ATP7A results in inhibition of LDL oxidation, at least in part through a cPLA2α-dependent pathway.
ATP7A primarily functions to egress copper from cells, thereby supplying this cofactor to secreted copper- containing proteins, including extracellular superoxide dismutase (8), peptidylglycine α-amidating monooxygenase (34), and tyrosinase (35), through a posttranslational mechanism. These actions are facilitated by the functional domains of ATP7A, which include the NH2-terminal metal binding domain, the transmembrane domain, the ATP-binding domain, and the COOH-terminal di-leucine motifs. In addition, ATP7A may contain a nuclear targeting sequence (36), suggesting that it is capable of regulating gene transcription. In this regard, ATP7A-deficient cells exhibited reduced mRNA expression of lysyl oxidase, elastin, and procollagen type I (37). Our finding that cPLA2α expression, and its promoter activity, are reduced following downregulation of ATP7A in macrophages further suggests a direct role for ATP7A in regulating gene transcription.
cPLA2α, also known as group IVA PLA2, is the major intracellular form of PLA2. This phospholipase is an 85 kDa protein that harbors a Ca2+-binding C2 domain, two catalytic domains, a putative pleckstrin homology domain, and a number of phosphorylation sites (38). cPLA2α preferentially hydrolyzes membrane phospholipids at the sn-2 position to release arachidonic acid, which serves as precursor for a wide spectrum of eicosanoids, including prostaglandins, thromboxanes, leukotrienes, hydroxy- and epoxy-fatty acids, lipoxins, and isoprostanes. cPLA2α is activated by increased intracellular Ca2+ concentration and by phosphorylation on multiple serine residues [see review by Leslie (39)]. In addition to posttranslational control, the expression of cPLA2α is regulated transcriptionally by a number of agents, including cytokines such as interleukin-1β (40), tumor necrosis factor-α, and interferon-γ, as well as thrombin (41) and platelet-derived growth factor-BB (42). The promoter for cPLA2α has been isolated from both humans (43) and rats (44). A number of putative binding sites for possible regulatory elements have been identified within the 5′-flanking region of cPLA2α, including AP-1 sites (43), nuclear factor κB sites (40), and glucocorticoid regulatory elements (43). Our finding that the mRNA level of cPLA2α is decreased after downregulation of ATP7A is further evidence of the importance of transcriptional regulation of cPLA2α. Because both cPLA2α and ATP7A are widely expressed in cells throughout the body, this finding could be of significance to normal cellular physiology and to the pathology of Menkes disease.
The mechanisms whereby endogenous ATP7A contributes to LDL oxidation by THP-1 macrophages remain to be definitively established. We recently reported that endogenous ATP7A functions to egress copper in PMA-stimulated THP-1 cells (9). As extracellular copper and other transition metals can oxidize LDL, it is conceivable that downregulation of ATP7A could modulate LDL oxidation simply by inhibiting copper efflux into the extracellular medium. However, if such were the case, we would expect to observe reduced LDL oxidation when incubated in conditioned medium from ATP7A-downregulated cells compared with control cells. As shown in Fig. 3, no such differences were detected. Rather, our data suggest that downregulation of ATP7A reduces oxidation of LDL by inhibiting intracellular oxidant-dependent mechanisms. To address this possibility, we examined the effects of ATP7A downregulation on the expression of enzymes implicated in macrophage-mediated oxidation of LDL. We observed that downregulation of ATP7A led to reduced expression and activity of cPLA2α, which is thought to be involved in LDL oxidation by supplying fatty acid substrate for LOX-mediated metabolism and by inducing membrane translocation and activation of NADPH oxidase in monocytes (45). Interestingly, treatment of a murine macrophage-like cell line with PMA resulted in increased lysosomal oxidant generation, presumably due to assembly and activation of NADPH oxidase in the lysosomal membrane (46). Moreover, human monocyte-derived macrophages were recently demonstrated to oxidize LDL intracellularly through a lysosomal-dependent mechanism (18). Thus, the cellular source of oxidants, the location of LDL oxidation, and the overall importance of the ATP7A-cPLA2α pathway in regulating LDL oxidation in macrophages remain to be determined.
In summary, we report that ATP7A is expressed in macrophages present within murine atherosclerotic lesions and that downregulation of ATP7A attenuates oxidation of LDL by THP-1-derived macrophages. The mechanism is likely related to reduced expression and activity of cPLA2, suggesting a novel link between this phospholipase and ATP7A. These findings suggest that ATP7A could play a previously unrecognized role in the pathogenesis of atherosclerosis.
Acknowledgments
We thank Dr. Christina C. Leslie (National Jewish Medical and Research Center) for providing the ECFP- cPLA2α plasmid, Dr. Tohru Fukai (University of Illinois at Chicago) for his helpful discussion, and Ahmer Kodvawala for his technical assistance.
Footnotes
Abbreviations:
- cPLA2α
- cytosolic phospholipase A2 α
- DHE
- dihydroethidium
- ECFP
- enhanced cyan fluorescent protein
- LDLR
- LDL receptor
- LOX
- lipoxygenase
- LRP1
- LDL receptor-related protein 1
- MDA
- malondialdehyde
- O2•−
- superoxide anion
- PMA
- phorbol-12-myristate-13-acetate
- siRNA
- small interfering RNA
- TBA
- thiobarbituric acid
This work was supported by HL-076684 and HL-62984 from the National Institutes of Health, a National Scientist Development Grant (0835268N) from the American Heart Association, and by a URC Faculty Development Award from the University of Cincinnati. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health or other granting agencies.
REFERENCES
- 1.Schaefer M., Gitlin J. D. 1999. Genetic disorders of membrane transport. IV. Wilson's disease and Menkes disease. Am. J. Physiol. 276: G311–G314. [DOI] [PubMed] [Google Scholar]
- 2.Lutsenko S., et al. 2007. Function and regulation of human copper-transporting ATPases. Physiol. Rev. 87: 1011–1046. [DOI] [PubMed] [Google Scholar]
- 3.La Fontaine S., Mercer J. F. 2007. Trafficking of the copper-ATPases, ATP7A and ATP7B: role in copper homeostasis. Arch. Biochem. Biophys. 463: 149–167. [DOI] [PubMed] [Google Scholar]
- 4.Hardman B., et al. 2004. Expression and localization of menkes and Wilson copper transporting ATPases in human placenta. Placenta. 25: 512–517. [DOI] [PubMed] [Google Scholar]
- 5.Paynter J. A., et al. 1994. Expression of the Menkes gene homologue in mouse tissues lack of effect of copper on the mRNA levels. FEBS Lett. 351: 186–190. [DOI] [PubMed] [Google Scholar]
- 6.Niciu M. J., et al. 2006. Developmental changes in the expression of ATP7A during a critical period in postnatal neurodevelopment. Neuroscience. 139: 947–964. [DOI] [PubMed] [Google Scholar]
- 7.Kaler S. G., et al. 2008. Neonatal diagnosis and treatment of Menkes disease. N. Engl. J. Med. 358: 605–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Qin Z., et al. 2006. Essential role for the Menkes ATPase in activation of extracellular superoxide dismutase: implication for vascular oxidative stress. FASEB J. 20: 334–336. [DOI] [PubMed] [Google Scholar]
- 9.Afton S., et al. 2009. Copper egress is induced by PMA in human THP-1 cells: a potential regulator of VEGFR1. Biometals. 22: 531–539. [DOI] [PubMed] [Google Scholar]
- 10.Andrews E. J., White W. J., Bullock L. P. 1975. Spontaneous aortic aneurysms in blotchy mice. Am. J. Pathol. 78: 199–210. [PMC free article] [PubMed] [Google Scholar]
- 11.Chisolm G. M., III, et al. 1999. The oxidation of lipoproteins by monocytes-macrophages. Biochemical and biological mechanisms. J. Biol. Chem. 274: 25959–25962. [DOI] [PubMed] [Google Scholar]
- 12.Ehrenwald E., Fox P. L. 1996. Role of endogenous ceruloplasmin in low density lipoprotein oxidation by human U937 monocytic cells. J. Clin. Invest. 97: 884–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rozenberg O., et al. 2003. Paraoxonase (PON1) deficiency is associated with increased macrophage oxidative stress: studies in PON1-knockout mice. Free Radic. Biol. Med. 34: 774–784. [DOI] [PubMed] [Google Scholar]
- 14.Carr A. C., McCall M. R., Frei B. 2000. Oxidation of LDL by myeloperoxidase and reactive nitrogen species: reaction pathways and antioxidant protection. Arterioscler. Thromb. Vasc. Biol. 20: 1716–1723. [DOI] [PubMed] [Google Scholar]
- 15.Rydberg E. K., et al. 2004. Hypoxia increases LDL oxidation and expression of 15-lipoxygenase-2 in human macrophages. Arterioscler. Thromb. Vasc. Biol. 24: 2040–2045. [DOI] [PubMed] [Google Scholar]
- 16.Sakashita T., et al. 1999. Essential involvement of 12-lipoxygenase in regiospecific andstereospecific oxidation of low density lipoprotein by macrophages. Eur. J. Biochem. 265: 825–831. [DOI] [PubMed] [Google Scholar]
- 17.Cathcart M. K. 2004. Regulation of superoxide anion production by NADPH oxidase in monocytes/macrophages: contributions to atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 24: 23–28. [DOI] [PubMed] [Google Scholar]
- 18.Xu W., et al. 2001. Low density lipoprotein receptor-related protein is required for macrophage-mediated oxidation of low density lipoprotein by 12/15-lipoxygenase. J. Biol. Chem. 276: 36454–36459. [DOI] [PubMed] [Google Scholar]
- 19.Li Q., Cathcart M. K. 1997. Selective inhibition of cytosolic phospholipase A2 in activated human monocytes. Regulation of superoxide anion production and low density lipoprotein oxidation. J. Biol. Chem. 272: 2404–2411. [DOI] [PubMed] [Google Scholar]
- 20.Wen Y., Leake D. S. 2007. Low density lipoprotein undergoes oxidation within lysosomes in cells. Circ. Res. 100: 1337–1343. [DOI] [PubMed] [Google Scholar]
- 21.Fukai T., et al. 1998. Vascular expression of extracellular superoxide dismutase in atherosclerosis. J. Clin. Invest. 101: 2101–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barker K. S., Liu T., Rogers P. D. 2005. Coculture of THP-1 human mononuclear cells with Candida albicans results in pronounced changes in host gene expression. J. Infect. Dis. 192: 901–912. [DOI] [PubMed] [Google Scholar]
- 23.Lagoumintzis G., et al. 2003. Pseudomonas aeruginosa slime glycolipoprotein is a potent stimulant of tumor necrosis factor alpha gene expression and activation of transcription activators nuclear factor kappa B and activator protein 1 in human monocytes. Infect. Immun. 71: 4614–4622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu L., et al. 2008. Activation of cytosolic phospholipase A2alpha through nitric oxide-induced S-nitrosylation. Involvement of inducible nitric-oxide synthase and cyclooxygenase-2. J. Biol. Chem. 283: 3077–3087. [DOI] [PubMed] [Google Scholar]
- 25.Tay A., et al. 1994. Isolation of promoter for cytosolic phospholipase A2 (cPLA2). Biochim. Biophys. Acta. 1217: 345–347. [DOI] [PubMed] [Google Scholar]
- 26.Van Putten V., et al. 2001. Induction of cytosolic phospholipase A2 by oncogenic Ras is mediated through the JNK and ERK pathways in rat epithelial cells. J. Biol. Chem. 276: 1226–1232. [DOI] [PubMed] [Google Scholar]
- 27.Wang J. H., et al. 2002. Induction of bacterial lipoprotein tolerance is associated with suppression of toll-like receptor 2 expression. J. Biol. Chem. 277: 36068–36075. [DOI] [PubMed] [Google Scholar]
- 28.Stewart A., et al. 2002. Enzymatic properties of human cytosolic phospholipase A(2)gamma. J. Biol. Chem. 277: 29526–29536. [DOI] [PubMed] [Google Scholar]
- 29.Noor S., et al. 2008. Activation of cytosolic phospholipase A2alpha in resident peritoneal macrophages by Listeria monocytogenes involves listeriolysin O and TLR2. J. Biol. Chem. 283: 4744–4755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pavicevic Z., Leslie C. C., Malik K. U. 2008. cPLA2 phosphorylation at serine-515 and serine-505 is required for arachidonic acid release in vascular smooth muscle cells. J. Lipid Res. 49: 724–737. [DOI] [PubMed] [Google Scholar]
- 31.Girotti M., et al. 2004. Cytosolic phospholipase A2 translocates to forming phagosomes during phagocytosis of zymosan in macrophages. J. Biol. Chem. 279: 19113–19121. [DOI] [PubMed] [Google Scholar]
- 32.Kim B. E., Smith K., Petris M. J. 2003. A copper treatable Menkes disease mutation associated with defective trafficking of a functional Menkes copper ATPase. J. Med. Genet. 40: 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qin Z., et al. 2008. Role of Menkes ATPase in angiotensin II-induced hypertension: a key modulator for extracellular superoxide dismutase function. Hypertension. 52: 945–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.El Meskini R., et al. 2003. Supplying copper to the cuproenzyme peptidylglycine alpha-amidating monooxygenase. J. Biol. Chem. 278: 12278–12284. [DOI] [PubMed] [Google Scholar]
- 35.Petris M. J., Strausak D., Mercer J. F. 2000. The Menkes copper transporter is required for the activation of tyrosinase. Hum. Mol. Genet. 9: 2845–2851. [DOI] [PubMed] [Google Scholar]
- 36.Reddy M. C., Majumdar S., Harris E. D. 2000. Evidence for a Menkes-like protein with a nuclear targeting sequence. Biochem. J. 350: 855–863. [PMC free article] [PubMed] [Google Scholar]
- 37.Gacheru S., et al. 1993. Expression and accumulation of lysyl oxidase, elastin, and type I procollagen in human Menkes and mottled mouse fibroblasts. Arch. Biochem. Biophys. 301: 325–329. [DOI] [PubMed] [Google Scholar]
- 38.Evans J. H., et al. 2001. Intracellular calcium signals regulating cytosolic phospholipase A2 translocation to internal membranes. J. Biol. Chem. 276: 30150–30160. [DOI] [PubMed] [Google Scholar]
- 39.Leslie C. C. 1997. Properties and regulation of cytosolic phospholipase A2. J. Biol. Chem. 272: 16709–16712. [DOI] [PubMed] [Google Scholar]
- 40.Roshak A. K., et al. 1996. Manipulation of distinct NFkappaB proteins alters interleukin-1beta-induced human rheumatoid synovial fibroblast prostaglandin E2 formation. J. Biol. Chem. 271: 31496–31501. [DOI] [PubMed] [Google Scholar]
- 41.Dronadula N., et al. 2005. STAT-3-dependent cytosolic phospholipase A2 expression is required for thrombin-induced vascular smooth muscle cell motility. J. Biol. Chem. 280: 3112–3120. [DOI] [PubMed] [Google Scholar]
- 42.Neeli I., et al. 2004. An essential role of the Jak-2/STAT-3/ cytosolic phospholipase A(2) axis in platelet-derived growth factor BB-induced vascular smooth muscle cell motility. J. Biol. Chem. 279: 46122–46128. [DOI] [PubMed] [Google Scholar]
- 43.Wu T., et al. 1994. Characterization of the promoter for the human 85 kDa cytosolic phospholipase A2 gene. Nucleic Acids Res. 22: 5093–5098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dolan-O'Keefe M., et al. 2000. Transcriptional regulation and structural organization of the human cytosolic phospholipase A(2) gene. Am. J. Physiol. Lung Cell. Mol. Physiol. 278: L649–L657. [DOI] [PubMed] [Google Scholar]
- 45.Zhao X., et al. 2002. Cytosolic phospholipase A2 (cPLA2) regulation of human monocyte NADPH oxidase activity. cPLA2 affects translocation but not phosphorylation of p67(phox) and p47(phox). J. Biol. Chem. 277: 25385–25392. [DOI] [PubMed] [Google Scholar]
- 46.Chen C. S. 2002. Phorbol ester induces elevated oxidative activity and alkalization in a subset of lysosomes. BMC Cell Biol. 3: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]