

Abstract
Regulation of platelet activation plays a central role in hemostasis and pathophysiological processes such as coronary artery disease. Thrombin is the most potent activator of platelets. Human platelets express two thrombin receptors, PAR1 and PAR4, both of which signal platelet activation. Evidence is lacking on the mechanism by which PAR1 and PAR4 may differentially signal platelet aggregation. Here we show that at the relatively high concentration of agonist most likely found at the site of a local thrombus, dual inhibition of the P2Y12 receptor and calcium mobilization result in a complete inhibition of PAR4-induced aggregation, while having no effect on either thrombin or PAR1-mediated platelet aggregation. Both PAR1- and PAR4-mediated aggregation are independent of calcium mobilization. Furthermore, we show that P2Y12 receptor activation is not required for protease-activated receptor-mediated aggregation at higher agonist concentrations and is only partially required for Rap1 as well as GPIIbIIIa activation. P2Y12 receptor inhibitors clinically in use such as clopidogrel are postulated to decrease platelet aggregation through partial inhibition of PAR1 signaling. Our data, however, indicate that at high local concentrations of thrombin, it is the signaling through PAR4 rather than PAR1 that may be regulated through purinergic feedback. Thus, our data identify an intra-platelet mechanism that may function as a future site for therapeutic intervention.
Vascular hemostasis is critical for normal physiological function (1). Thrombin is the most potent activator of prothrombotic hemostatic functions such as fibrin activation (2), endothelial gap formation (3), and activation of platelets (4, 5) as well as initiation of the antithrombotic pathway involving the activation of protein C (5, 6). Thrombin signaling has been extensively studied in numerous cell types (7) resulting in a greater understanding of how this enzyme is able to regulate such a broad range of physiological effects (8). These studies have led to the discovery of a family of G protein-coupled receptors that are activated via protease cleavage of the receptor revealing a new tethered ligand and are appropriately termed the protease-activated receptor (PAR)2 family (9). Human platelets express two such PARs, PAR1 and PAR4 (10). Research in the field has indicated that PAR1 is a high affinity thrombin receptor that signals through at least three classes of G proteins (Gα12, Gαq, and Gαi/o) (11, 12), whereas PAR4 has been shown to signal through at least two G protein signaling pathways (Gα12 and Gαq but not to Gαi/o) (13).
Mouse models have been essential for identifying important steps in platelet signaling downstream of thrombin such as receptor activation of PAR3 and PAR4 (10, 14, 15) and Rap1 signaling of GPIIbIIIa activation (16). Because mouse platelets do not express PAR1, the PAR-mediated signaling pathways may not be identical in human models to that postulated for the mouse. For this reason, it is important to gain an understanding of how thrombin regulates PAR-mediated signaling in the human platelet.
PAR activation of platelets results in the secretory release of a number of small molecule and protein modulators of platelet function, including ATP and ADP (17). Once released, ADP further stimulates the platelet in an autocrine fashion through purinergic receptors on the platelet surface (18). In the mouse model, purinergic stimulation is crucial to platelet activation, whereas human platelets appear to be able to bypass this pathway. It has also been shown that a synergistic link may exist between one of the purinergic receptors, P2Y12, and PAR signaling (19), although the convergence of the P2Y12 and PAR4 pathways has yet to be fully elucidated. Furthermore, because PAR1 has been shown to be activated before PAR4 (20), PAR1 may be activated at low levels of circulating thrombin, whereas PAR4 may become active following the initial clot formation when the local thrombin concentration is significantly increased (7). Based on published data (10, 20), it is plausible to postulate that PAR1 and PAR4 signal through a different complement of G proteins in the human platelet (21). Our findings identify for the first time a discrete signaling difference between PAR1 and PAR4 in human platelets. Understanding these signaling differences may provide insight into targets that lend themselves to the development of better anti-platelet therapies having less pronounced bleeding liabilities than current therapeutic approaches (22, 23).
EXPERIMENTAL PROCEDURES
Human Platelets
Human platelets were obtained from healthy volunteers from within the Vanderbilt University community. These studies were approved by the Vanderbilt University institutional review board. Informed consent was obtained from all individuals prior to platelet donation.
Materials
Human α-thrombin (2700 NIH units/mg) was purchased from Enzyme Research Laboratories (South Bend, IN). Activating peptides for PAR1 (PAR1-AP; SFLLRN) and PAR4 (PAR4-AP; AYPGKF) were purchased from GL Biochem (Shanghai, China). Purified human fibrinogen, 2-methylthioadenosine monophosphate (2-MeSAMP), and ADP were purchased from Sigma. Anti-Rap1 Ab was purchased from Santa Cruz Biotechnology. Blocking buffer and anti-rabbit IRDYE 800 were purchased from LI-COR Biosciences (Lincoln, NE). Anti-PAC1-FITC Ab was purchased from Pharmingen. Aggregometry cuvettes and stir bars were purchased from Chrono-Log Corp. (Havertown, PA). BAPTA-AM was purchased from Calbiochem. MRS-2179 was purchased from Tocris Pharmaceuticals (Ellisville, MO). Fura2-AM was purchased from Molecular Probes. U-73122 and U-73343 were purchased from Biomol (Plymouth Meeting, PA).
Platelet Aggregation
Platelet aggregations were measured using washed platelets. Briefly, blood was centrifuged in a Forma 400-ml GP centrifuge at 170 × g for 15 min at room temperature. The platelet-rich plasma was placed into 15-ml conical tubes containing a 10% acid citrate dextrose solution (39 mM citric acid, 75 mm sodium citrate, and 135 mm glucose; pH 7.4) and centrifuged at 800 × g for 10 min at room temperature. The pelleted platelets were resuspended in Tyrode’s buffer (12 mm NaHCO3, 127 mm NaCl, 5 mm KCl, 0.5 mm NaH2PO4, 1 mm MgCl2, 5 mm glucose, 10 mm HEPES) and adjusted to a concentration of 3 × 108 platelets/ml using a Coulter counter. Some platelets were treated with 20 μm BAPTA-AM for 10 min, 4 μm U-73122 or U-73343 for 5 min, 50 μm 2-MeSAMP for 5 min, or 100 μm MRS-2179 for 5 min. Following stimulation with either 10 nm thrombin, 20 μm PAR1-AP, or 200 μm PAR4-AP, the change in light transmission was monitored with an aggregometer (Chrono-Log Corp.).
Measurement of RAP1 Activity
Rap1 activity was measured using GST-RalGDS-Rap1-binding domain (RalGDS-RBD) that specifically pulls down activated Rap1 (24, 25). The RalGDS-RBD was coupled at 4 °C for 1.5 h with 0.3 ml of glutathione-Sepharose 4B, washed twice with lysis buffer, and aliquoted into 1.5-ml centrifuge tubes. Following stimulation, platelets were lysed with 2 × lysis buffer (100 mm Tris-HCl, pH 7.4, 2% Triton X-100, 150 mm NaCl, 2% IGEPAL, 1% sodium deoxycholate, 0.05% SDS, 2 mm Na3VO4, 2 mm phenylmethylsulfonyl fluoride, 2 μg/ml leupeptin, 2 μg/ml aprotinin), centrifuged, and incubated with RalGDS-RBD for 1 h at 4 °C. After several washes, precipitated Rap1 was boiled in Laemmli buffer for 10 min and run on SDS-PAGE followed by immunoblotting with the anti-Rap1 Ab. To control for protein loading, total platelet lysates were loaded and immunoblotted to confirm equal protein loading for each sample. Some platelets were pretreated with BAPTA-AM, 2-MeSAMP, or both BAPTA-AM and 2-MeSAMP. Following pretreatment, cells were stimulated for 5 min with thrombin, PAR1-AP, or PAR4-AP and analyzed for Rap1 activation.
GPIIbIIIa Activation
Human platelet GPIIbIIIa activation was assessed using fluorescence-activated cell sorting (FACS) (26). Platelets were pretreated with BAPTA-AM, Me2SO, 2-MeSAMP, or both BAPTA-AM and 2-MeSAMP followed by activation with PAR1-AP or PAR4-AP. Platelets were then incubated for 15 min with a FITC-conjugated PAC1 Ab that only recognizes the active form of GPIIbIIIa followed by flow cytometric measurement of active surface expression of GPIIbIIIa.
Calcium Mobilization
Washed platelets were incubated with Fura-2-AM (2.5 μm) at 37 °C for 30 min. Following incubation, 10% acid citrate dextrose was added to the suspended platelets, and they were centrifuged 2 times for 10 min at 800 × g at room temperature and resuspended in Tyrode’s buffer. Some platelets were pretreated with BAPTA-AM or U-73122. Thrombin, PAR1-AP, or PAR4-AP was added immediately prior to fluorescent analysis using a Varian Eclipse fluorometer.
Statistical Analysis
Comparison between experimental groups was made using a paired t test program using Prism software. Differences in mean values were considered significant at p < 0.05.
RESULTS
PAR-specific Affinity for Platelet Aggregation
PAR1 and PAR4 have been reported to have significantly different EC50 values for platelet aggregation. This parameter is dependent on many factors, including platelet preparation (platelet-rich plasma versus washed) and types of inhibitors used (i.e. aspirin or apyrase) to pretreat the platelets prior to stimulation. The dose-response curves for both PAR1-AP and PAR4-AP are shown in Fig. 1. Washed human platelets were suspended in Tyrode’s buffer in the absence of inhibitors such as aspirin, apyrase, or P2Y receptor inhibitors. The EC50 for PAR1-AP and PAR4-AP under these conditions was 3 and 80 μm, respectively. Based on these curves, PAR1-AP and PAR4-AP were used in all subsequent experiments at a concentration of 20 and 200 μm, respectively, in order to ascertain the effect of inhibiting several different signaling pathways on the ability of PAR1/PAR4 to initiate platelet aggregation.
FIGURE 1. Dose-dependent PAR-induced aggregation of human platelets.

Washed human platelets were stimulated with increasing concentrations of PAR1-AP or PAR4-AP. Maximal aggregation was measured in response to agonist stimulation. PAR1-AP aggregation response curve ranged from 0.1 to 100 μm, whereas the PAR4-AP aggregation response curve ranged from 1 to 800 μm. Each point on the curve is representative of the average of at least three independent experiments ± S.E. EC50 for PAR1-AP and PAR4-AP was 3 and 80 μm, respectively.
PAR-induced Calcium Effects on Platelet Aggregation
The role calcium plays in PAR-mediated regulation of human platelets is not well defined. To determine whether chelation of Ca2+ by BAPTA was able to fully inhibit intra-platelet calcium mobilization, free calcium was measured following stimulation with thrombin, PAR1-AP, or PAR4-AP with or without pretreatment with 20 μm BAPTA (Fig. 2, A and B). A transient increase in calcium mobilization was observed following treatment with all three PAR agonists. To determine whether calcium plays a necessary role in PAR-mediated platelet aggregation, calcium was chelated with BAPTA-AM, and intra-platelet calcium was monitored fluorometrically with Fura2-AM (Fig. 2, A and B, respectively). Following pretreatment, calcium mobilization was completely inhibited. Under conditions where calcium mobilization was fully attenuated, platelet aggregation was minimally affected (Fig. 4). To determine whether the PAR-induced calcium mobilization was downstream of phospholipase C activation, phospholipase C was blocked with U-73122 (but not its inactive isomer U-73343), and similar results were obtained (supplemental Fig. 1).
FIGURE 2. Calcium regulation of PAR-mediated platelet aggregation.

A, platelets were treated with or without 20 μm BAPTA-AM for 10 min and stimulated with 10 nm thrombin, 20 μm PAR1-AP, or 200 μm PAR4-AP, and fluorimetric changes were observed to identify intra-platelet calcium mobilization. B, Ca2+ mobilization increase relative to Ca2+ levels in the unstimulated condition (n = 3).
FIGURE 4. Dual inhibition blocks PAR4-mediated platelet aggregation.
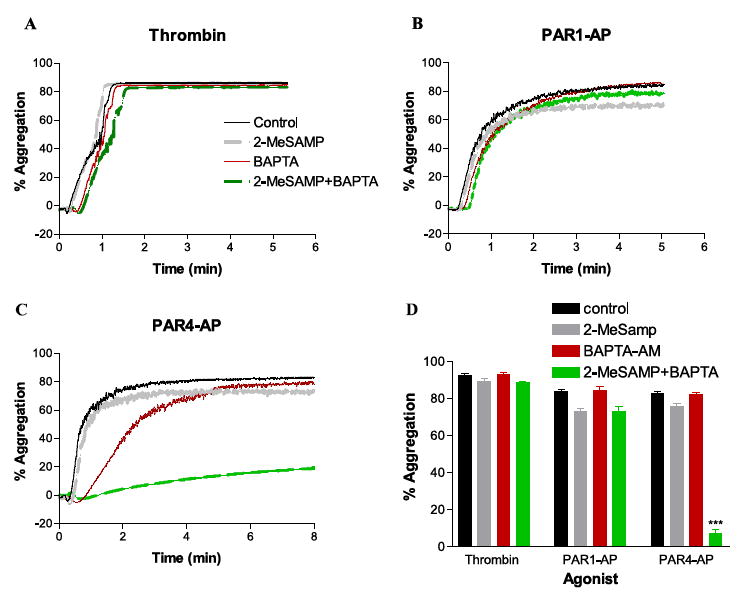
A, platelets were pretreated with 50 μm 2-MeSAMP, 20 μm BAPTA-AM, or both 2-MeSAMP and BAPTA-AM followed by stimulation with 10 nm thrombin (A), 20 μm PAR1-AP (B), or 200 μm PAR4-AP (C). D, percent aggregation with thrombin (n = 9), PAR1-AP (n = 10), or PAR4-AP (n = 8) after pretreatment with the 2-MeSAMP, BAPTA-AM, or the combination of 2-MeSAMP + BAPTA-AM (***; p < 0.0001).
Purinergic Regulation of Platelet Aggregation
It has been well documented that purinergic agonists such as ADP are able to induce platelet aggregation. Furthermore, platelet activation in the mouse is thought to involve both the PAR and purinergic receptor signaling pathways. Therefore, we wanted to determine which purinergic receptor, P2Y1 (thought to specifically signal through Gαq) or P2Y12 (thought to specifically signal through the Gαi/o family), might be involved in PAR-induced human platelet aggregation. Platelets pretreated with MRS-2179 (P2Y1 receptor antagonist), 2-MeSAMP (P2Y12 receptor antagonist), or both were stimulated with 40 μm ADP, and the resulting platelet aggregation was measured (Fig. 3A). Inhibition of the P2Y1 receptor was unable to inhibit ADP-induced platelet aggregation (but did induce a delay in the rate of aggregation calculated as the percentage of aggregation/s in the control and MRS-2179-treated conditions, 0.862 ± 0.12 and 0.314 ± 0.17, respectively). Inhibiting the P2Y12 receptor resulted in a 90% inhibition of platelet aggregation, and inhibiting both receptors resulted in a complete block of platelet aggregation in response to ADP. To confirm that neither P2Y1 activation nor its signaling through Gαq is a requisite component of PAR-mediated platelet aggregation, platelets were pretreated with MRS-2179 with or without BAPTA-AM followed by stimulation with either PAR1-AP or PAR4-AP. Neither PAR1-AP- nor PAR4-AP-mediated platelet aggregation was affected by inhibition of the P2Y1 receptor indicating that P2Y12 and not P2Y1 activation may be required for PAR-mediated platelet aggregation (Fig. 3, B and C).
FIGURE 3. Role of ADP receptors in PAR-induced platelet activation.

A, to determine the importance of PAR-mediated purinergic regulation of platelet activation, washed platelets were treated with or without 100 μm MRS-2179, 50 μm 2-MeSAMP, or both. After pretreatment, platelet aggregation was measured following ADP stimulation. Inhibition of the P2Y1 receptor was unable to inhibit ADP-induced platelet aggregation. Inhibiting the P2Y12 receptor resulted in a 90% inhibition of platelet aggregation, and inhibiting both receptors resulted in a complete block of platelet aggregation in response to ADP (n = 3). B, washed platelets were treated with or without MRS-2179, 20 μm BAPTA-AM, or both followed by stimulation with 20 μm PAR1-AP. Aggregation was then measured for each of the conditions up to 10 min post-stimulation (n = 3). C, washed platelets were treated with or without MRS-2179, 20 μm BAPTA-AM, or both followed by stimulation with 200 μm PAR4-AP. Aggregation was then measured for each of the conditions up to 10 min post-stimulation (n = 3).
Dual Inhibition of PAR-mediated Platelet Activation
Identification of signaling differences between PARs may require simultaneous inhibition of multiple pathways as has been eluded to in mouse platelets (19, 27). To test this hypothesis, platelets were pretreated with 2-MeSAMP, BAPTA, or 2-Me-SAMP + BAPTA. Aggregation was then measured following stimulation with thrombin, 20 μm PAR1-AP, or 200 μm PAR4-AP (Fig. 4). There was no observable decrease in maximum platelet aggregation observed with BAPTA pretreatment. However, a delay in the rate ofaggregation was observed following pretreatment with BAPTA (calculated as percentage aggregation/s in the control and BAPTA-AM treated conditions, respectively) in the thrombin (1.4 ± 0.11; 0.93 ± 0.11), PAR1-AP (1.71 ± 0.15; 1.28 ± 0.09), and PAR4-AP (1.72 ± 0.14; 0.7 ± 0.16) conditions. Thrombin and PAR1-AP-stimulated platelet aggregation was not inhibited by any of the combinations of inhibitors (Fig. 4, A, B, and D). PAR4-AP-stimulated platelet aggregation, although not being greatly affected by inhibition of either calcium mobilization or P2Y12 signaling, was completely blocked by dual inhibition with 2-MeSAMP and BAPTA and was significantly attenuated following pretreatment with 2-MeSAMP (Fig. 4, C and D). Similarly, pretreatment with U-73122 alone had little effect on either PAR1AP- or PAR4AP-induced maximal platelet aggregation. However, pretreatment with both U-73122 and 2-MeSAMP resulted in an 80% inhibition of PAR4AP-induced platelet aggregation, whereas PAR1AP-induced aggregation was not significantly affected.
Because the ability of low concentrations of agonists to induce aggregation may be attenuated through inhibition of specific signaling pathways (because of partial activation of the surface receptors), we looked at the ability of increasing concentrations of agonist to either PAR1 or PAR4 to overcome these inhibitory effects (Figs. 5 and 6). BAPTA and 2-MeSAMP appeared to attenuate the level of aggregation at lower concentrations of agonist. However, Fig. 5 indicates that treatment with PAR1-AP overcame the inhibitory effect of dual inhibition at higher concentrations of agonist (when the PARs are fully activated), whereas Fig. 6 indicates that PAR4-AP was unable to induce aggregation even at very high levels of agonist (7.5-fold EC50 and 10-fold EC50 for PAR1-AP and PAR4-AP, respectively) even though PAR4-AP was able to induce full platelet aggregation under BAPTA or P2Y12 conditions at the highest agonist concentrations tested, indicating the existence of differential signaling pathways between these two receptors.
FIGURE 5. PAR1 dose-response curves to dual inhibitory effect of BAPTA and 2-MeSAMP.
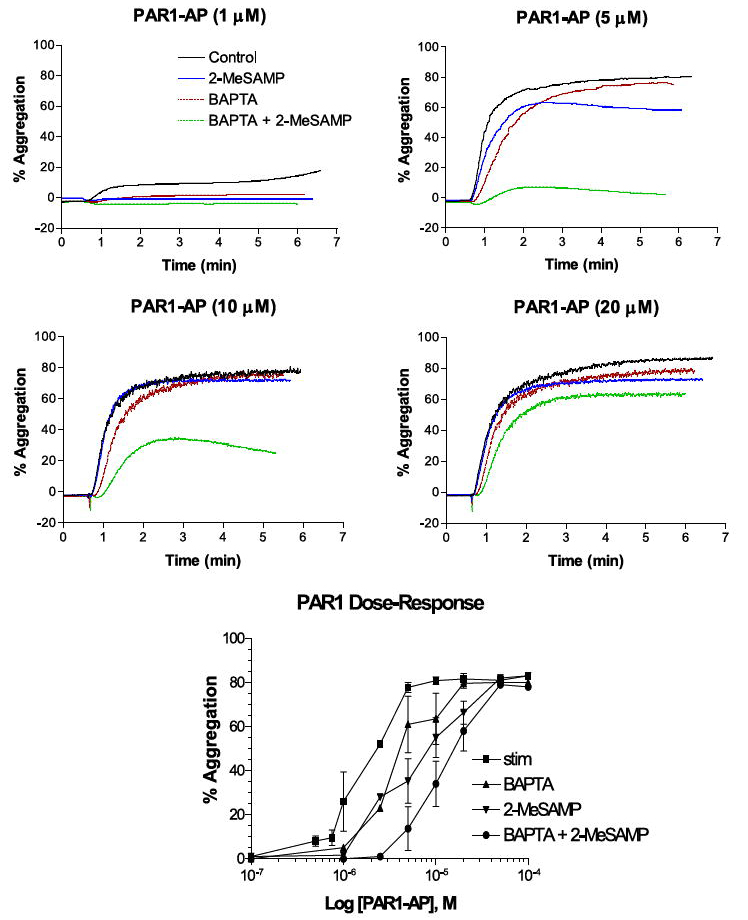
Representative dose-response curves for PAR1-AP-induced aggregation at different concentrations of PAR1-AP (1–20 μm) with or without 20 μm BAPTA, 50 μm 2-MeSAMP, or both (n = 3). Bottom panel indicates composite effective dose-response curves for each condition tested. stim, stimulated.
FIGURE 6. PAR4 dose-response curves to dual inhibitory effect of BAPTA and 2-MeSAMP.

Representative dose-response curves for PAR4-AP-induced aggregation at different concentrations of PAR4-AP (50–800 μm) with or without 20 μm BAPTA, 50 μm 2-MeSAMP, or both (n = 3). Bottom panel indicates composite effective dose-response curves for each condition tested.
Mouse studies have indicated that calcium plays an important role in the activation of Rap1 following both purinergic and thrombin stimulation (28, 29). To ascertain what role calcium or P2Y12 activation plays in PAR-induced Rap1 activation in human platelets, GTP-bound Rap1 was measured in platelets pretreated with inhibitors to these pathways (supplemental Fig. 2). 2-MeSAMP partially blocked PAR-induced Rap1 activation, whereas calcium inhibition had no apparent effect on PAR-mediated Rap1 activation.
Role of Fibrinogen in PAR-mediated Platelet Aggregation
As all of the experiments use washed platelets (to maintain a similar treatment of platelets for all experiments in the study), it is possible that the differences observed in inhibition may be due to the fact that the majority of fibrinogen has been removed prior to the experiment. To test for this possibility, platelets were pretreated with or without 2-MeSAMP + BAPTA-AM. Following pretreatment, the platelets were supplemented with or without 100 μg/ml fibrinogen and stimulated with thrombin, PAR1-AP, or PAR4-AP (Fig. 7, A and B). There was no observable difference in aggregation between platelets supplemented with or without fibrinogen.
FIGURE 7. Fibrinogen is not essential for PAR-mediated platelet aggregation.

Washed platelets were supplemented with or without 100 μg/ml fibrinogen after pretreatment with or without 2-MeSAMP and BAPTA-AM (n = 3). A, fibrinogen had no apparent effect on platelet aggregation mediated by thrombin, PAR1-AP, or PAR4-AP. B, there was also no observed change in the level of aggregation either with or without supplemented fibrinogen. stim, stimulated.
PAR-mediated Activation of GPIIbIIIa
Glycoprotein IIbIIIa is the integrin receptor thought to play a crucial role in platelet aggregation (30, 31). To determine what role GPIIbIIIa plays in PAR-mediated platelet aggregation, platelets were pretreated with BAPTA-AM, Me2SO (as the vehicle control), 2-MeSAMP, or BAPTA-AM and 2-MeSAMP. Following pretreatment, platelets were stimulated with PAR1-AP (Fig. 8, A–C) or PAR4-AP (Fig. 8, D–F). Surface activation of GPIIbIIIa was measured through FACS analysis with the aid of a FITC-tagged antibody that only recognizes active GPIIbIIIa (PAC1). BAPTA-AM pretreatment had no effect on PAR-mediated GPIIbIIIa activation (Fig. 8, A and D). Pretreatment with 2-MeSAMP caused a substantial decrease in the number of active GPIIbIIIa molecules following both PAR1-AP as well as PAR4-AP stimulation (Fig. 8, B and E). Similarly, dual inhibition with BAPTA-AM and 2-MeSAMP showed a decrease in the number of active GPIIbIIIa molecules following stimulation (Fig. 8, C and F). The relative decrease in GPIIbIIIa activation was similar between PAR1-AP and PAR4-AP stimulation (Fig. 8G). These data are similar to the findings that Rap1 activation is partially, but not completely, inhibited by 2-MeSAMP, whereas BAPTA-AM has relatively no effect on either Rap1 or GPIIbIIIa activation (supplemental Fig. 2).
FIGURE 8. GPIIbIIIa activation is dependent on P2Y12R activation.
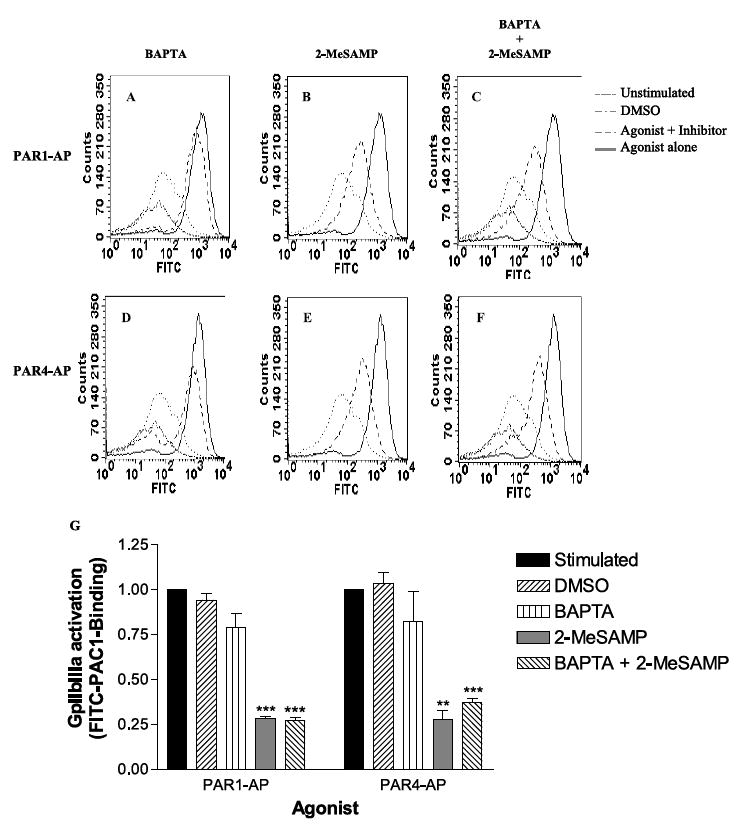
GPIIbIIIa activation was measured through FACS analysis using FITC-conjugated anti-PAC1, which only recognizes the active form of GPIIbIIIa (n = 3). Platelets were treated with either 20 μm PAR1-AP (A–C) or 200 μm PAR4-AP (D–F). Platelets were pretreated with 20 μm BAPTA-AM (A and C), 50 μm 2-MeSAMP (B and E), or both (C and F). Me2SO (DMSO) was used as a control vector for BAPTA-AM (A, C, D, and F). G, relative changes in GPIIbIIIa activation. **, p < 0.0022; ***, p < 0.0001.
DISCUSSION
This study focused on how PAR4 differs from PAR1 in the way it signals platelet aggregation. We observed that at high levels PAR4-AP-mediated aggregation, which mostly likely occurs at the site of a local thrombus, was severely attenuated following the dual inhibition of calcium mobilization and P2Y12 receptor signaling, whereas at high concentrations of thrombin and PAR1-AP signaling was unaffected. Furthermore, we showed that calcium mobilization was not required in order for thrombin, PAR1-AP, or PAR4-AP to induce Rap1 activation, GPIIbIIIa activation, or platelet aggregation. Interestingly, although Rap1 and GPIIbIIIa are thought to play important roles in platelet activation, we indicate here that other as yet undetermined components must play a critical role in PAR-mediated aggregation as well as Rap1 and GPIIbIIIa activation because none of the conditions tested were able to completely block either of these important signaling components. Hence, this report provides evidence that physiological effects observed downstream of PAR1 and PAR4 are most likely not because of redundant signaling pathways.
Increases in intra-platelet calcium mobilization were observed following thrombin, PAR1, and PAR4 stimulation (Fig. 2). Calcium is thought to play a role in Rap1 activation, secretion, and possibly aggregation; however, its requirement for these processes has not been thoroughly investigated in human platelets. Differences in the reported requirement for calcium mobilization downstream of PAR activation (24, 29) may be based on the receptor being studied as well as the concentration of agonist used in these studies. Our results indicate that because PARs signal to multiple G proteins, calcium is not required for PAR-mediated aggregation in human platelets. Agonist concentration also plays a role in platelet aggregation as local concentrations of thrombin at the site of vascular injury may be quite high. Therefore, this study investigated the effects of thrombin and PARs at high concentrations (as described in Fig. 1) to better understand PAR signaling at the site of a thrombus. Figs. 5 and 6 demonstrate that although both PAR1-AP- and PAR4-AP-induced aggregation are severely attenuated at sub-maximal concentrations of each agonist, at high concentrations (above the EC100: EC50 for PAR1-AP and PAR4-AP was 3 and 80 μm, respectively) PAR1-AP but not PAR4-AP was able to overcome this inhibition. This is in line with the general observation that at higher concentrations some agonists are able to recruit additional G protein signaling pathways. The results support the notion that calcium-dependent signaling is not required for Rap1 activation (supplemental data), and subsequent platelet aggregation as these PAR-mediated effects may involve several complex signaling pathways downstream of PAR1 and PAR4 (21).
Work on PAR4 signaling within the mouse model indicates that PAR4 may signal synergistically with P2Y12 in order to induce platelet activation (32, 33). Furthermore, it has been well documented that ADP may play a role in PAR-induced effects on both the human as well as mouse model for platelet activation (19, 21). Data presented here not only concur with this hypothesis (Fig. 4) but additionally indicate that not all PARs require this synergy, because at higher concentrations PAR1- but not PAR4-induced platelet aggregation shows little attenuation under any of the conditions studied. These findings support the hypothesis that Gαi/o, which has only been shown to couple to PAR1 and not to PAR4, may play an important role in signaling platelet aggregation, and that calcium mobilization together with Gαi/o (through P2Y12 or PAR1) activation is necessary for aggregation to occur. Blocking either calcium mobilization or P2Y12 activation had no effect on PAR4 (indicating that neither PAR4- nor P2Y12-mediated calcium activation is required for PAR4-mediated platelet activation), whereas blocking both of these signaling pathways completely inhibited the ability of PAR4-AP (even at 10-fold EC50 for PAR4-AP as indicated in Fig. 6, whereas PAR1-AP fully responds to the dual inhibition at 6-fold its EC50 as indicated in Fig. 5), but not thrombin or PAR1-AP, to induce platelet aggregation. It is difficult to directly assess the involvement of Gαi/o as a signaling partner in platelets because there is no mechanism by which pertussis toxin can move to the intra-platelet surface to inhibit Gαi/o activation. Some published work in the field indicating that PAR1 does not signal through Gαi/o relies on the indirect downstream measurement of cAMP levels to determine Gαi/o activation (34). It has been well established that inhibition of cAMP is not the only pathway in which Gi/o signals its effects. For example, Gαo can regulate Rap1 through Rap1GAP activity (35, 36). Furthermore, the βγ subunits of Gi/o have been shown to induce multiple downstream signaling pathways that are independent of cAMP levels (37, 38). Hence, we suggest that the critical Gαi/o component may arise through PAR1 stimulation directly at higher levels of PAR1-AP, but in PAR4 signals through secretory feedback and activation of P2Y12 (a receptor which specifically signals through Gαi/o).
Under no conditions tested was Rap1 or GPIIbIIIa activation completely blocked, indicating that although P2Y12 activation plays an important upstream regulatory role for Rap1 activation (29), other PAR-mediated signaling pathways must regulate Rap1 activation independent of P2Y12 (16, 36, 39, 40). A recent finding that Rap1 regulates cross-talk between several integrins and is regulated itself by these receptors further illustrates the complexity involved in Rap1 signaling in platelets (41). Our findings give strong evidence that there is a limited correlation between GPIIbIIIa activity and platelet aggregation because partial inhibition of GPIIbIIIa following 2-MeSAMP resulted in a minimal attenuation of platelet aggregation.
PAR1 has a higher affinity for thrombin than PAR4 and thus is most likely the first PAR activated when thrombin is formed (10, 42). Because PAR4 signaling is prolonged (43), it is thought to play a role in the late phase of the platelet aggregation (8, 20), establishing a differential physiological function for the two receptors and indicating a potentially crucial role for PAR4 signaling in mediating irreversible platelet activation resulting in subsequent occlusion of the vessel. Furthermore, PAR1, but not PAR4, has been shown to couple to the Gαi/o family of G proteins (11, 13). Our findings provide direct evidence that PAR1 and PAR4 signaling differ not only in their efficacy to thrombin but also in their individual signaling networks. With this in mind, it is plausible to hypothesize that PAR1 activation at low concentrations of thrombin is necessary to form the initial platelet clot following vascular injury. The lower PAR4 affinity for thrombin may indicate that PAR4 is activated within the local environment of the initial platelet plug and plays a role in the late (irreversible) phase of platelet activation. If this is the case, modulation of PAR4 activity may play an essential role in regulating the probability of vessel occlusion following a vascular injury. P2Y12 receptor antagonists (clopidogrel and ticlopidine) are currently in clinical use. The CAPRIE study indicated that clopidogrel in particular significantly decreased the incidence of vascular occlusive diseases associated with platelet activation, such as myocardial infarction and ischemic stroke (44, 45), as well as reocclusion following percutaneous coronary intervention (46, 47). Furthermore, other studies indicate that clopidogrel was more effective than treatment with aspirin alone (44-46, 48). However, under all of the conditions tested in these clinical studies, thrombin maintained the ability to signal platelet aggregation indicating that complex signaling mediated through multiple signaling pathways is involved. Our data indicate that the multiple receptor activation observed following thrombin is not redundant. Furthermore, our findings elucidate which signaling pathways may be critical for platelet activation. We showed that signaling through the P2Y12 but not P2Y1 receptor plays at least a partial role in PAR4-mediated signaling, whereas no observable dependence on P2Y12 was measured following stimulation with maximal concentrations of PAR1-AP (20 μm). Therefore, inhibition of signaling by ADP through P2Y12 may function to affect platelet aggregation by attenuating the PAR4 signaling pathway (33) while having little effect on PAR1-mediated thrombin signaling. If this is the case, a specific inhibitor of PAR4 signaling may result in a more desirable side effect profile as compared with clopidogrel. Future studies will be aimed at elucidating at what point in the signaling pathway calcium converges downstream of PAR4 with the P2Y12 signaling pathway as this may identify a better potential site for therapeutic intervention.
We have proposed a model of how thrombin induces human platelet activation through its receptors (Fig. 9). In this model, thrombin fully activates both PAR1 and PAR4 following vascular injury. PAR1 (signaling through Gα12, Gαq, and Gαi/o) and PAR4 (signaling through Gα12 and Gαq) both induce secretion of the α-granules and dense granules, Rap1, and GPIIbIIIa activation, as well as platelet aggregation. PAR1 does not directly or indirectly require P2Y12 receptor activation because PAR1 directly activates Gαi/o. PAR4 signaling, however, converges downstream with P2Y12 signaling to mediate platelet aggregation. Although our data, as well as others’, indicate that P2Y12 receptor activation is important for Rap1 activation, inhibition of the P2Y12 receptor does not fully eliminate PAR-mediated Rap1 activation indicating another mechanism for partial activation of Rap1 and GPIIbIIIa. These findings are the first to identify how PAR1 signaling differs from that of PAR4 (8, 10, 11, 20, 49). Furthermore, PAR4-mediated intra-platelet calcium mobilization plays a role in ADP activation of purinergic receptors and is thought to be partially regulated by PAR1 (20). These data, combined with our current findings, identify a potential physiological link between the complex activity observed in platelet activation (1, 7, 20, 21) and the mechanism by which thrombin regulates platelet activity (through PAR1 and PAR4).
FIGURE 9. Model of thrombin-induced platelet activation through PAR1 and PAR4.
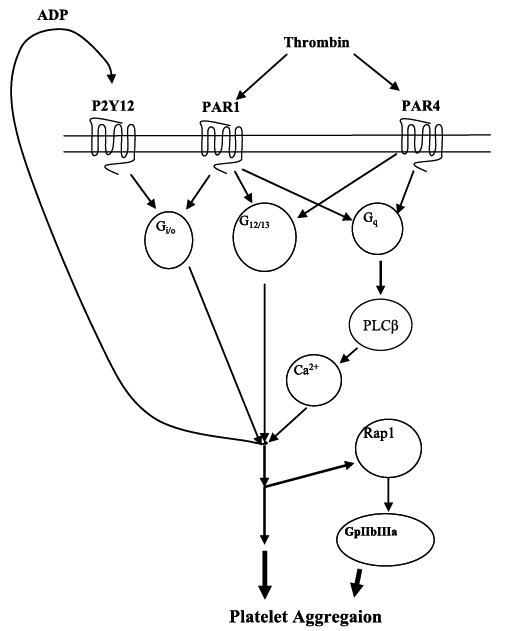
Thrombin activates both PAR1 and PAR4 following vascular injury. PAR1 activates Gαi/o, Gαq, and Gα12/13, whereas PAR4 activates Gαq and Gα12/13. All three of these G proteins play a role in platelet aggregation. Additionally, through secretion of the dense granules, ADP is released and acts as an autocrine signal on the P2Y12 receptor. PAR4 signaling calcium mobilization downstream of phospholipase Cβ through the Gαq pathway converges with signaling through the P2Y12 pathway to induce platelet aggregation. Finally, Rap1 activation is partially dependent on P2Y12 signaling but may also be activated directly through PAR1 or PAR4 most likely through signaling in the Gαq or Gα12/13 pathway.
Supplementary Material
Acknowledgments
We thank Connie Childers for coordinating blood donations for this study. We also thank Drs. John Oates and David Adler for a critical reading of the manuscript and helpful discussion of the results.
Footnotes
This work was supported by National Institutes of Health Grants HL084388 and EY010291 (to H. E. H.) and National Research Service Awards HL-076133-02 and HL-082068-01 (to M. H.).
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1 and 2.
The abbreviations used are: PAR, protease-activated receptor; BAPTA-AM, 1,2-Bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetra(acetoxymethyl) ester; 2-MeSAMP, 2-methylthioadenosine monophosphate; Ab, antibody; FITC, fluorescein isothiocyanate; FACS, fluorescence-activated cell sorting.
References
- 1.Rosenberg RD, Aird WC. N Engl J Med. 1999;340:1555–1564. doi: 10.1056/NEJM199905203402007. [DOI] [PubMed] [Google Scholar]
- 2.Mosesson MW. J Thromb Haemostasis. 2005;3:1894–1904. doi: 10.1111/j.1538-7836.2005.01365.x. [DOI] [PubMed] [Google Scholar]
- 3.Laposata M, Dovnarsky DK, Shin HS. Blood. 1983;62:549–556. [PubMed] [Google Scholar]
- 4.Jamieson GA. Thromb Haemostasis. 1997;78:242–246. [PubMed] [Google Scholar]
- 5.Davey MG, Luscher EF. Nature. 1967;216:857–858. doi: 10.1038/216857a0. [DOI] [PubMed] [Google Scholar]
- 6.Espana F, Medina P, Navarro S, Zorio E, Estelles A, Aznar J. Curr Med Chem. 2005;3:119–131. doi: 10.2174/1568016053544336. [DOI] [PubMed] [Google Scholar]
- 7.Brass LF. Chest. 2003;124:S18–S25. [Google Scholar]
- 8.Ossovskaya VS, Bunnett NW. Physiol Rev. 2004;84:579–621. doi: 10.1152/physrev.00028.2003. [DOI] [PubMed] [Google Scholar]
- 9.Vu TK, Hung DT, Wheaton VI, Coughlin SR. Cell. 1991;64:1057–1068. doi: 10.1016/0092-8674(91)90261-v. [DOI] [PubMed] [Google Scholar]
- 10.Kahn ML, Zheng YW, Huang W, Bigornia V, Zeng D, Moff S, Farese RV, Jr, Tam C, Coughlin SR. Nature. 1998;394:690–694. doi: 10.1038/29325. [DOI] [PubMed] [Google Scholar]
- 11.Coughlin SR. Proc Natl Acad Sci U S A. 1999;96:11023–11027. doi: 10.1073/pnas.96.20.11023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Macfarlane SR, Seatter MJ, Kanke T, Hunter GD, Plevin R. Pharmacol Rev. 2001;53:245–282. [PubMed] [Google Scholar]
- 13.Faruqi TR, Weiss EJ, Shapiro MJ, Huang W, Coughlin SR. J Biol Chem. 2000;275:19728–19734. doi: 10.1074/jbc.M909960199. [DOI] [PubMed] [Google Scholar]
- 14.Scase TJ, Heath MF, Evans RJ. Blood. 1997;90:2113–2114. [PubMed] [Google Scholar]
- 15.Ishihara H, Connolly AJ, Zeng D, Kahn ML, Zheng YW, Timmons C, Tram T, Coughlin SR. Nature. 1997;386:502–506. doi: 10.1038/386502a0. [DOI] [PubMed] [Google Scholar]
- 16.Chrzanowska-Wodnicka M, Smyth SS, Schoenwaelder SM, Fischer TH, White GC., II J Clin Investig. 2005;115:680–687. doi: 10.1172/JCI22973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reed GL. Semin Thromb Hemostasis. 2004;30:441–450. doi: 10.1055/s-2004-833479. [DOI] [PubMed] [Google Scholar]
- 18.Dorsam RT, Kunapuli SP. J Clin Investig. 2004;113:340–345. doi: 10.1172/JCI20986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dorsam RT, Tuluc M, Kunapuli SP. J Thromb Haemostasis. 2004;2:804–812. doi: 10.1111/j.1538-7836.2004.00692.x. [DOI] [PubMed] [Google Scholar]
- 20.Covic L, Gresser AL, Kuliopulos A. Biochemistry. 2000;39:5458–5467. doi: 10.1021/bi9927078. [DOI] [PubMed] [Google Scholar]
- 21.Graff J, Klinkhardt U, Harder S. Thromb Res. 2004;113:295–302. doi: 10.1016/j.thromres.2004.03.014. [DOI] [PubMed] [Google Scholar]
- 22.Maroo A, Lincoff AM. Semin Thromb Hemostasis. 2004;30:329–336. doi: 10.1055/s-2004-831045. [DOI] [PubMed] [Google Scholar]
- 23.Ali A, Hashem M, Rosman HS, Moser L, Rehan A, Davis T, Romanelli M, LaLonde T, Yamasaki H, Barbish B, Michael J, Ali SA, Schreiber TL, Gardin JM. J Clin Pharmacol. 2004;44:1328–1332. doi: 10.1177/0091270004269559. [DOI] [PubMed] [Google Scholar]
- 24.Franke B, Akkerman JW, Bos JL. EMBO J. 1997;16:252–259. doi: 10.1093/emboj/16.2.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Triest M, de Rooij J, Bos JL. Methods Enzymol. 2001;333:343–348. doi: 10.1016/s0076-6879(01)33068-9. [DOI] [PubMed] [Google Scholar]
- 26.Shattil SJ, Cunningham M, Hoxie JA. Blood. 1987;70:307–315. [PubMed] [Google Scholar]
- 27.Adam F, Verbeuren TJ, Fauchere JL, Guillin MC, Jandrot-Perrus M. J Thromb Haemostasis. 2003;1:798–804. doi: 10.1046/j.1538-7836.2003.00138.x. [DOI] [PubMed] [Google Scholar]
- 28.Crittenden JR, Bergmeier W, Zhang Y, Piffath CL, Liang Y, Wagner DD, Housman DE, Graybiel AM. Nat Med. 2004;10:982–986. doi: 10.1038/nm1098. [DOI] [PubMed] [Google Scholar]
- 29.Woulfe D, Jiang H, Mortensen R, Yang J, Brass LF. J Biol Chem. 2002;277:23382–23390. doi: 10.1074/jbc.M202212200. [DOI] [PubMed] [Google Scholar]
- 30.Boekholdt SM, Peters RJ, de Maat MP, Zwinderman AH, van Der Wallqq EE, Reitsma PH, Jukema JW, Kastelein JJ. Am Heart J. 2004;147:181–186. doi: 10.1016/j.ahj.2003.07.008. [DOI] [PubMed] [Google Scholar]
- 31.Fuster V, Badimon L, Badimon JJ, Chesebro JH. N Engl J Med. 1992;326:310–318. doi: 10.1056/NEJM199201303260506. [DOI] [PubMed] [Google Scholar]
- 32.Woulfe D, Jiang H, Morgans A, Monks R, Birnbaum M, Brass LF. J Clin Investig. 2004;113:441–450. doi: 10.1172/JCI20267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van der Meijden PE, Feijge MA, Giesen PL, Huijberts M, van Raak LP, Heemskerk JW. Thromb Haemostasis. 2005;93:1128–1136. doi: 10.1160/TH04-09-0597. [DOI] [PubMed] [Google Scholar]
- 34.Kim S, Foster C, Lecchi A, Quinton TM, Prosser DM, Jin J, Cattaneo M, Kunapuli SP. Blood. 2002;99:3629–3636. doi: 10.1182/blood.v99.10.3629. [DOI] [PubMed] [Google Scholar]
- 35.Schultess J, Danielewski O, Smolenski AP. Blood. 2005;105:3185–3192. doi: 10.1182/blood-2004-09-3605. [DOI] [PubMed] [Google Scholar]
- 36.Bertoni A, Tadokoro S, Eto K, Pampori N, Parise LV, White GC, Shattil SJ. J Biol Chem. 2002;277:25715–25721. doi: 10.1074/jbc.M202791200. [DOI] [PubMed] [Google Scholar]
- 37.Hamm HE. J Biol Chem. 1998;273:669–672. doi: 10.1074/jbc.273.2.669. [DOI] [PubMed] [Google Scholar]
- 38.Clapham DE, Neer EJ. Annu Rev Pharmacol Toxicol. 1997;37:167–203. doi: 10.1146/annurev.pharmtox.37.1.167. [DOI] [PubMed] [Google Scholar]
- 39.Larson MK, Chen H, Kahn ML, Taylor AM, Fabre JE, Mortensen RM, Conley PB, Parise LV. Blood. 2003;101:1409–1415. doi: 10.1182/blood-2002-05-1533. [DOI] [PubMed] [Google Scholar]
- 40.Franke B, van Triest M, de Bruijn KM, van Willigen G, Nieuwenhuis HK, Negrier C, Akkerman JW, Bos JL. Mol Cell Biol. 2000;20:779–785. doi: 10.1128/mcb.20.3.779-785.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bernardi B, Guidetti GF, Campus F, Crittenden JR, Graybiel AM, Balduini C, Torti M. Blood. 2006;107:2728–2735. doi: 10.1182/blood-2005-07-3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu WF, Andersen H, Whitmore TE, Presnell SR, Yee DP, Ching A, Gilbert T, Davie EW, Foster DC. Proc Natl Acad Sci U S A. 1998;95:6642–6646. doi: 10.1073/pnas.95.12.6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murugappan S, Shankar H, Bhamidipati S, Dorsam RT, Jin J, Kunapuli SP. Blood. 2005;106:550–557. doi: 10.1182/blood-2004-12-4866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tendera M, Wojakowski W. Thromb Res. 2003;110:355–359. doi: 10.1016/j.thromres.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 45.CAPRIE Steering Committee. Lancet. 1996;348:1329–1339. doi: 10.1016/s0140-6736(96)09457-3. [DOI] [PubMed] [Google Scholar]
- 46.Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK. N Engl J Med. 2001;345:494–502. doi: 10.1056/NEJMoa010746. [DOI] [PubMed] [Google Scholar]
- 47.Leon MB, Baim DS, Popma JJ, Gordon PC, Cutlip DE, Ho KK, Giambartolomei A, Diver DJ, Lasorda DM, Williams DO, Pocock SJ, Kuntz RE. N Engl J Med. 1998;339:1665–1671. doi: 10.1056/NEJM199812033392303. [DOI] [PubMed] [Google Scholar]
- 48.Steinhubl SR, Berger PB, Mann JT, III, Fry ET, DeLago A, Wilmer C, Topol EJ. J Am Med Assoc. 2002;288:2411–2420. doi: 10.1001/jama.288.19.2411. [DOI] [PubMed] [Google Scholar]
- 49.Coughlin SR. Nature. 2000;407:258–264. doi: 10.1038/35025229. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.