

Abstract
We investigated a family from northern Sweden in which three of four siblings have congenital chylomicronemia. LPL activity and mass in pre- and postheparin plasma were low, and LPL release into plasma after heparin injection was delayed. LPL activity and mass in adipose tissue biopsies appeared normal. [35S]Methionine incorporation studies on adipose tissue showed that newly synthesized LPL was normal in size and normally glycosylated. Breast milk from the affected female subjects contained normal to elevated LPL mass and activity levels. The milk had a lower than normal milk lipid content, and the fatty acid composition was compatible with the milk lipids being derived from de novo lipogenesis, rather than from the plasma lipoproteins. Given the delayed release of LPL into the plasma after heparin, we suspected that the chylomicronemia might be caused by mutations in GPIHBP1. Indeed, all three affected siblings were compound heterozygotes for missense mutations involving highly conserved cysteines in the Ly6 domain of GPIHBP1 (C65S and C68G). The mutant GPIHBP1 proteins reached the surface of transfected Chinese hamster ovary cells but were defective in their ability to bind LPL (as judged by both cell-based and cell-free LPL binding assays). Thus, the conserved cysteines in the Ly6 domain are crucial for GPIHBP1 function.
Keywords: compound heterozygote, lipoprotein lipase, milk lipids, mammary gland, glycosylphosphatidylinositol-anchored high density lipoprotein-binding protein 1, endothelial cells
LPL hydrolyzes triglycerides in plasma lipoproteins, making fatty acids available for use in cells (1, 2). LPL also mediates the binding of lipoproteins to cell surfaces and receptors (3–5). Reduced LPL activity from mutations in LPL or its cofactor apolipoprotein CII lead to a striking accumulation of triglyceride-rich lipoproteins in the plasma (type I hyperlipoproteinemia) (6). Clinical symptoms and signs include abdominal pain with or without pancreatitis, eruptive xanthomas, and hepatosplenomegaly. Recently, mutations in the gene for apolipoprotein AV have been uncovered in some patients with unexplained chylomicronemia (7).
LPL is synthesized primarily in parenchymal cells in skeletal muscle, heart, and adipose tissue (1, 2) but then finds its way into the lumen of capillaries, where it participates in the processing of the plasma lipoproteins. LPL is also synthesized in the mammary gland and appears in breast milk after parturition (8, 9). Its function within the milk is unknown, but there is little doubt that LPL-mediated processing of lipoproteins within the capillaries of the mammary gland is important for providing the lipid nutrients to produce milk fat.
Levels of LPL activity in the plasma are normally extremely low (10–12), but they increase more than 100-fold after intravenous injection of heparin. It was long assumed that LPL binds to proteoglycans along the luminal surface of endothelial cells (13–15), but the discovery of glycosylphosphatidylinositol-anchored HDL-binding protein 1 (GPIHBP1) is beginning to reshape that view (16, 17). GPIHBP1 binds LPL avidly and is expressed in capillary endothelial cells of heart, skeletal muscle, and adipose tissue, leading Beigneux et al. (18) to suggest that GPIHBP1 could be an “important platform for the LPL-mediated processing of chylomicrons in capillaries.” Adult GPIHBP1-deficient mice manifest severe chylomicronemia, closely resembling the phenotype of mice lacking LPL (18). GPIHBP1-deficient mice have normal stores of LPL in their tissues, but the entry of LPL into the plasma is delayed after an injection of heparin (19). These observations led to the proposal that GPIHBP1 could be important for the proper localization of LPL within capillaries (18).
Recently, a human subject with lifelong chylomicronemia was shown to be homozygous for a missense mutation (Q115P) in the Ly6 domain of GPIHBP1 (20). A mutant GPIHBP1 protein harboring this amino acid substitution lacked the ability to bind LPL.4
Here, we report a family from northern Sweden in which three of four siblings have chylomicronemia. Early biochemical analyses of these patients revealed low levels of LPL activity and mass in pre- and postheparin plasma. However, both LPL activity and mass in adipose tissue appeared normal. Moreover, in breast milk from the affected female subjects, LPL mass and activity levels were normal. None of the subjects had deleterious mutations in the gene for LPL. This constellation of findings suggested that the defect had nothing to do with the integrity of the LPL gene or the ability to produce enzymatically active LPL but perhaps was due to reduced LPL on the surface of capillaries.
In this study, we show that all three affected siblings were compound heterozygotes for two mutant GPIHBP1 alleles, each harboring a missense mutation involving a highly conserved Cys within the Ly6 domain of GPIHBP1. The mutant GPIHBP1 proteins were able to reach the surface of transfected cells, but they lacked the ability to bind LPL.
MATERIALS AND METHODS
Subjects
The three patients, born in 1969 (II-2), 1974 (II-3), and 1985 (II-4), are the youngest of four siblings. The older son (II-1), born in 1967, is healthy, as are the mother (I-2), born in 1944, and the father (I-1), born in 1940 (Fig. 1). There was no history of consanguinity in the family, although both parents are from the same village in the rural area of northern Sweden. The parents of the father were from this area, but the parents of the mother moved from the central and southern parts of Sweden. There was no family history of hyperlipidemia. The Ethics Committee on Research Involving Human Subjects of the Faculty of Medicine, Umeå University, approved the study, although most of the samples were taken from the family for diagnostic purposes. Informed consent was obtained for all studies on patients and controls.
Fig. 1.

Pedigree in three generations of the family. Filled symbols denote family members with chylomicronemia. Birth years for the second generation are shown.
Proband II-2 was referred to the pediatric clinic at 9 months of age because of hepatosplenomegaly, which was noted incidentally during an evaluation for a respiratory tract infection. There was no history of abdominal pain or other illnesses. She had been breast fed for the first 4 months and thereafter consumed a typical diet for her age without restriction. Blood sampling revealed lipemic serum (plasma triglycerides were 22–57 mmol/l). Weight gain, linear growth, and psychomotor development were normal. During the next few years, she had a few episodes of abdominal pain, occasionally accompanied by reduced appetite and vomiting. At age 27, she gave birth to a healthy child (the first of three) after 39 weeks of gestation. Because she had plasma triglyceride levels above 25 mmol/l, they were monitored once or twice a week during the second and third trimesters. To prevent bouts of pancreatitis, she reduced dietary fat to <15% of energy intake. After parturition, the patient's plasma triglyceride levels increased further; when they reached 42 mmol/l, plasmapheresis was initiated. During subsequent pregnancies, she underwent prophylactic plasmaphereses during the second and third trimesters.
The affected son (II-3) was referred at 10 years of age. He was fatigued and had lipemic serum. Except for an episode of croup at 4 years of age, he had never been ill and never had episodes of abdominal pain. His plasma triglyceride level was 19.5 mmol/l, and his cholesterol level was 5.5 mmol/l. Physical examination revealed mild splenomegaly. After an 18 h fast, the triglyceride level fell to 10.0 mmol/l, and the cholesterol level was 5.7 mmol/l. The patient was advised to follow a low-fat diet. Subsequently, he has been asymptomatic, with plasma triglycerides of 7.2–9.9 mmol/l.
Patient II-4 had gastroenteritis at 16 months of age. Her plasma was severely lipemic (triglycerides, 48.5 mmol/l; cholesterol, 10.5 mmol/l). Her growth and development were normal. She followed the same low-fat diet as her siblings (initially 17% fat, later reduced to 10% fat, with supplements of n-6 and n-3 fatty acids to avoid essential fatty acid deficiency). During the ensuing years, she had a few episodes of pancreatitis confirmed by increased levels of amylase in serum. After treatment with fish oil (15 ml/day), she had no further attacks of abdominal pain. She has continued to take fish oil, primarily as a source of essential fatty acids. Arguments for dietary supplement with medium-chain triglycerides and fish oil for patients with hyperchylomicronemia due to unknown reasons were previously discussed (21). No systematic studies were made on patient II-4 on the effect of fish oil on plasma triglyceride levels or other variables. At age 10, her fasting triglyceride level was typically ∼20 mmol/l. She had severe hypertriglyceridemia during pregnancy with her first child (in year 2009), but she did not require plasmapheresis.
Tissue biopsies
Adipose tissue was obtained by needle biopsy of the upper outer quadrant of the buttock with the Vacutainer technique (22). In patients II-2 and II-3, adipose tissue was also obtained by surgical incision on the upper lateral part of the thigh. Adipose tissue from two unrelated healthy controls was obtained during elective surgery.
Plasma lipid determinations
Fasting blood samples were obtained. After centrifugation, sera were collected and total cholesterol, HDL-cholesterol, and triglycerides were analyzed at the Department of Clinical Chemistry, Umeå University Hospital. Total cholesterol was measured with an automated enzymatic colorimetric test (HiCo Cholesterol; Boehringer Mannheim, Manheim, Germany). HDL-cholesterol was analyzed by the same method after precipitating apolipoprotein-B-containing lipoproteins with sodium phosphotungstate and magnesium chloride. Triglycerides were measured with an automated enzymatic colorimetric test (Triglycerides GPO-PAP; Boehringer Mannheim).
Analysis of the LPL gene
DNA was isolated from white blood cells, and each of the 10 exons of LPL was amplified by PCR and analyzed by single-strand conformation polymorphism and DNA sequencing (23). The promoter and exon 1 from patients II-3 and II-4 were PCR amplified with primer pairs T7LPL-2 (5′-TAATACGACTCACTATAGGGTTTATGTGCATGCCTCTTATCC-3′, positions –994 to –973) and MFLPL-1 (5′-TGTAAAACGACGGC CAGTGCTAGAAGTGGGCAGCTTTC, positions 37–56) and prLPL-8 (5′-GTGTTTGGTGCTTAGACAGG, positions –258 to –239) together with LPL-7 (5′-AGGGGAGT TTGCGCGCAAA-3′, positions 283–301). The PCR products were separated on 1.5% agarose gels in 1× TAE buffer. The fragments were run into a well containing 2.5× TAE, precipitated, and dissolved in 20 μl of water. Both strands of the purified DNA were sequenced by dye terminator cycle sequencing with an ABI/PE 373A sequencer (Applied Biosystems, Carlsbad, CA) according to the manufacturer's instructions. The region covering the Asn291Ser polymorphism in LPL was amplified by PCR with forward primer 5′-GCTCCATTCATCTCTTCATC-3′ and reverse primer 5′-TTTCCTTATTTACAACAGTCT-3′. The PCR product was purified with a Qiagen kit and sequenced with the same primers used for PCR amplification. For DNA sequencing, the concentrations of the PCR product and primers were 5 ng/μl and 2 pmol/μl, respectively.
Lipase activity measurements
LPL was measured with a phospholipid-stabilized emulsion of triglycerides (Intralipid; KABI-Pharmacia Parenterals, Stockholm, Sweden) into which 3H-labeled triolein had been incorporated by sonication (24). HL activity was measured with a gum Arabic-stabilized emulsion of triolein in the presence of 1.0 M NaCl. These assays have been described in detail elsewhere (25). One milliunit of lipase activity corresponds to release of 1 nmol of fatty acid per min at pH 8.5 and 25°C.
For measurements of LPL activity in adipose tissue, subcutaneous fat samples were homogenized in 25 mmol/l ammonia, pH 8.2, containing (per ml) 0.1 mg heparin, 10 μg of leupeptin, 1 μg of pepstatin, 25 kallikrein inhibitor units of aprotinin, 5 mmol EDTA, 1% (w/v) Triton X-100, and 0.1% SDS (24). After centrifugation, 2–5 μl aliquots were used for each assay. LPL activity was normalized to the DNA content of the tissue homogenate (26) in some experiments and to the wet weight of the tissue in one experiment.
To investigate whether the patients’ plasma contained normal amounts of apolipoprotein CII, an incubation system with Intralipid (10%) was used as substrate, and the fatty acids released were determined by manual titration with NaOH. EDTA plasma was added to an incubation system containing 5 mg of triglycerides from Intralipid in a total volume of 1 ml. After 15 min at 25°C, the incubations were started by adding purified bovine LPL. To determine whether the plasma contained an inhibitor, purified bovine LPL was added directly to fresh plasma and incubated at 37°C. A series of 1 ml samples were withdrawn, and free fatty acids were determined by titration. For comparison, Intralipid was added to normal plasma to give a similar concentration of triglycerides (10 mmol/l). The incubations were started within minutes after blood samples were drawn.
Milk samples
Breast milk from nursing mothers was obtained from spontaneously dripping breasts during a breast feed, immediately frozen at −20°C, and analyzed for LPL activity and mass within 2 weeks. Lipids were extracted from 250 µl of human milk according to Folch, Lees, and Sloane Stanley (27) as modified by Dodge and Phillips (28), using chloroform/methanol (2:1, v/v, methanol containing butylhydroxytoluene at a concentration of 50 mg/l). C:17:0 was added as an internal standard before the extraction. Fatty acid methyl esters, prepared with boron trifluoride-methanol (29), were extracted with light petroleum, evaporated under nitrogen, and dissolved in dichloromethane. Fatty acid methyl esters were separated and quantified with a Perkin-Elmer gas chromatograph (AutoSystem GC) attached to a Perkin-Elmer integrator (model 1020). A fused silica capillary column (30 m, 0.32 mm ID, 0.25 μm film thickness) was used (Omegawax 320; Supelco). Helium at a pressure of 12.0 p.s.i. was used as the carrier gas. Individual fatty acids were identified by comparing their retention times with reference standards (Larodan Fine Chemicals, Malmö, Sweden).
Immunoassay for LPL
LPL mass was determined with an ELISA (10, 11, 24) using microtiter plates coated with an affinity-purified chicken antibody against bovine LPL. Bound LPL was detected with a monoclonal antibody against bovine LPL (5D2, provided by Dr. John Brunzell, University of Washington) and horseradish peroxidase-labeled goat anti-mouse IgG. As indicated in the legends to the figures and tables, we used human LPL isolated from postheparin plasma as the standard in earlier experiments (10, 11), while more recently LPL purified from bovine milk was used. With the human standard, values for LPL mass in plasma and milk samples were about 4-fold higher than with the bovine LPL standard.
Heparin-Sepharose chromatography
Small (3 ml) heparin-Sepharose columns (30) were equilibrated at 4°C in 20 mmol/l Tris-Cl, pH 7.4, containing 20% glycerol, 0.1% Triton X-100, and 1 mg/ml BSA. Postheparin plasma was applied to the columns, which were then washed with 30 ml of the equilibration buffer (flow rate, 0.5 ml/min). The columns were eluted with a linear gradient of 0–1.6 M NaCl in the equilibration buffer. Fractions of 5 ml were collected.
Met incorporation and immunoprecipitation
Met incorporation and immunoprecipitation studies were performed on adipose tissue as described (31). The incorporation time in one experiment was 2 h to allow maximal incorporation of radioactivity. In an experiment to assess the glycosylation pattern, the time was reduced to 30 min. Immunoprecipitations were performed with a rabbit antiserum against LPL that had been purified from human milk by chromatography on heparin-Sepharose and on N-desulfated, N-acetylated heparin coupled to Sepharose (9, 32). A corresponding preimmune serum was used as a control. The precipitated proteins were size fractionated by SDS-PAGE. After fluorography, bands were cut out and counted for radioactivity. LPL synthesis was calculated as percentage of the total TCA-precipitable radioactivity. Radioactivity (in the same position on the gel) from control samples was subtracted. For glycosylation studies, samples were treated with endoglycosidase H as described (33).
Analyses of GPIHBP1
Genomic DNA was isolated from whole blood with Blood and Cell Culture DNA Midi kits (Qiagen). The coding regions of GPIHBP1 along with 5′- and 3′-flanking sequences were amplified by PCR with an annealing temperature of 60°C and three primer pairs: 5′-TTGAGGGCATTGACTGTGT-3′ and 5′-TACTACCCTACTCACCCTA-3′ for exon 1; 5′-TAGGGTGAGTAGGGTAGTA-3′ and 5′-CTGCCTGGTGAACTCCTATT-3′ for exon 2; and 5′-AATCCCTTGCCCCCTAAACA-3′ and 5′-ATCGCCCAAGACACTCCAAA-3′ for exons 3 and 4. The amplified fragments were 873, 604, and 812 bp, respectively. The PCR products were purified, and both strands were sequenced. Previously published oligonucleotide primers for GPIHBP1 were used as sequencing primers (34).
Expression of wild-type and mutant GPIHBP1 in Chinese hamster ovary pgsA-745 cells
Expression vectors for S-protein-tagged human GPIHBP1 and the soluble form of mouse GPIHBP1 (G198×) have been described (20, 35). The C65S and C68G mutations were introduced by site-directed mutagenesis (Stratagene). All constructs were expressed in Chinese hamster ovary (CHO) pgsA-745 cells as described (35). The expression of wild-type GPIHBP1 in nonpermeabilized and permeabilized cells was monitored by immunofluorescence microscopy with an FITC-conjugated goat antiserum against the S-protein tag (1:400) (20, 35). Fore LPL-binding studies, GPIHBP1-expressing cells were incubated with a V5-tagged human LPL (20, 35). The amount of LPL bound in the absence and presence of heparin was assessed by Western blotting (18, 20, 35). In some experiments, GPIHBP1 was released from the cell surface by incubating with a phosphatidylinositol-specific phospholipase C (PIPLC). GPIHBP1 in cell extracts and culture medium was assessed by Western blotting.
Binding of LPL to GPIHBP1 captured by monoclonal antibody-coated agarose beads
Agarose beads were coated with a rat monoclonal antibody against mouse GPIHBP1 (antibody 11A12) (35) and incubated with cell culture medium containing soluble wild-type mouse GPIHBP1 with a G198× mutation (designed to prevent the addition of a GPI anchor) or variants of the GPIHBP1 construct with C63S and C66G mutations (note that the residue numbers are slightly shifted in the mouse sequence compared with the human). V5-tagged human LPL was also added. The beads were washed three times, and soluble GPIHBP1 and GPIHBP1-bound LPL were eluted with 0.1 M glycine, pH 2.5. The amounts of GPIHBP1 and LPL in the fractions were assessed by Western blotting (35).
RESULTS
Clinical history
Three siblings (II-2, II-3, and II-4) in one family (Fig. 1) had severe hypertriglyceridemia that was partially responsive to a low-fat diet. None had ever had eruptive xanthomas. The two affected females had hepatosplenomegaly, episodes of mild pancreatitis, and, during pregnancies, very severe hypertriglyceridemia. Table 1 shows plasma lipid levels in 1987, when the investigation of this kindred was initiated. Not surprisingly, the affected subjects had low HDL-cholesterol levels. Both parents were normolipidemic.
TABLE 1.
Plasma lipid concentrations in serum samples from the entire family except the older son in November 1987
| Triglycerides (mmol/l) | Cholesterol (mmol/l) |
||||
|---|---|---|---|---|---|
| Subject | Age (Years) | Gender | Total Plasma | In HDL | |
| I-1 | 47 | M | 0.73 | 5.7 | 1.63 |
| I-2 | 42 | F | 0.85 | 4.5 | 1.58 |
| II-2 | 18 (22) | F | 18.1 (10.4) | 4.9 (5.1) | 0.12 (0.20) |
| II-3 | 13 (17) | M | 9.10 (7.16) | 3.2 (2.6) | 0.31 (0.17) |
| II-4 | 2 (6) | F | 29.3 (12.4) | 7.5 (4.5) | 0.14 (0.16) |
Values in plasma samples from March 1991 are shown in parentheses
Assessing apolipoprotein CII deficiency and LPL inhibition
Initially, we suspected that the patients had low levels of apolipoprotein CII, the protein activator of LPL. In lipolysis assays with Intralipid as substrate, half-maximal lipolysis was obtained with smaller amounts of the patients’ plasma than of plasma from the parents or controls, indicating that the patients had substantial amounts of apolipoprotein CII in their plasma (data not shown).
To study whether the patients had an inhibitor for LPL, we added exogenous bovine LPL to freshly drawn plasma and followed lipolysis in vitro at 37°C. Similar incubations were carried out with plasma from a normal subject and from both parents. Intralipid was added to the normolipidemic plasma samples to yield triglyceride concentrations similar to those of the three patients (10 mmol/l). Lipolysis was linear with time for 40 min (data not shown). The patients’ plasma triglycerides were hydrolyzed at the same rate as the Intralipid in the plasma of healthy controls (∼170 mU/μg LPL), strongly suggesting that there were no LPL inhibitors in their plasma.
Studies of the LPL gene
No rearrangements in the LPL gene were detected in the affected siblings, as judged by Southern blotting (data not shown). Single-strand conformation polymorphism studies revealed a band shift in exon 6 of the proband (II-2). Subsequently, exon 6 was sequenced, and an A-to-G transition was identified in the second base of codon 291, resulting in an Asn (AAT) to Ser (AGT) substitution. Screening for Asn291Ser was carried out in the rest of the family; the mother (I-2) and all four siblings, including the healthy older son (II-1), were heterozygous for this polymorphism. In patient II-2, sequencing of all exons of the LPL gene revealed no other DNA changes. Also, no sequence changes were detected in the 972 bp of the proximal LPL promoter in patients II-3 and II-4, and no dinucleotide (CC) insertions were detected in the 5′ untranslated region of exon 1 (36).
LPL mass in pre- and postheparin plasma
LPL mass in preheparin plasma from the parents was ∼100 ng/ml (Table 2). The mean value in 20 normal male volunteers was 69.8 ± 6.6 ng/ml (11). In the three siblings with chylomicronemia, however, LPL mass levels were 5–14% of those in the parents and were close to the detection limit of the assay. After heparin infusion, LPL mass in the parent's plasma increased about 10-fold. A similar increase was observed in all three affected siblings, but the absolute levels remained extremely low, about 5% of those in the parents (Table 2).
TABLE 2.
LPL mass in plasma samples obtained in September 1993
| LPL Mass (ng/ml) |
||
|---|---|---|
| Subject | Preheparin | Postheparin |
| I-1 | 92 | 1,075 |
| I-2 | 119 | 984 |
| II-2 | 13 | 56 |
| II-3 | 4.5 | 52 |
| II-4 | 7.0 | 42 |
Human LPL was used as standard for the ELISA.
Plasma lipase activities
LPL activity in preheparin plasma was 1.4 and 1.0 mU/ml in the father and mother, respectively (Table 3). Patients II-2 and II-3 had lower activities, about 0.1 mU/ml, while patient II-4, at 3 years of age, had an activity of 1.1 mU/ml plasma. After heparin injection, LPL activity was 261 mU/ml in the father and 130 mU/ml in the mother (Table 3). These values were on the low side of the normal range (11, 25). The increase in LPL activity after heparin was markedly lower in the patients (6.3, 11, and 7.0 mU/ml; Table 3). Similar results were obtained 6 years later (Table 3). The time course for the release seemed delayed for the two older affected siblings compared with the father (data not shown). HL activity was in the normal range for all family members.
TABLE 3.
LPL and HL activities in plasma obtained in November 1987
| LPL (mU/ml) |
HL (mU/ml) |
|||
|---|---|---|---|---|
| Subject | Preheparin | Postheparin | Preheparin | Postheparin |
| I-1 | 1.4 (0.85) | 261 (229) | (1.06) | 267 (271) |
| I-2 | 1.0 (1.25) | 130 (198) | (0.30) | 74 (117) |
| II-2 | 0.1 (0.14) | 6.3 (8.2) | (0.42) | 193 (117) |
| II-3 | 0.1 (0.59) | 11 (13) | (0.50) | 236 (238) |
| II-4 | 1.1 (0.24) | 7.0 (6.8) | (1.04) | 455 (245) |
Values in plasma samples from September 1993 are shown in parentheses.
Time-course studies of LPL and HL release after heparin injection were repeated several years later (in 2007) in proband II-2 and an age-matched healthy female control subject (Fig. 2). LPL activity and mass in the proband's plasma increased slowly and continuously over 60 min, but in control plasma peaked at 10 min. In both, however, HL activity peaked as early as at 4.5 min.
Fig. 2.

Release of LPL activity and mass into plasma after heparin injection in patient II-2 and a healthy, age-matched control subject. A single intravenous injection of heparin (100 U/ kg body weight) was given at time 0. Values are means of triplicate measurements. A: LPL activity measured after immunoinhibition of HL activity in the samples and HL activity measured in an assay containing 1 M NaCl to inactivate LPL. B: LPL mass in patient II-2 and a control subject measured with bovine LPL as standard for the ELISA.
Chromatography of postheparin plasma
Postheparin plasma from the father (I-1) and the affected son (II-3), taken 10 min after heparin injection, was separated by chromatography on heparin-Sepharose (Fig. 3). LPL from the father's plasma showed a normal elution pattern with an initial peak of inactive LPL protein followed by a peak of HL activity (HL gave about 25% activity in the assay for LPL) and finally a peak of LPL activity coeluting with a peak of LPL mass at around 0.9 M NaCl. The mass ratio of the peaks of active and inactive LPL was about three. In the son (II-3), HL activity was similar to that in the father, but LPL mass was only about 5% of the father's level (note the difference in scale on the y axis). On chromatography of the son's plasma, two peaks of LPL mass eluted, at similar salt concentrations as for the father's plasma. The mass ratio of the second and the first peak was around 1. The expected LPL activity in the second peak fraction should have been around 13 mU/ml. Since we were unable to detect this level, it is likely that some inactivation had occurred between the end of the chromatography and assay of the fractions for LPL activity. The high concentration of NaCl from the gradient prevented analyses of larger sample volumes; therefore, the expected activity was near the detection limit of our assay.
Fig. 3.

Separation of lipases in postheparin plasma by chromatography on heparin-Sepharose. Fresh postheparin plasma (9 ml) was applied to columns with 3 ml of heparin-Sepharose. A: Postheparin plasma from the father (I-1). B: Postheparin plasma from the affected son (II-3). HL displayed activity in this assay (about 25% of the activity in the assay optimized for measurements of HL with 1 M NaCl to inactivate LPL). Values for activity are means of duplicate determinations. In this experiment, human LPL was used as standard for the ELISA.
LPL activities in adipose tissue biopsies
The levels of LPL activity in adipose tissue were similar in two patients and the controls (Table 4). LPL activity (expressed per mg DNA) in homogenates of the needle biopsies from adipose tissue varied considerably. Activity was lowest in the father and highest in one of the control subjects (Table 4). To some extent, these differences may reflect differences in the cellular composition of the biopsies. In all study subjects, [35S]methionine radioactivity was incorporated at similar levels into immunoprecipitable LPL protein (0.014–0.044%). Similar Met incorporation data were reported in another study (37).
TABLE 4.
LPL activity and incorporation of [35S]methionine into immunoprecipitable LPL in needle biopsies of adipose tissue
| Subject | LPL Activity (mU/mg DNA) | LPL Synthesis (% of Total Protein Synthesis) |
|---|---|---|
| Control, male | 198 | 0.044 |
| Control, female | 55 | 0.014 |
| I-1 | 23 | 0.020 |
| I-2 | 90 | 0.038 |
| II-2 | 57 | 0.032 |
| II-3 | 62 | 0.014 |
Samples of adipose tissue from needle biopsies were labeled with [35S]methionine for 2 h. Homogenates were prepared and LPL activity was immediately analyzed. The rest of the remaining samples were divided in two equal parts for immunoprecipitation, with one-half incubated with an antiserum against LPL and the other half with nonimmune serum. The controls were 35 years (male) and 24 years (female) of age and the experiment was performed in 1987, when patient II-2 was 18 years old and patient II-3 was 13 years old.
To further study LPL activity and LPL synthesis rate in the two probands, we repeated the experiments using surgical biopsies of subcutaneous adipose tissue from two of the patients and two unrelated healthy controls. The LPL activities were higher than in the previous experiment (Table 5), probably because the surgical biopsies were richer in adipocytes than the needle biopsies, especially those from the probands. There were no striking differences in LPL activity in tissue or in medium between the two patients and the two controls. Heparin in the medium had little effect on release of LPL activity. Adipose tissue taken at surgery was pulse-labeled with [35S]methionine for 30 min and then chased for 60 min, with and without heparin in the medium. Radioactivity recovered in LPL ranged from 0.016–0.024% of total protein radioactivity, and there were no obvious differences between the patients and the controls. Biopsies of adipose tissue were taken again in 1993 from the father and the son (II-3) after an overnight fast. LPL activity was in the normal range in both the father (9.3 mU/g) and the son (4.5 mU/g).
TABLE 5.
LPL activity and the amount of radioactivity in adipose tissue pieces and in culture media after a pulse-chase experiment
| LPL Activity |
35S-Radioactivity (cpm) |
|||||
|---|---|---|---|---|---|---|
| (mU/mg DNA) |
In LPL |
|||||
| Subject | Heparin | Tissue | Medium | In Total Proteins | Tissue | Medium |
| Control 1 | – | 203 | 71 | 16.0 × 106 | 3,673 | 479 |
| + | 196 | 79 | n.d. | 491 | ||
| Control 2 | – | 138 | 43 | 21.8 × 106 | 3,480 | 333 |
| + | 127 | 50 | n.d. | 317 | ||
| II-2 | – | 185 | 43 | 21.7 × 106 | 4,548 | 548 |
| + | 108 | 51 | n.d. | 520 | ||
| II-3 | – | 107 | 57 | 26.7 × 106 | 4,483 | 183 |
| + | 154 | 61 | n.d. | 108 | ||
The controls were two unrelated healthy adults undergoing elective surgery. Tissue pieces were pulse labeled for 30 min in medium containing [35S]methionine with or without heparin (100 IU/ml) and then chased for 60 min in medium without [35S]methionine. After the chase period, both LPL activity and radioactivity were measured in tissue homogenates and in the medium. n.d., not determined.
To study possible disturbances in the intracellular lipase processing, immunoprecipitates of tissue extracts from both the pulse and after the chase (Table 5) were dissolved, treated with endoglucosidase H, and analyzed by SDS-PAGE. At the end of the pulse, virtually all of the labeled LPL from control subjects was transformed to a band with higher mobility by endoglycosidase digestion (data not shown). After the chase, the lipase had been converted to a form whose mobility changed less. Thus, at least one oligosaccharide chain had been processed to a digestion-resistant form. LPL from one of the probands yielded a similar pattern, indicating no major disturbance in LPL glycosylation.
LPL activity and mass in breast milk from the affected siblings
Breast milk samples collected from probands II-2 and II-4 early after parturition contained normal to elevated levels of LPL activity and LPL protein mass compared with milk from healthy mothers (Fig. 4A). Thus, LPL was produced by the mammary gland and secreted into the milk. However, the total lipid content of the milk samples was approximately one-third that of milk from normal mothers (data not shown), and the fatty acid composition showed a shift toward more medium-chain fatty acids and a higher fraction of saturated than of mono- and polyunsaturated fatty acids (Fig. 4B).
Fig. 4.

LPL activity and mass in milk samples. A: LPL activity and mass in milk from subjects II-2 and II-4 were compared with two healthy mothers. From patient II-2, three milk samples were analyzed from days 3, 10, and 18 postpartum. From patient II-4, eight milk samples were taken during days 14–24. From control subject 1, a total of 37 milk samples were taken during days 18–22 postpartum, and from control subject 2, a total of 42 milk samples were taken from a similar time period. Human LPL was used as standard for the ELISA, and data are mean values ± SD. B: Milk fatty acid composition. Values are mean ± SD from seven samples taken at days 3–60 postpartum from subject II-2 and of samples from five healthy mothers taken <1 month postpartum (control subjects, mean of three samples from each mother ± SD).
Identification of missense mutations in the gene for GPIHBP1
Because GPIHBP1 is crucial for the function of LPL (17, 18), we next investigated GPIHBP1 in family members. DNA sequence analysis of subject II-2 (GPIHBP1 coding region, 5′ and 3′ flanking sequences) revealed two heterozygous mutations in exon 3, which were later identified in all three affected siblings (Fig. 5). A G-to-C transition at the second base of codon 65 changes a Cys (TGC) to a Ser (TCC), and a G-to-T transversion in the first base of codon 68 changes a Cys (TGC) to Gly (GGC). Both mutations involve highly conserved Cys residues in the Ly6 domain. The father was heterozygous for the C65S mutation, and the mother was heterozygous for the C68G substitution. Exon 3 was sequenced in 50 randomly selected healthy controls; none had either of these two amino acid substitutions.
Fig. 5.

Identification of mutations in GPIHBP1. Filled symbols indicate family members with chylomicronemia. Black circles within the symbols indicate heterozygous carriers of GPIHBP1 mutations. Question mark indicates the lack of DNA sequencing in the son (II-1). Electropherograms including the two mutations, C65S and C68G, in exon 3 of GPIHBP1 are shown below the symbols to the right. Arrows indicate heterozygous changes.
Cell expression and LPL binding properties of GPIHBP1 mutants
To determine whether the mutant GPIHBP1 proteins reached the cell surface, we electroporated expression vectors for wild-type GPIHBP1 and the mutant GPIHBP1 proteins (C65S and C68G) into CHO pgsA-745 cells (20, 35). Both wild-type GPIHBP1 and the mutant GPIHBP1 proteins reached the surface of transiently transfected cells, as judged by immunofluorescence microscopy on nonpermeabilized cells (Fig. 6). Also, the mutant proteins were readily released from the surface of cells with a PIPLC (Fig. 7). As a control, we tested the ability of PIPLC to release GPIHBP1 from the surface of cells that expressed a mutant version of GPIHBP1 (N76Q) known to be retained in the endoplasmic reticulum (38) (Fig. 7). With the N76Q mutant, PIPLC released lower amounts of GPIHBP1 into the medium.
Fig. 6.

Expression of wild-type and mutant forms of the GPIHBP1 protein in CHO pgsA-745 cells. The cells were electroporated with an empty vector, a construct encoding an S-tagged human GPIHBP1, or the mutants C65S and C68G. GPIHBP1 in nonpermeabilized (A) and permeabilized (B) cells was assessed by immunofluorescence microscopy with an antiserum against the S-protein tag (green). Cell nuclei were visualized with 4’,6-diamidino-2-phenylindole (blue).
Fig. 7.
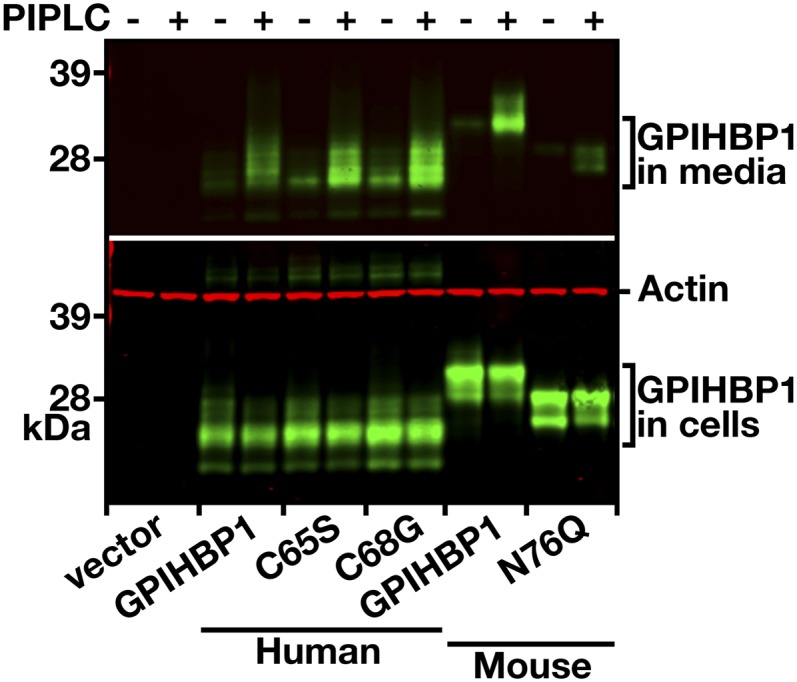
Release of wild-type and mutant GPIHBP1 proteins from the cell surface with PIPLC. CHO pgsA-745 cells were electroporated with expression vectors encoding wild-type mouse GPIHBP1, a mutant mouse GPIHBP1 (N76Q) that eliminates the sole N-linked glycosylation site (38), wild-type human GPIHBP1, and mutant human GPIHBP1-C65S or GPIHBP1-C68G proteins. All of the GPIHBP1 constructs contained an S-protein tag. The amounts of GPIHBP1 released into the medium by PIPLC (5 U/ml for 1 h at 37°C) were assessed by Western blotting.
We next tested the ability of the electroporated cells to bind to LPL. Transiently transfected cells were incubated with a human V5-tagged LPL in the presence or absence of heparin (500 U/ml). After extensive washing of the cells, extracts were prepared, and the amount of LPL was assessed by Western blotting. As expected, cells expressing wild-type GPIHBP1 bound LPL avidly, and the binding was largely blocked by heparin (Fig. 8). In contrast, LPL did not bind to cells expressing GPIHBP1-C65S or GPIHBP1-C68G. Consistent with previously published studies (20), LPL did not bind to cells expressing GPIHBP1-Q115P.
Fig. 8.

Binding of LPL to cells expressing wild-type and mutant GPIHBP1. CHO pgsA-745 cells were electroporated with constructs encoding S-protein-tagged wild-type GPIHBP1 or the mutant forms C65S, C68G, or Q115P. Twenty-four hours later, the cells were incubated with V5-tagged human LPL in the presence or absence of heparin (500 U/ml). The cells were washed six times, cell extracts were prepared, and the level of LPL bound to the cells was assessed by Western blotting with a V5-specific antibody. Simultaneously, the level of GPIHBP1 in cell extracts was assessed by Western blotting with an antibody against the S-protein tag. Actin was used as a loading control.
We next used a cell-free assay (35) to assess LPL binding to soluble versions (i.e., proteins without a GPI anchor, truncated at residue 198) of wild-type mouse GPIHBP1 and mouse GPIHBP1 containing mutations corresponding to those in the human patients (C65S and C68G). Soluble mouse GPIHBP1 and V5-tagged human LPL were incubated with agarose beads coated with antibody 11A12, a rat monoclonal antibody that binds to the C terminus of mouse GPIHBP1 (35). The beads were washed, and soluble GPIHBP1 (along with any LPL bound to the GPIHBP1) was eluted with 0.1 M glycine, pH 2.5. The amounts of GPIHBP1 and LPL in each fraction were assessed by Western blotting. When antibody 11A12-coated beads were incubated with soluble wild-type mouse GPIHBP1 and LPL, the captured GPIHBP1 bound LPL, and GPIHBP1 and LPL eluted together (Fig. 9). In contrast, the mutant versions of GPIHBP1 bound to the antibody-coated agarose beads but did not bind LPL (i.e., only GPIHBP1 was eluted) (Fig. 9).
Fig. 9.

Elution of human LPL together with soluble wild-type GPIHBP1 or the soluble forms of the mutants C65S and C68G from agarose beads coated with anti-GPIHBP1 monoclonal antibody (11A12). V5-tagged human LPL was mixed with the different forms of soluble mouse GPIHBP1, along with antibody 11A12-coated beads. After three washes, the bound proteins were eluted with 0.1 M glycine, pH 2.5. GPIHBP1 and LPL in the starting material, the unbound fraction, the washes, and elution fractions were detected by Western blotting. In all cases, most of the GPIHBP1 protein was captured by the antibody 11A12-coated beads, but LPL was coeluted only in the presence of wild-type GPIHBP1 (A). With the GPIHBP1 mutants (B and C), no LPL was detectable in the eluates.
DISCUSSION
We explored the biochemical basis for chylomicronemia in three siblings from northern Sweden. Initially, we thought they might have classic LPL deficiency, given the low levels of LPL mass and activity in their postheparin plasma. The mother and all four siblings, including the symptom-free oldest son, were heterozygote carriers for the Asn291Ser mutation in the LPL gene. In light of the meta-analysis of Hu et al. (39), it is unlikely that the Asn291Ser mutant would contribute more than marginally to the hyperchylomicronemia seen in the affected siblings. Other findings also pointed away from a diagnosis of LPL deficiency. First, the adipose tissue of affected subjects contained normal amounts of LPL activity. Second, normal amounts of enzymatically active LPL were synthesized by adipose tissue. Third, enzymatically active LPL entered the plasma, albeit with delayed kinetics, after an injection of heparin. Fourth, entirely normal amounts of LPL were present in milk. This constellation of findings led us to predict that the three siblings might have defects in GPIHBP1, an LPL-binding protein on the surface of capillary endothelial cells (16–18). Mice lacking GPIHBP1 have severe chylomicronemia, normal tissue stores of LPL, and delayed entry of LPL into the plasma after a heparin injection. Our prediction about GPIHBP1 mutations in the three siblings was upheld. All three were compound heterozygotes for missense mutations involving highly conserved Cys residues in GPIHBP1’s Ly6 domain (C65S and C68G). The parents, both of whom carried only one of the Cys mutations, were asymptomatic and normolipidemic, demonstrating that GPIHBP1 is normally not rate limiting. This is similar to findings in GPIHBP1 heterozygote mice (18) and in individuals with heterozygote mutations in LPL or the activator protein apolipoprotein CII (6).
There is little doubt that the two Cys substitutions were functionally important. First, the Cys residues of Ly6 proteins are disulfide bonded (40, 41), and disulfide bonds are crucial for forming a three-fingered structural motif. Interfering with any of the disulfide bonds would be predicted to induce gross alterations in protein structure. Second, Cys residues in two other Ly6 proteins, CD59 and SLURP-1, are also critical for protein function. In SLURP-1, missense mutations involving conserved Cys residues of the Ly6 domain cause a human disease, Mal de Meleda (a recessive palmoplantar keratoderma) (42). Third, both of the missense mutations, C65S and C68G, abolished the ability of GPIHBP1 to bind LPL. LPL bound avidly to cells expressing wild-type GPIHBP1 but did not bind to cells expressing the C65S and C68G mutants, despite the fact that both mutants reached the cell surface without difficulty. The mutants also could not bind LPL in a cell-free binding assay. A soluble (secreted) version of wild-type mouse GPIHBP1 bound LPL avidly, while soluble versions of GPIHBP1 proteins with either Cys mutation did not. The inability of the Cys mutants to bind LPL is consistent with results in a very recent study by Beigneux et al. (35), who found that Cys-to-Ala substitutions in Ly6 Cys residues interfere with LPL binding.
Previously, Beigneux and coworkers (20) identified a homozygous Q115P mutation in a young man with lifelong chylomicronemia. Like the two missense mutations reported here, the Q115P mutation virtually abolished the ability of GPIHBP1 to bind LPL. Together, these three mutations show that the structural integrity of the Ly6 domain is critical for LPL binding.
In an earlier study of GPIHBP1-deficient mice (19), LPL was released into the plasma after a bolus of heparin, but entered the plasma with abnormal kinetics. Plasma LPL levels were abnormally low during the first few minutes after the heparin injection, but virtually normal after 15 min. These findings were thought to suggest that there are two pools of heparin-releasable LPL, one bound to GPIHBP1 within capillaries, and a second, largely irrelevant to lipolysis of lipoproteins, within a subendothelial compartment. The authors suggested that the intravascular pool would be rapidly released into the plasma by heparin and that the delayed entry of LPL into the plasma after heparin reflected the absence of the intravascular GPIHBP1-bound pool of LPL.
We observed a similar abnormality in our patients with GPIHBP1 defects. In normal subjects, an injection of heparin (100 IU/kg) promptly released LPL into the plasma, and both LPL activity and mass levels peaked within 10 or 20 min. In the patient II-2, plasma LPL activity and mass levels increased continuously up to the final time point in the study (60 min). A likely explanation for the delayed release of LPL in the affected patient—and for the apparent absence of an early burst of LPL release—is reduced amounts of intravascular LPL (i.e., GPIHBP1-bound LPL). Of course, this would also explain the patients’ severe hypertriglyceridemia.
There was a noteworthy difference between the GPIHBP1-deficient mice and the three affected human patients. By 15 min after heparin injection, LPL levels were normal in the mice (19) but were low in the affected patients. In patient II-2, the LPL activity and mass levels at 60 min were only 15–30% of those in a control subject. These differences might indicate that the subendothelial pool of LPL in humans is more tightly attached to its binding sites and more resistant to release by heparin. A second possibility is that the heparin doses (units/kg) given to the mice were far greater than those given to the humans.
A new and intriguing finding was that the levels of both activity and mass were normal, or even elevated, in milk from the two affected females. No one knows why LPL is secreted into the milk in mammals. The enzyme has no known function in milk or in the intestines of nursing offspring. LPL in the milk is bound to casein micelles or to milk fat droplets and can be found on fat droplets in secretory vesicles, but the enzyme does not hydrolyze the milk lipids (9). That LPL levels were normal in milk, but very low in postheparin plasma, strongly suggests that entry of LPL into milk, unlike entry of LPL into the capillary lumen, is independent of GPIHBP1.
The breast milk of the affected siblings contained low levels of milk fat—to the degree that pediatricians recommended that breast feeding should be terminated. It is interesting that the fatty acid content of the milk was abnormal, with a higher content of medium-chain fatty acids and a higher fraction of saturated than of mono- and polyunsaturated fatty acids. These findings are in agreement with a defect in the production of milk fats from lipoprotein-derived fats. Such a defect would not be completely unexpected; effects on milk fat quality have been described in patients with LPL deficiency (43, 44).
In conclusion, we identified two mutations affecting highly conserved Cys residues in GPIHBP1 in three siblings with chylomicronemia. Both mutations abolished the ability of the protein to bind LPL in cell-based and cell-free assays. GPIHBP1 defects in humans lead to very low levels of LPL in postheparin plasma, presumably because GPIHBP1 interferes with the ability of LPL to reach the capillary lumen. However, the GPIHBP1 mutations had no impact on the entry of LPL into maternal milk.
Acknowledgments
The authors thank all family members for patience and courage. We thank the late Ann-Sofie Jakobsson for technical assistance during the earlier part of this study, Else-Britt Lundström for running gas chromatography separations of fatty acids, Solveig Nilsson for analyses of LPL, Yvonne Andersson for collection of samples and data analyses, Valentina Sukonina for measuring LPL transcripts, and Dr. John Brunzell (University of Washington, Seattle, WA) for monoclonal antibody 5D2.
Footnotes
Abbreviations:
- CHO
- Chinese hamster ovary
- GPIHBP1
- glycosylphosphatidylinositol-anchored high density lipoprotein-binding protein 1
- PIPLC
- phosphatidylinositol-specific phospholipase C
This work was supported by grants from Swedish Medical Research Council, the Bank of Sweden Tercentenary Foundation, the Kempe Foundations, the King Gustav V and Queen Victoria's research fund, the Swedish Heart-Lung foundation, the Västerbotten County Fund for Clinical Research, the Karolinska Institutet Funds, the Canadian Medical Research Council, the Heart and Stroke Foundation of British Columbia and Yukon, a Scientist Development Award from the American Heart Association, National Office, and grants from the National Institutes of Health (R01 HL094732, P01 HL090553, and R01 HL087228).
REFERENCES
- 1.Wang H., Eckel R. H. 2009. Lipoprotein lipase: from gene to obesity. Am. J. Physiol. Endocrinol. Metab. 297: E271–E288. [DOI] [PubMed] [Google Scholar]
- 2.Olivecrona T., Olivecrona G. 2009. The ins and outs of adipose tissue. In Cellular Lipid Mmetabolism Ehnholm C., editor Springer, Heidelberg, Germany: 315–369. [Google Scholar]
- 3.Krieger M., Herz J. 1994. Structures and functions of multiligand lipoprotein receptors: macrophage scavenger receptors and LDL receptor-related protein (LRP). Annu. Rev. Biochem. 63: 601–637. [DOI] [PubMed] [Google Scholar]
- 4.Beisiegel U. 1995. Receptors for triglyceride-rich lipoproteins and their role in lipoprotein metabolism. Curr. Opin. Lipidol. 6: 117–122. [DOI] [PubMed] [Google Scholar]
- 5.Gliemann J. 1998. Receptors of the low density lipoprotein (LDL) receptor family in man. Multiple functions of the large family members via interaction with complex ligands. Biol. Chem. 379: 951–964. [PubMed] [Google Scholar]
- 6.Fojo S. S., Brewer H. B. 1992. Hypertriglyceridaemia due to genetic defects in lipoprotein lipase and apolipoprotein C–II. J. Intern. Med. 231: 669–677. [DOI] [PubMed] [Google Scholar]
- 7.Kluger M., Heeren J., Merkel M. 2008. Apoprotein A-V: an important regulator of triglyceride metabolism. J. Inherit. Metab. Dis. 31: 281–288. [DOI] [PubMed] [Google Scholar]
- 8.Hernell O., Olivecrona T. 1974. Human milk lipases. I. Serum-stimulated lipase. J. Lipid Res. 15: 367–374. [PubMed] [Google Scholar]
- 9.Olivecrona T., Vilaro S., Olivecrona G. 2003. Lipases in milk. In Advanced Dairy Chemistry Fox P. F., McSweeney P. L. H., Kluwer Academic, New York: 473–494. [Google Scholar]
- 10.Vilella E., Joven J., Fernandez M., Vilaro S., Brunzell J. D., Olivecrona T., Bengtsson-Olivecrona G. 1993. Lipoprotein lipase in human plasma is mainly inactive and associated with cholesterol-rich lipoproteins. J. Lipid Res. 34: 1555–1564. [PubMed] [Google Scholar]
- 11.Tornvall P., Olivecrona G., Karpe F., Hamsten A., Olivecrona T. 1995. Lipoprotein lipase mass and activity in plasma and their increase after heparin are separate parameters with different relations to plasma lipoproteins. Arterioscler. Thromb. Vasc. Biol. 15: 1086–1093. [DOI] [PubMed] [Google Scholar]
- 12.Zambon A., Schmidt I., Beisiegel U., Brunzell J. D. 1996. Dimeric lipoprotein lipase is bound to triglyceride-rich plasma lipoproteins. J. Lipid Res. 37: 2394–2404. [PubMed] [Google Scholar]
- 13.Braun J. E., Severson D. L. 1992. Regulation of the synthesis, processing and translocation of lipoprotein lipase. Biochem. J. 287: 337–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Olivecrona T., Bengtsson G., Marklund S. E., Lindahl U., Hook M. 1977. Heparin-lipoprotein lipase interactions. Fed. Proc. 36: 60–65. [PubMed] [Google Scholar]
- 15.Goldberg I. J. 1996. Lipoprotein lipase and lipolysis: central roles in lipoprotein metabolism and atherogenesis. J. Lipid Res. 37: 693–707. [PubMed] [Google Scholar]
- 16.Young S. G., Davies B. S., Fong L. G., Gin P., Weinstein M. M., Bensadoun A., Beigneux A. P. 2007. GPIHBP1: an endothelial cell molecule important for the lipolytic processing of chylomicrons. Curr. Opin. Lipidol. 18: 389–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beigneux A. P., Weinstein M. M., Davies B. S., Gin P., Bensadoun A., Fong L. G., Young S. G. 2009. GPIHBP1 and lipolysis: an update. Curr. Opin. Lipidol. 20: 211–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beigneux A. P., Davies B. S., Gin P., Weinstein M. M., Farber E., Qiao X., Peale F., Bunting S., Walzem R. L., Wong J. S., et al. 2007. Glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1 plays a critical role in the lipolytic processing of chylomicrons. Cell Metab. 5: 279–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weinstein M. M., Yin L., Beigneux A. P., Davies B. S., Gin P., Estrada K., Melford K., Bishop J. R., Esko J. D., Dallinga-Thie G. M., et al. 2008. Abnormal patterns of lipoprotein lipase release into the plasma in GPIHBP1-deficient mice. J. Biol. Chem. 283: 34511–34518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beigneux A. P., Franssen R., Bensadoun A., Gin P., Melford K., Peter J., Walzem R. L., Weinstein M. M., Davies B. S., Kuivenhoven J. A., et al. 2009. Chylomicronemia with a mutant GPIHBP1 (Q115P) that cannot bind lipoprotein lipase. Arterioscler. Thromb. Vasc. Biol. 29: 956–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rouis M., Dugi K. A., Previato L., Patterson A. P., Brunzell J. D., Brewer H. B., Santamarina-Fojo S. 1997. Therapeutic response to medium-chain triglycerides and omega-3 fatty acids in a patient with the familial chylomicronemia syndrome. Arterioscler. Thromb. Vasc. Biol. 17: 1400–1406. [DOI] [PubMed] [Google Scholar]
- 22.Beynen A. C., Katan M. B. 1985. Rapid sampling and long-term storage of subcutaneous adipose-tissue biopsies for determination of fatty acid composition. Am. J. Clin. Nutr. 42: 317–322. [DOI] [PubMed] [Google Scholar]
- 23.Gagne E., Genest J., Jr., Zhang H., Clarke L. A., Hayden M. R. 1994. Analysis of DNA changes in the LPL gene in patients with familial combined hyperlipidemia. Arterioscler. Thromb. 14: 1250–1257. [DOI] [PubMed] [Google Scholar]
- 24.Holm C., Olivecrona G., Ottosson M. 2001. Assays of lipolytic enzymes. Methods Mol. Biol. 155: 97–119. [DOI] [PubMed] [Google Scholar]
- 25.Karpe F., Olivecrona T., Walldius G., Hamsten A. 1992. Lipoprotein lipase in plasma after an oral fat load: relation to free fatty acids. J. Lipid Res. 33: 975–984. [PubMed] [Google Scholar]
- 26.Labarca C., Paigen K. 1980. A simple, rapid, and sensitive DNA assay procedure. Anal. Biochem. 102: 344–352. [DOI] [PubMed] [Google Scholar]
- 27.Folch J., Lees M., Sloane Stanley G. H. 1957. A simple method for the isolation and purification of total lipids from animal tissues. J. Biol. Chem. 226: 497–509. [PubMed] [Google Scholar]
- 28.Dodge J. T., Phillips G. B. 1967. Composition of phospholipids and of phospholipid fatty acids and aldehydes in human red cells. J. Lipid Res. 8: 667–675. [PubMed] [Google Scholar]
- 29.Morrison W. R., Smith L. M. 1964. Preparation of fatty acid methyl esters and dimethylacetals from lipids with boron fluoride–methanol. J. Lipid Res. 5: 600–608. [PubMed] [Google Scholar]
- 30.Iverius P. H. 1971. Coupling of glycosaminoglycans to agarose beads (sepharose 4B). Biochem. J. 124: 677–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Semb H., Olivecrona T. 1986. Lipoprotein lipase in guinea pig tissues: molecular size and rates of synthesis. Biochim. Biophys. Acta. 878: 330–337. [DOI] [PubMed] [Google Scholar]
- 32.Wallinder L., Bengtsson G., Olivecrona T. 1982. Purification and properties of lipoprotein lipase in guinea pig milk. Biochim. Biophys. Acta. 711: 107–113. [DOI] [PubMed] [Google Scholar]
- 33.Semb H., Olivecrona T. 1989. The relation between glycosylation and activity of guinea pig lipoprotein lipase. J. Biol. Chem. 264: 4195–4200. [PubMed] [Google Scholar]
- 34.Wang J., Hegele R. A. 2007. Homozygous missense mutation (G56R) in glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1 (GPI-HBP1) in two siblings with fasting chylomicronemia (MIM 144650). Lipids Health Dis. 6: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beigneux A. P., Gin P., Davies B. S., Weinstein M. M., Bensadoun A., Fong L. G., Young S. G. 2009. Highly conserved cysteines within the Ly6 domain of GPIHBP1 are crucial for the binding of lipoprotein lipase. J. Biol. Chem. 284: 30240–30247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang W. S., Nevin D. N., Iwasaki L., Peng R., Brown B. G., Brunzell J. D., Deeb S. S. 1996. Regulatory mutations in the human lipoprotein lipase gene in patients with familial combined hyperlipidemia and coronary artery disease. J. Lipid Res. 37: 2627–2637. [PubMed] [Google Scholar]
- 37.Ottosson M., Vikman-Adolfsson K., Enerback S., Olivecrona G., Bjorntorp P. 1994. The effects of cortisol on the regulation of lipoprotein lipase activity in human adipose tissue. J. Clin. Endocrinol. Metab. 79: 820–825. [DOI] [PubMed] [Google Scholar]
- 38.Beigneux A. P., Gin P., Davies B. S., Weinstein M. M., Bensadoun A., Ryan R. O., Fong L. G., Young S. G. 2008. Glycosylation of Asn-76 in mouse GPIHBP1 is critical for its appearance on the cell surface and the binding of chylomicrons and lipoprotein lipase. J. Lipid Res. 49: 1312–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hu Y., Liu W., Huang R., Zhang X. 2006. A systematic review and meta-analysis of the relationship between lipoprotein lipase Asn291Ser variant and diseases. J. Lipid Res. 47: 1908–1914. [DOI] [PubMed] [Google Scholar]
- 40.Llinas P., Le Du M. H., Gardsvoll H., Dano K., Ploug M., Gilquin B., Stura E. A., Menez A. 2005. Crystal structure of the human urokinase plasminogen activator receptor bound to an antagonist peptide. EMBO J. 24: 1655–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang Y., Fedarovich A., Tomlinson S., Davies C. 2007. Crystal structure of CD59: implications for molecular recognition of the complement proteins C8 and C9 in the membrane-attack complex. Acta Crystallogr. D Biol. Crystallogr. 63: 714–721. [DOI] [PubMed] [Google Scholar]
- 42.Favre B., Plantard L., Aeschbach L., Brakch N., Christen-Zaech S., de Viragh P. A., Sergeant A., Huber M., Hohl D. 2007. SLURP1 is a late marker of epidermal differentiation and is absent in Mal de Meleda. J. Invest. Dermatol. 127: 301–308. [DOI] [PubMed] [Google Scholar]
- 43.Steiner G., Myher J. J., Kuksis A. 1985. Milk and plasma lipid composition in a lactating patient with type I hyperlipoproteinemia. Am. J. Clin. Nutr. 41: 121–128. [DOI] [PubMed] [Google Scholar]
- 44.Berger G. M., Spark A., Baillie P. M., Huskisson J., Stockwell G., van der Merwe E. 1983. Absence of serum-stimulated lipase activity and altered lipid content in milk from a patient with type I hyperlipoproteinaemia. Pediatr. Res. 17: 835–839. [DOI] [PubMed] [Google Scholar]